
Abstract
Background
High-titer anti-interferon (IFN)-γ autoantibodies are strongly associated with intracellular pathogens such as nontuberculous mycobacteria and Talaromyces marneffei, but they are not as commonly associated with Talaromyces marneffei co-infected with Mycobacterium tuberculosis.
Case presentation
Herein, we report a case of an HIV-negative Chinese man with a severe, disseminated co-infection of Talaromyces marneffei and Mycobacterium tuberculosis, who had a high-titer of anti IFN-γ autoantibodies and a CFI heterozygous nonsense gene mutation. The patient rapidly developed sepsis and died. Through by flow cytometry for CD4+ T cells’ intracellular phosphorylated STAT-1 and Th1 cells (CD4+ IFN-γ+ cells), we found that the patient’s serum can inhibited IFN γ-induced CD4+ T cells’ STAT-1 phosphorylation and Th1 cell differentiation in normal peripheral blood mononuclear cells, but this phenomenon was not observed in normal control’s serum. In addition, the higher serum concentration in the culture medium, the more obvious inhibition of Th1 cell differentiation.
Conclusions
For HIV-negative individuals with relapsing, refractory, fatal double or multiple intracellular pathogen infections, especially Talaromyces marneffei, clinicians should be aware that if they might be dealing with adult-onset immunodeficiency syndrome due to high-titer anti-IFN-γ autoantibodies. Systematic genetic and immunological investigations should also be performed.
Similar content being viewed by others


Background
Adult-onset immunodeficiency syndrome due to high-titer anti-interferon (IFN)-γ autoantibodies is considered to be a susceptibility factor for intracellular pathogens infection, especially nontuberculous mycobacteria and Talaromyces marneffei in Southeast Asia [1,2,3]. However, the specific mechanism of immune deficiency of anti-IFN-γ autoantibodies is still unclear. In addition, anti-IFN-γ autoantibodies have not widely been cognizant in tuberculosis [4]. Thus, we report a case of a 56-year-old HIV-negative Chinese man, with a high levels of anti-IFN-γ autoantibodies, simultaneously diagnosed disseminated Mycobacterium tuberculosis and Talaromyces marneffei co-infection by using metagenomics next-generation sequencing (mNGS), aims to attract clinical attention and attempts to study the possible immune deficiency mechanism of anti-IFN-γ autoantibodies.
Case presentation
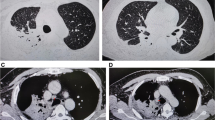
A 56-year-old Chinese man with coronary atherosclerotic heart disease was admitted to the local hospital on June 22, 2019 for a 4-month period due to expectoration, fever (body temperature: 39–40 °C), weight loss, and multiple lymphadenopathy. The patient also had significantly increased white blood cell (WBC) and neutrophil (N) counts as well as an increased erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) and procalcitonin (PCT) levels (Fig. 1A). He was nonresponsive to intermittent antibacterial therapy for 2 months (comprising cefoperazone sulbactam and moxifloxacin), and his condition deteriorated due to neck, armpit, and groin lymphadenopathy, jaundice, and proteinuria. Lymphocyte subset counts and percentages were normal. The patient had low levels of circulating factor I at 43% (normal value: 70–120%), but normal complement C3 and C4 levels. Thrombocytopenia, jaundice, and acute renal failure rapidly manifested later (Fig. 1A). HIV, antinuclear antibody, and anti-double stranded DNA tests were all negative. Chest computerized tomography (CT) revealed bilateral pulmonary infiltration with mediastinal lymphadenopathy, multiple bone destruction, and pleural and pericardial effusions (Fig. 1B). The emission CT showed a significantly increased uptake in multiple bones (Fig. 1C). A yellowish pleural effusion and its exudative manifestations and a marked increase in protein content and predominantly high levels of neutrophils (85%) were observed. The concentration of pleural fluid adenosine deaminase was 3.4 U/L. Histopathology of the lymph node and pulmonary lesions revealed granulomatous inflammation. Furthermore, no evidence of organisms or malignancy was identified in the bronchoscopy alveolar lavage fluid (BALF), blood, pleural effusion, bone marrow, lymphatic, or lung tissues when tested using microbial smears (negative acid-fast bacilli), cultures, and pathological examinations. However, Mycobacterium tuberculosis (TB) and Talaromyces marneffei were identified using Metagenomic next-generation sequencing (mNGS) [5] from the bronchoscopy analysis of the alveolar lavage fluid and cervical lymph nodes. The sputum and bone marrow were also analyzed for pathogen cultures using mNGS. Following a 3-day regimen of anti-tuberculosis treatment and antifungal therapy (amphotericin B liposome combined with voriconazole), the patient died of septic shock. On the 3rd day after his death, Talaromyces marneffei was isolated from the sputum and Mycobacterium tuberculosis was identified in the bone marrow using mNGS.

A Inflammatory markers, liver, and kidney function: white blood cell (WBC) and neutrophil (N) counts, and C-reactive protein (CRP), procalcitonin (PCT), urea and total bilirubin levels rapidly increased; platelets (PLT), hemoglobin (HGB) level, and creatinine clearance decreased rapidly. B Computed tomography dynamic monitoring series: pulmonary lesions, pleural effusions, pericardial effusions and osteolysis. C Emission computed tomography: significantly increased uptake in multiple ribs and vertebrae, left sacroiliac spine, and left acetabulum. D The patient’s anti-IFN-γ autoantibody titer increased significantly as the condition worsened during the disease course. E Multiplex screening of serum from the patient and 7 normal control plasmas for cytokines and anti-IFN-γ autoantibodies. F Pedigree tree. Whole-genome sequencing of the proband (patient) and his mother revealed a heterozygous nonsense mutation (c.559 C>T; p. Arg187*) in CFI
The patient, his mother, his two healthy daughters, and seven healthy controls, were recruited from the First Affiliated Hospital of Guangxi Medical University between June 2019 and July 2019. All subjects provided written informed consent. This study was approved by the Ethical Review Committee of the First Affiliated Hospital of Guangxi Medical University (2020.KY-E-032). Detailed methods for Anti-IFN-γ autoantibody assay, Bio-Plex™ 25 cytokine assay, flow cytometry, DNA extraction, DNA libraries and sequencing, and bioinformatics analysis are provided in Additional file 1.
The patient’s serum showed a remarkably high-titer of anti-IFN-γ autoantibody, and the titer increased significantly as the condition worsened during the course of the disease. The patient’s serum IL-17 A, IL-2, IL-6, IL-8, TNF-α, MIP-1α, MIP-1β, MCP-1, G-CSF, IL-10, and IL-1Rα levels were also increased significantly than the seven healthy controls’ (Fig. 1D). However, the IFN-γ levels were in the same range as those in the healthy controls. Whole exome sequencing (WES) of the proband and his mother revealed a heterozygous CFI nonsense mutation (c.559 C>T; p. Arg187*; Figs. 1F and 2). The CFI gene (CFI, OMIM*217030) comprises 13 exons localized on chromosome 4q25 and its deficiency is inherited in an autosomal recessive manner. WES verification revealed that the patient’s mother was also a carrier of the heterozygous variant.

Whole-exome sequencing indicate a CFI heterozygous nonsense gene mutation (c.559 C>T; p. Arg187*) found in the patient and his mother
Through by flow cytometry for CD4+ T cells’ intracellular phosphorylated STAT-1 and Th1 cells (CD4+ IFN-γ+ cells), we found that the patient’s serum can remarkably inhibited IFN γ-induced CD4+ T cells’ STAT-1 phosphorylation and Th1 cell differentiation in normal peripheral blood mononuclear cells, but this phenomenon was not observed in normal control’s serum. However, when the patient PBMCs were washed free of the patient’s serum, they demonstrated normal IFN γ-induced STAT-1 phosphorylation in CD4+ T cells and Th1 cell differentiation (Fig. 3). In addition, the higher serum concentration in the culture medium, the more obvious inhibition of Th1 cell differentiation.

The expression of IFN-γ+ CD4+ T (Th1), and pSTAT-1+CD4+ T (CD4+ T cells intracellular phosphorylated STAT-1) cells in peripheral blood by Flow cytometry (n = 4). A Lymphocytes were identified based on their characteristic properties shown in by FSC and SSC. CD4+ T cells were gated from lymphocytes and total numbers of CD4+T cells were more than 25,000. B The dot plots of CD4+ T pSTAT-1 staining (CD4+ T cells intracellular phosphorylated STAT-1) cells in peripheral blood before and after stimulated with 1000 U/mL IFN γ and non-stimulated condition. C The histogram of CD4+ T pSTAT-1 staining (CD4+ T cells intracellular phosphorylated STAT-1) cells in peripheral blood before and after stimulated with 1000 U/mL IFN γ and non-stimulated condition. D The representative flow cytometric dot plots of IFN-γ+ CD4+ T cells (Th1). E The CD4+ T cells intracellular phosphorylated STAT-1 under stimulated with 1000 U/mL IFN γ and non-stimulated in three condition. F The CD4+ T cells intracellular phosphorylated STAT-1 under stimulated with 1000 U/mL IFN γ. G Th1 cells in the normal PBMCs and patients PBMCs co-culture with patient or normal serum. The patient’s serum but not the control serum inhibited IFN γ-induced CD4+ T cells STAT-1 phosphorylation in the normal peripheral blood mononuclear cells (PBMCs), whereas when the patient’s PBMCs were washed free of autologous serum they demonstrated normal IFN γ-induced STAT-1 phosphorylation. The patient but not control serum inhibited Th1 cell differentiation in the normal PBMCs. Data are expressed as median with interquartile range. n = 4 for each group. Statistical comparisons were made using the Kruskal–Wallis test followed by Bonferroni test. *P < 0.05. **P < 0.01. FSC forward scatter, SSC side scatter
Discussion and conclusion
A high-titer of anti-IFN-γ autoantibodies is strongly associated with intracellular pathogens especially in Nontuberculous mycobacteria and Talaromyces marneffei [1]. Cao et al. found that there was a high prevalence of neutralizing anti-IFN-γ autoantibodies (94.8%) in 58 HIV-negative adults with severe Talaromyces marneffei infections who were otherwise healthy [5]. In our previously researches, we found that it showed that adult-onset immunodeficiency syndrome, due to high-titer of anti-IFN-γ autoantibodies were the most common underlying immunodeficiency in HIV-negative Talaromyces marneffei infection patients [6]. In addition, the ratio of actual value to cut-off value of anti-IFN-γ autoantibodies was an independent risk factor of TM infection [2]. However, the immune deficiency mechanism of anti-IFN-γ autoantibodies in Talaromyces marneffei infection is less clear.
Lightly increased levels of anti-IFN-γ autoantibodies were found in patients with severe pulmonary tuberculosis [1]. However, the neutralization or biological activity of lightly increased levels of anti-IFN-γ autoantibodies in these pulmonary tuberculosis were not proved [7]. Meanwhile, remarkable high titer of anti-IFN-γ autoantibodies found in tuberculosis is rare reported. Before our case, there was only one Thai woman with a disseminated tuberculosis single infection and a dramatic paradoxical inflammatory response after treatment initiation, was found to have a high-titer of neutralizing anti-IFN-γ autoantibodies [4]. However, our patient had disseminated tuberculosis and a TM co-infection combined with a high level of anti-IFN-γ autoantibodies, phenomena that triggered an inflammatory storm, resulting in rapid death. Moreover, our patient’s serum could inhibit IFN γ-induced CD4+ T cells’ STAT-1 phosphorylation and Th1 cell differentiation in normal PBMCs. These observations suggest that the immunodeficiency mechanism of the anti-IFN-γ autoantibody may include inhibition of the CD4+ T cells’ IFN-γ/pSTAT-1/Th1 pathway, ultimately leading to a severely compromised Th1 response. Thus, Th1 cells immunodeficiency due to anti-IFN-γ autoantibodies was the cause of the severe and fatal multiple intracellular pathogen infections in this HIV-negative adult. Furthermore, the levels of proinflammatory cytokines were increased significantly in our patient, especially IL-17 A, IL-6, IL-8, TNF-α, G-CSF, and IL-10.
Browne, et al. identified a major epitope using anti-interferon-γ autoantibodies in patients with mycobacterial disease that showed a homology to the Aspergillus protein and found that anti-IFN-γ autoantibodies cross-reacted with Aspergillus and Mycobacterium intracellulare Noc2 [8]. These results suggest that when a patient is infected with TM and TB, these forms of molecular mimicry may trigger the production of these autoantibodies. Thus, the anti-IFN-γ autoantibody titer increased significantly as the condition worsened during the disease course.
In conclusion, clinicians should be aware that HIV-negative individuals with relapsing, refractory, fatal multiple intracellular pathogen infections, may have multiple immunodeficiencies, including primary immunodeficiency and/or secondary immunodeficiency, that are unrecognized. Thus, systematic genetic testing and immunological investigations should be performed for such patients.
Availability of data and materials
The DNA sequencing of the patient’s data were submitted to the NCBI GenBank database (The GenBank accession number for the patient’s nucleotide sequence: OL537177). The other data generated or analyzed during this study are included in this published article.
Abbreviations
- IFN-γ:
-
Interferon-γ
- PBMCs:
-
Peripheral blood mononuclear cells
- mNGS:
-
Metagenomics next-generation sequencing
- WBC:
-
White blood cell
- N:
-
Neutrophil
- ESR:
-
Erythrocyte sedimentation rate
- CRP:
-
C-reactive protein
- PCT:
-
Procalcitonin
- CT:
-
Computerized tomography
- BALF:
-
Bronchoscopy alveolar lavage fluid
- EDTA:
-
Ethylene diamine tetraacetic acid
- PBS:
-
Phosphate buffer saline
- ELISA:
-
Enzyme-linked immunosorbent assay
- MFI:
-
Mean fluorescent intensity
- PMA:
-
Phorbol myristate acetate
- DNB:
-
DNA Nanoballs
- PMDB:
-
Pathogens Metagenomics Database
- WES:
-
Whole exome sequencing
References
Browne SK, Burbelo PD, Chetchotisakd P, et al. Adult-onset immunodeficiency in Thailand and Taiwan. N Engl J Med. 2012;367(8):725–34.
Zeng W, Qiu Y, Tang S, Zhang J, Pan M, Zhong X. Characterization of Anti-interferon-gamma antibodies in hiv-negative patients infected with disseminated Talaromyces marneffei and Cryptococcosis. Open Forum Infect Dis. 2019;6(10):ofz208.
Qiu Y, Huang J, Li Y, et al. Talaromyces marneffei and nontuberculous mycobacteria co-infection in HIV-negative patients. Sci Rep. 2021;11(1):16177.
Xie YL, Rosen LB, Sereti I, et al. Severe paradoxical reaction during treatment of disseminated tuberculosis in a patient with neutralizing anti-IFNγ autoantibodies. Clin Infect Dis. 2016;62(6):770–3.
Guo J, Ning XQ, Ding JY, et al. Anti-IFN-γ autoantibodies underlie disseminated Talaromyces marneffei infections. J Exp Med. 2020;217(12):e20190502.
Qiu Y, Feng X, Zeng W, et al. Immunodeficiency disease spectrum in HIV-negative individuals with Talaromycosis. J Clin Immunol. 2021;41(1):221–3.
Madariaga L, Amurrio C, Martín G, et al. Detection of anti-interferon-gamma autoantibodies in subjects infected by Mycobacterium tuberculosis. Int J Tuberc Lung Dis. 1998;2:62–8.
Lin CH, Chi CY, Shih HP, et al. Identification of a major epitope by anti-interferon-gamma autoantibodies in patients with mycobacterial disease. Nat Med. 2016;22(9):994–1001.
Acknowledgements
The authors thank Meng Li, Professor of Microbiology, Department of Microbiology laboratory, the First Affiliated Hospital of Guangxi Medical University.
Funding
This work was supported by Grants from the Natural Science Foundation of China [NSFC81760010 and 82060364] and the Science and Technology Department of Guangxi Zhuang Autonomous Foundation of Guangxi Key Research and Development Program (No. GuikeAB20238025) from Jianquan Zhang for analysis and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
JZ conceived of the study, critically revised the manuscript for important intellectual content. YQ conceived and designed the study, acquired, analyzed, and interpreted the data; and drafted the manuscript. MP, ZY and WZ analyzed and interpreted the data. HZ conceived of the study and modified the manuscript. ZL acquired and analyzed the data and agreed to be accountable for all aspects of the work for ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethical Review Committee of the First Affiliated Hospital of Guangxi Medical University (2020.KY-E-023). As the patient has died, written informed consent was obtained from the patients’ daughter. Copies of the written consents are available for review.
Consent for publication
The patient’s next-of-kin gave written consent for their relative’s personal or clinical details along with any identifying images to be published in this study.
Competing interests
All authors declare that there are no reported conflicts of interest. All authors had access to the data and a role in writing the manuscript. All authors have read and approved the manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Detailed methods for Anti-IFN-γ autoantibody assay, Bio-Plex™ 25 cytokine assay, flow cytometry, DNA extraction, DNA libraries and sequencing, and bioinformatics analysis are provided in Additional file.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qiu, Y., Pan, M., Yang, Z. et al. Talaromyces marneffei and Mycobacterium tuberculosis co-infection in a patient with high titer anti-interferon-γ autoantibodies: a case report. BMC Infect Dis 22, 98 (2022). https://doi.org/10.1186/s12879-021-07015-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-021-07015-5