
Abstract
Background
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome of excessive inflammation. We aimed to describe the clinical and laboratory findings of HLH patients secondary to Visceral leishmaniasis (VL) and their treatment outcome during a 4-year follow-up period compared to primary HLH.
Method
Forty children with primary HLH confirmed by genetic study and 20 children with HLH secondary to VL confirmed by a blood or bone marrow polymerase chain reaction from 2014 to 2018 in Shiraz, Fars province, Southern Iran, were enrolled.
Results
The median age at diagnosis was 11.5 months (range 1–170), and 56.7% were male. Fever and splenomegaly were the most frequent clinical presentations. 93.3% of the subjects had an HScore > 169, which had a good correlation with HLH-2004 criteria (r = 0.371, P = 0.004). Patients with primary HLH experienced more thrombocytopenia (P = 0.012) and higher alanine transaminase (P = 0.016), while patients with VL-associated HLH had higher ferritin (P = 0.034) and erythrocyte sedimentation rate (P = 0.011). Central nervous system (CNS) involvement occurred in 38.3% of patients. The mortality rate was higher in patients with CNS disease (61% vs. 35%, P = 0.051). The 3-yr overall survival rate was 35.9%. (24% in primary HLH and 100% in VL-associated HLH, P < 0.001). In Cox regression analysis, platelet count < 100,000/ μ l (hazard ratio 4.472, 95% confidence interval 1.324–15.107, P = 0.016) correlated with increased mortality in patients with primary HLH.
Conclusion
VL is a potential source of secondary HLH in regions with high endemicity. Treatment of the underlying disease in VL-associated HLH is sufficient in most cases, with no need to start etoposide-based chemotherapy.
Similar content being viewed by others

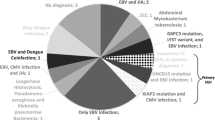

Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome of excessive inflammation and tissue destruction due to abnormal immune activation. The absence of normal downregulation by activated macrophages and lymphocytes is possibly the cause of hyper inflammation and dysregulated immune state [1]. Infants are most commonly affected, with the highest incidence in those younger than 3 months [2].
HLH presents as a febrile illness associated with multiple organ involvement. Thus, initial signs and symptoms of HLH can mimic common infections, fever of unknown origin, hepatitis, or encephalitis [3]. Therefore, the diagnosis of HLH is primarily based on fulfilling at least 5 out of 8 criteria based on the published diagnostic criteria used in the HLH-2004 trial [4].. Similarly, homozygosity or compound heterozygosity for verified HLH-associated mutations will confirm the diagnosis [5]. Moreover, a scoring system has been developed to generate a diagnostic score referred to as “HScore,” which estimates the probability of HLH (Table 1). A score higher than 169 was shown to predict the risk of HLH with 93% sensitivity and 86% specificity [6].
On the other hand, secondary (sporadic, acquired) HLH is generally used to describe patients without a known familial mutation who typically have a clear trigger for developing HLH. It usually follows an infectious disease caused by organisms, including Epstein-Barr virus (EBV), cytomegalovirus (CMV), hepatitis viruses, varicella infection, leishmania, Mycobacterium tuberculosis, among many others. Moreover, autoimmune and rheumatologic disorders, malignancies, and metabolic disorders may trigger the cytokine storm [7].
A few small case series of primary or secondary HLH have been reported in Iran [8,9,10]. Additionally, despite a high prevalence of Visceral leishmaniasis (VL) in Southern Iran [11], there are few reports of VL-associated HLH in our region [12, 13]. Therefore, in this prospective study, we aimed to describe the clinical and laboratory findings and the outcome of children diagnosed with VL-associated HLH during a 4-year follow-up period compared to patients with primary HLH.
Methods and materials
Study population and data acquisition
This prospective observational study was conducted in a tertiary oncology referral center in Shiraz, Fars province in Southern Iran, 2014–2018. During the study period, patients with a confirmed diagnosis of HLH were included. HLH was diagnosed based on the diagnostic criteria proposed by the Histiocyte Society in 2004 [14].
Patients with a positive family history or with disease-causing mutations in the genes encoding perforin (FHL2), Munc 13–4 (FHL3), Syntaxin 11 (FHL4), Munc 18–2 (FHL5), Lyst (Chediak-Higashi syndrome), and Rab 27A (Griscelli syndrome type 2), signaling lymphocyte activation molecule associated protein (XLP1) and X-linked inhibitor of apoptosis (XLP2) were defined as primary HLH. Patients with a confirmed VL diagnosis with a blood or bone marrow polymerase chain reaction (PCR) or indirect fluorescent antibody (IFA) test who fulfilled HLH-2004 criteria were defined as VL-associated HLH. In addition, information on demographic characteristics, clinical, laboratory, and radiological findings at presentation, treatment response, and survival outcomes were collected. HScore was calculated in each patient to estimate the probability of HLH in our patients and compare it with HLH-2004 diagnostic criteria. Participants in this study were assented to come in the survey according to the local Ethics Committee of the Shiraz University of Medical Sciences (IR.sums.med.rec. 1396.s195). A written informed consent was signed by the parents or the legal guardians of the participants.
Initial diagnostic workup
Based on our HLH-management protocol, the initial diagnostic workup included serological tests for EBV, CMV, human immunodeficiency virus, Brucella, Salmonella, and VL. In any suspected case of viral-associated HLH or leishmania-associated HLH, an additional molecular test with real-time polymerase chain reaction (RT-PCR) was requested to quantify viral or parasite load. RT-PCR provides valuable information about baseline virologic or parasitic status and is very useful for monitoring and response to treatment. The molecular diagnosis of disease was assessed with whole-exome sequencing with the next-generation sequencing (NGS) method in most study patients. The mutations found by NGS were confirmed by Sanger method.
Soluble CD25 (sCD25) measurement
sCD25 was measured using a commercial ELISA kit (Human sCD25 ELISA Ready-SET-Go, e-bioscience, USA) (sensitivity: 4 pg/ml) based on the manufacturer’s instructions. We determined the levels of sCD25 within the sample by setting up a standard curve of known target protein concentrations provided in the kit. Normal ranges for sCD25 in this study were 1.9–13.1 pg/ml.
CNS involvement
CNS involvement was defined if the patients had neurologic signs and symptoms including seizure, altered level of consciousness, or focal neurologic signs in addition to one of these two conditions: 1. pleocytosis and/or proteinosis in cerebrospinal fluid or 2. radiologic abnormalities on magnetic resonance imaging (MRI) such as high signal intensity lesions, hemorrhage, atrophy, and leptomeningeal enhancement.
Treatment protocol
All of our study patients with primary HLH were treated with HLA-94-based immune-chemotherapy. Briefly, the induction regimen lasted 8 weeks, including etoposide 150 mg/m2/day twice a week for 2 weeks and then once weekly for the next 6 weeks. Besides, dexamethasone 10 mg/m2/day for 2 weeks was started. The dose was halved every 2 weeks and tapered and discontinued on the last week of induction therapy. Patients with evidence of CNS disease were additionally treated with intrathecal methotrexate if the symptoms did not improve with systemic treatment. The continuation therapy included etoposide 150 mg/m2/day every other week with pulses of dexamethasone 10 mg/m2/day for 3 days and oral cyclosporine 6 mg/kg/day aiming at blood trough level around 200 mcg/L. The treatment continued for up to 40 weeks unless hematopoietic stem cell transplantation from a matched donor was available. Most of the patients with leishmaniasis-associated HLH (94.7%) were only treated with liposomal amphotericin B (total cumulative dose: 20 mg/kg divided into 4 or 5 doses administered in 4 or 5 days, respectively with no need to start HLH-specific therapy [15].. A minority were treated with dexamethasone (0.15 mg/kg/dose three times a day) for 3–5 days to control the cytokine storm.
Statistical analysis
Data were analyzed by the Statistical Package for the Social Sciences (SPSS Inc., Chicago, Illinois, USA, version 23). Descriptive data were presented as median, interquartile range (IQR), and percentages. The comparison of qualitative variables among different groups was made by the Chi-square test or the Fisher exact test. Quantitative variables with non-normal distribution were compared with the non-parametric Mann-Whitney U test between two groups. The Pearson correlation tested the correlation between the 2004-HLH diagnosis criteria and HScore. Univariate analysis was used to test possible covariates associated with outcome measures, including CNS involvement, relapse, and death. Those variables with a P value less than 0.2 were entered in multivariable analysis. Binary logistic regression was used to determine independent variables associated with CNS involvement and relapse. The survival curve was illustrated by the Kaplan-Mayer method. The Log-rank test evaluated a comparison of survival curves between two groups of patients. The Cox regression model was used to determine the independent variables associated with the outcome. The associations were reported as hazard ratio (HR) and 95% confidence interval (CI). A P-value of less than 0.05 was considered statistically significant.
Results
Demographic data
Sixty patients with a confirmed diagnosis of HLH were enrolled. The clinical features and laboratory findings of patients at their initial presentation are shown in Table 2. In total, 34 patients were male with male/ female ratio = 1.30. The median age at diagnosis was 11.5 months (IQR 4–35.7 months), and 42 patients (70%) were younger than 24 months. Twenty patients (33.3%) had VL-associated HLH.
Clinical and laboratory data
Fever and splenomegaly comprised the most frequent initial clinical findings, with 100 and 96.7% prevalence, respectively. Similarly, high serum ferritin (> 500 ng/ml) and high serum lactic dehydrogenase (LDH) (> 450 U/dL) were found exclusively in all patients. The study population’s median serum ferritin was 4825 ng/ml (range 912–72,000). Extreme hyperferritinemia (> 10,000 ng/ml) was detected in 16 (26.7%) of individuals. sCD25 was measured in one-third of the study population. It was increased in 14 (70%) out of 20 individuals.
The clinical and laboratory characteristics of patients with primary and VL-associated secondary HLH were compared in Table 2. Patients with primary HLH had a more frequently low platelet count of less than 100,000/μl (P = 0.012) and increased alanine transaminase (ALT) (P = 0.016) compared to secondary HLH. Moreover, the risk of death was significantly higher in primary HLH (P < 0.001). On the other hand, patients with VL-associated HLH had higher serum ferritin (P = 0.034). Moreover, they included a higher proportion of patients with high ESR (> 20 mm/hr) than primary HLH (P = 0.011). None of the patients in this group showed skin eruptions, bleeding tendency, or bone marrow hemophagocytosis. Nobody experienced disease relapse, and all of them survived.
HScore
Fifty-six (93.3%) patients achieved a score ≥ of 169, with no difference in the two groups of primary and secondary HLH (P > 0.99). HScore significantly correlated with HLH-2004 criteria (r = 0.371, P = 0.004).
CNS involvement
CNS involvement occurred in 23 (38.3%) patients. The proportion of patients with CNS involvement was not significantly different between primary and secondary HLH groups (P = 0.133). In univariate analysis, age < 2 years (P = 0.024), bone marrow hemophagocytosis (P = 0.027), and bicytopenia (P = 0.02) were associated with CNS involvement (Table 3). In subgroup analysis, age < 2 years remained a risk factor in primary HLH (P = 0.016). However, no risk factor was found in patients with VL-associated HLH. Binary logistic regression revealed that only age < 2 years (Odds ratio (OR) 6.446, 95% CI 1.538–27.012, P = 0.011) increased the odds of CNS involvement. On the other hand, patients with bicytopenia had less chance of neurologic disease (OR 0.073, 95% CI 0.008–0.838, P = 0.018). Patients with CNS disease had a worse outcome than those without CNS involvement (death rate 61% vs. 35%) with marginal statistical significance (P = 0.051).
Treatment and outcomes
During the study period, 33 patients (55%) showed a treatment response. Twenty-nine patients (87%) finished their treatment successfully. They were all alive and off-treatment at the end of the study. Treatment was ongoing in 4 more patients with an excellent initial response to treatment. Twenty-six (43.3%) patients died during treatment that all of them had primary HLH.
Fig. 1 shows the Kaplan-Meier survival curve of the study population during the 40 months follow-up period. The patients experienced a 3-year overall survival (OS) rate of 35.9%, which was significantly lower in the primary HLH than the secondary HLH group (24% vs. 100%; P < 0.001).

Kaplan-Meier survival curve of patients with hemophagocytic lymphohistiocytosis
Table 4 demonstrates a univariate analysis of possible covariates associated with the outcome in patients with HLH, assessed by the log-rank test. Among the tested variables, platelet count < 100,000 /μl (P = 0.001), total bilirubin > 1 mg/dL (P = 0.008), and erythrocyte sedimentation rate (ESR) ≤ 20 mm/hr. (P = 0.02) were associated with mortality. In Cox regression analysis, platelet count < 100,000/ μ l (HR 4.472, 95% CI 1.324–15.107, P = 0.016) remained an independent risk factor of mortality.
During the 40 months follow-up period, six patients (10%) experienced disease relapse. All of them occurred in boys with primary HLH. In multivariable analysis, no independent risk factor was found.
Discussion
The current study represents the first study on HLH patients in a large referral oncology center in Southern Iran. It is also one of the most extensive case series of secondary HLH associated with VL. It was reported that VL is associated with about 2% of all cases of HLH [16]. However, we encountered a much higher prevalence due to VL’s high endemicity in Southern Iran, where this study was undertaken. It is good to know that Fars province is one of the central endemic regions of visceral leishmaniasis in southern Iran, with an estimated incidence rate of 0.01/10,000 in 2015 [11]. A recent meta-analysis reported a pooled prevalence of 2.1% (95% CI 1.4–2.8%) of VL in Southern Iran [17]. The first report of a benign hemophagocytic syndrome associated with VL in our region was published by Kumar et al. in 1994 [12]. Mokhtari and Kumar reported the first case series of VL-associated HLH among 13 patients and described their bone marrow characteristics. Hypercellular marrow with erythroid hyperplasia, megaloblastic changes, foamy macrophages, activated macrophages with cytoplasmic vacuoles and elongated cytoplasmic process, intra- and extracellular amastigotes, cytoplasmic fragments (blue bodies), plasma cells with inclusions and hemophagocytic cells, in addition to Leishman bodies (amastigotes) were the most common microscopic findings of the studied patients. The most common hematological abnormalities were anemia (88.8%), thrombocytopenia (50%,) and pancytopenia (55.5%) [13].
The clinical sign and symptoms and laboratory parameters in HLH associated with VL were very similar to primary HLH. Fever and splenomegaly were the most consistent presenting signs and symptoms detected in nearly all patients. Serum ferritin at diagnosis was higher than 900 ng/ml in all patients, and about 30% of them had extreme hyperferritinemia (> 10,000 ng/ml). However, it did not correlate with disease severity or adverse outcomes such as death or relapse. Allen et al. reported that ferritin level over 10,000 ng/ml was 90% sensitive and 96% specific for HLH [18].
On the other hand, a recent review concluded that HLH is a rare cause of extreme hyperferritinemia, and more common causes such as liver disease and malignancies should be ruled out first [19]. Since timely diagnosis and treatment are directly related to disease outcome, high serum ferritin should raise suspicion to HLH diagnosis if other criteria are met, especially in critically ill patients.
Patients with VL-associated HLH experienced higher serum ferritin and ESR. Though the underlying infection may be a simple explanation, enhanced ferroportin-mediated iron efflux due to overexpression of growth differentiation factor 15 may play a role [20]. On the other hand, primary HLH was associated with more severe thrombocytopenia, higher ALT, and worse outcome than secondary HLH. Koh et al. reported that splenomegaly, bicytopenia, Hb < 9 g/dL, and platelets < 100,000/μl were more frequently encountered in patients with primary HLH. They also reported a lower 5-yr OS in primary HLH compared to secondary HLH [21]. Some of the mentioned features, such as bicytopenia and thrombocytopenia, can be regarded as markers of disease severity, directly linked to poor survival. We came across a similar result in which thrombocytopenia served as an independent risk factor of death, especially in patients with primary HLH.
Nearly 40% of our patients had evidence of CNS involvement. The rate of CNS involvement in HLH varies in different studies ranging from 18 to 73% [22]. Some reported previously that patients with CNS disease had lower ferritin, AST, and ALT than those without CNS involvement [23]. We did not find such an association in our patients except that children younger than 2 years had more than a 6-fold higher chance of CNS disease than older patients. Moreover, bicytopenia decreased the probability of CNS involvement by 93%.
Many reports are indicating that CNS involvement might be associated with worse outcomes (19). Although the overall mortality rate was higher in those with CNS events, this association was not shown in multivariable analysis. It is generally agreed that CNS involvement poses a higher risk of relapse unless hematopoietic stem cell transplantation (HSCT) is undergone early after starting systemic therapy [22].
HScore had a good correlation with HLH-2004 criteria for the diagnosis of HLH in our study. It was reported that HScore carries a diagnostic sensitivity of 90% and specificity of 79% for HLH [6]. A cutoff of 169 had 93% sensitivity and 86% specificity for HLH [24]. Similarly, more than 90% of our confirmed cases of HLH had HScore ≥169. Given the unavailability of sCD25 and natural killer (NK) cell activity in some centers, HScore can be utilized to make early diagnosis and treatment in case of incomplete criteria.
The treatment response was 55% in our study cohort with a 3-yr OS rate of 36%. It was much lower in patients with primary HLH than secondary HLH (24% vs. 100%). All patients with VL-associated HLH were successfully treated with liposomal amphotericin. Nobody needed to be treated with etoposide or cyclosporine. Dexamethasone was initially started in a few patients with severe disease but was tapered and discontinued after a few weeks. The outcome was excellent, with no report of death or HLH relapse during the 40 months follow-up. Targeted treatment of the etiologic pathogen usually is the only treatment strategy recommended in secondary infection-associated HLH such as brucella and leishmania. Specific antimicrobial therapy usually is associated with complete recovery in most cases [25,26,27,28,29,30,31].
High-dose intravenous gamma-globulin (IVIG) has been used successfully in viral-associated hemophagocytic syndrome [32]; however, none of our cases were treated with IVIG.
Platelet count less than 100,000/μl was independently associated with more than four times the increased risk of death in our patients with primary HLH. The outcome of patients with HLH and associated prognostic factors differ among different studies. A recent study in Japan reported a much better outcome in their patients with a 3-yr OS rate of 74% [33]. Koh et al. reported a 5-yr OS rate of 68% in their study cohort, which was worse (38%) in primary HLH patients. They concluded that lower age at diagnosis, severe transaminasemia, and coagulation abnormality were independent risk factors of survival [21]. In other studies, decreased serum albumin level, LDH ≥ 3707 U/L, IL-10 ≥ 456 pg/ml, neutrophil count < 500/μl, and total bilirubin > 2× normal upper limit was associated with a higher mortality rate [34,35,36].
Our study faced some limitations. Firstly, some specific laboratory tests, such as NK cell function, were unavailable in our center. Besides, sCD25 was measured in a portion of our patients since it was available in our center in 2017. These tests’ unavailability might lead to a delayed diagnosis and poor outcome, evident as a lower 3-yr OS rate in our study cohort than many other reports.
Conclusion
VL is a potential source of secondary HLH in regions with high endemicity. The main clinical findings and complications such as CNS involvement are the same as primary HLH. It is associated with higher serum ferritin and ESR but a lower incidence of thrombocytopenia and transaminitis than primary HLH. Treatment of the underlying disease in VL-associated HLH is sufficient in most cases, with no need to start etoposide-based chemotherapy. The prognosis is excellent, with no reported death or recurrence of disease in our case series.
Availability of data and materials
The data presented in this study are available on request from the corresponding author.
Abbreviations
- HLH:
-
Hemophagocytic lymphohistiocytosis
- VL:
-
Visceral leishmaniasis
- CNS:
-
Central nervous system
- EBV:
-
Epstein-Barr virus
- CMV:
-
Cytomegalovirus
- RT-PCR:
-
Real-time polymerase chain reaction
- sCD25:
-
Soluble CD25
- MRI:
-
Magnetic resonance imaging
- SPSS:
-
Statistical Package for the Social Sciences
- IQR:
-
Interquartile range
- CI:
-
Confidence interval
- HR:
-
Hazard ratio
- ALT:
-
Alanine transaminase
- AST:
-
Aspartate transaminase
- OR:
-
Odds ratio
- NK cell:
-
Natural killer cell
- OS:
-
Overall survival
- LDH:
-
Lactate dehydrogenase
- IL-10:
-
Interleukin 10
- HSCT:
-
Hematopoietic stem cell transplantation
- ESR:
-
Erythrocyte sedimentation rate
- ULN:
-
Upper limit of normal
- BM:
-
Bone marrow
- HIV:
-
Human immunodeficiency virus
- aPTT:
-
Activated partial thromboplastin time
- PT:
-
Prothrombin time
- SGOT:
-
Serum glutamic-oxaloacetic transaminase
- HB:
-
Hemoglobin
- WBC:
-
White blood cell
- PLT:
-
Platelet
- ANC:
-
Absolute neutrophil count
- CRP:
-
C-reactive protein
References
Filipovich A, McClain K, Grom A. Histiocytic disorders: recent insights into pathophysiology and practical guidelines. Biol Blood Marrow Transplant. 2010;16(1 Suppl):S82–9. https://doi.org/10.1016/j.bbmt.2009.11.014.
Henter JI, Elinder G, Söder O, Ost A. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand. 1991;80(4):428–35. https://doi.org/10.1111/j.1651-2227.1991.tb11878.x.
Ricaurte FR, Kewan T, Chadalavada P, Misbah S, Daw H. Hemophagocytic Lymphohistiocytosis associated with natural T-cell leukemia. Cureus. 2019;11(2):e4107. https://doi.org/10.7759/cureus.4107.
Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118(15):4041–52. https://doi.org/10.1182/blood-2011-03-278127.
Larroche C. Hemophagocytic lymphohistiocytosis in adults: diagnosis and treatment. Joint Bone Spine. 2012;79(4):356–61. https://doi.org/10.1016/j.jbspin.2011.10.015.
Debaugnies F, Mahadeb B, Ferster A, Meuleman N, Rozen L, Demulder A, et al. Performances of the H-score for diagnosis of Hemophagocytic Lymphohistiocytosis in adult and pediatric patients. Am J Clin Pathol. 2016;145(6):862–70. https://doi.org/10.1093/ajcp/aqw076.
Esteban YM, de Jong JLO, Tesher MS. An overview of Hemophagocytic Lymphohistiocytosis. Pediatr Ann. 2017;46(8):e309–13. https://doi.org/10.3928/19382359-20170717-01.
Shamsian BS, Rezaei N, Alavi S, Hedayat M, Amin Asnafi A, Pourpak Z, et al. Primary hemophagocytic lymphohistiocytosis in Iran: report from a single referral center. Pediatr Hematol Oncol. 2012;29(3):215–9. https://doi.org/10.3109/08880018.2012.657338.
Ghanaie R, Shiari R, KARIMI AE, Armin S, Fahimzad A, Shiva F, et al. A case series report of Iranian children, hemophagocytic lymphohistiocytosis syndrome; 2012.
Shamsian BS, Nikoufar M, Esfahani SA, Shamshiri AR, Arzanian MT, Alavi S, et al. A 10-year single center survey of pediatric patients with histiocytic disorders in Iran. Turk J Pediatr. 2011;53(1):34–42.
World Health Organization. Leishmaniasis. 2020;. https://www.whoint/leishmaniasis/burden/Leishmaniasis_Iran/en/ Accessed 11 Apr 2020.
Alborzi A, POULADFAR GR, AALAMI MH. Visceral leishmaniasis; literature review and Iranian experience. Iran J of Clin Infect Dis. 2007;2(2):99–108.
Mokhtari M, Kumar PV. Visceral leishmaniasis-associated hemophagocytosis: a single center experience. Arch Iran Med. 2013;16(8):471–3.
Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–31. https://doi.org/10.1002/pbc.21039.
Krepis P, Argyri I, Krepi A, Syrmou A, Spyridis N, Tsolia M. Short-course regimens of liposomal amphotericin B for the treatment of Mediterranean visceral Leishmaniasis in children: an 11-year retrospective study at a tertiary care center. Pediatr Infect Dis J. 2017;36(9):849–54. https://doi.org/10.1097/INF.0000000000001602.
Bode SF, Bogdan C, Beutel K, Behnisch W, Greiner J, Henning S, et al. Hemophagocytic lymphohistiocytosis in imported pediatric visceral leishmaniasis in a nonendemic area. J Pediatr. 2014;165(1):147–153.e141.
Rostamian M, Bashiri H, Yousefinejad V, Bozorgomid A, Sohrabi N, Raeghi S, et al. Prevalence of human visceral leishmaniasis in Iran: A systematic review and meta-analysis. Comp Immunol Microbiol Infect Dis. 2021;75:101604.
Allen CE, Yu X, Kozinetz CA, McClain KL. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;50(6):1227–35. https://doi.org/10.1002/pbc.21423.
Suman S, Ahmed A. Extreme hyperferritinemia does not equal to HLH. Int J Clin Rheumatol. 2019;14(4):134–5.
Wu JR, Yuan LX, Ma ZG, Chen XX, Gu L, Gao J. GDF15-mediated upregulation of ferroportin plays a key role in the development of hyperferritinemia in children with hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2013;60(6):940–5. https://doi.org/10.1002/pbc.24373.
Koh KN, Im HJ, Chung NG, Cho B, Kang HJ, Shin HY, et al. Clinical features, genetics, and outcome of pediatric patients with hemophagocytic lymphohistiocytosis in Korea: report of a nationwide survey from Korea Histiocytosis working party. Eur J Haematol. 2015;94(1):51–9. https://doi.org/10.1111/ejh.12399.
Horne A, Wickström R, Jordan MB, Yeh EA, Naqvi A, Henter JI, et al. How to treat involvement of the central nervous system in Hemophagocytic Lymphohistiocytosis? Curr Treat Options Neurol. 2017;19(1):3. https://doi.org/10.1007/s11940-017-0439-4.
Kim MM, Yum MS, Choi HW, Ko TS, Im HJ, Seo JJ, et al. Central nervous system (CNS) involvement is a critical prognostic factor for hemophagocytic lymphohistiocytosis. Korean J Hematol. 2012;47(4):273–80. https://doi.org/10.5045/kjh.2012.47.4.273.
Soy M, Atagündüz P, Atagündüz I, Sucak GT. Hemophagocytic lymphohistiocytosis: a review inspired by the COVID-19 pandemic. Rheumatol Int. 2021;41(1):7–18. https://doi.org/10.1007/s00296-020-04636-y.
Cançado GG, Freitas GG, Faria FH, de Macedo AV, Nobre V. Hemophagocytic lymphohistiocytosis associated with visceral leishmaniasis in late adulthood. Am J Trop Med Hyg. 2013;88(3):575–7. https://doi.org/10.4269/ajtmh.12-0563.
Matnani R, Ganapathi KA. Hemophagocytic lymphohistiocytosis associated with visceral leishmaniasis. Blood. 2016;127(4):513. https://doi.org/10.1182/blood-2015-10-678862.
Tapisiz A, Belet N, Ciftçi E, Ince E, Dogru U. Hemophagocytic lymphohistiocytosis associated with visceral leishmaniasis. J Trop Pediatr. 2007;53(5):359–61. https://doi.org/10.1093/tropej/fmm024.
Scalzone M, Ruggiero A, Mastrangelo S, Trombatore G, Ridola V, Maurizi P, et al. Hemophagocytic lymphohistiocytosis and visceral leishmaniasis in children: case report and systematic review of literature. J Infect Dev Ctries. 2016;10(1):103–8. https://doi.org/10.3855/jidc.6385.
Rajagopala S, Dutta U, Chandra KS, Bhatia P, Varma N, Kochhar R. Visceral leishmaniasis associated hemophagocytic lymphohistiocytosis--case report and systematic review. J Inf Secur. 2008;56(5):381–8.
El Euch M, Kaabar MY, Bouaziz R, Mahfoudhi M, Jaziri F, Kefi A, et al. Successful resolution of Hemophagocytic lymphohistiocytosis associated to brucellosis in the adult. Tunis Med. 2018;96(7):458–61.
Yaman Y, Gözmen S, Özkaya AK, Oymak Y, Apa H, Vergin C, et al. Secondary hemophagocytic lymphohistiocytosis in children with brucellosis: report of three cases. J Infect Dev Ctries. 2015;9(10):1172–6. https://doi.org/10.3855/jidc.6090.
Larroche C, Bruneel F, André MH, Bader-Meunier B, Baruchel A, Tribout B, et al. Intravenously administered gamma-globulins in reactive hemaphagocytic syndrome. Multicenter study to assess their importance, by the immunoglobulins group of experts of CEDIT of the AP-HP. Ann Med Interne (Paris). 2000;151(7):533–9.
Yanagisawa R, Nakazawa Y, Matsuda K, Yasumi T, Kanegane H, Ohga S, et al. Outcomes in children with hemophagocytic lymphohistiocytosis treated using HLH-2004 protocol in Japan. Int J Hematol. 2019;109(2):206–13. https://doi.org/10.1007/s12185-018-02572-z.
Oguz MM, Sahin G, Altinel Acoglu E, Polat E, Yucel H, Oztek Celebi FZ, et al. Secondary hemophagocytic lymphohistiocytosis in pediatric patients: a single center experience and factors that influenced patient prognosis. Pediatr Hematol Oncol. 2019;36(1):1–16. https://doi.org/10.1080/08880018.2019.1572253.
Luo ZB, Chen YY, Xu XJ, Zhao N, Tang YM. Prognostic factors of early death in children with hemophagocytic lymphohistiocytosis. Cytokine. 2017;97:80–5. https://doi.org/10.1016/j.cyto.2017.03.013.
Bin Q, Gao JH, Luo JM. Prognostic factors of early outcome in pediatric hemophagocytic lymphohistiocytosis: an analysis of 116 cases. Ann Hematol. 2016;95(9):1411–8. https://doi.org/10.1007/s00277-016-2727-6.
Acknowledgments
This study was relevant to the fellowship thesis of H. Mottaghipisheh with project No. 13253. We would like to thank the Shiraz University of Medical Sciences for financial support. We appreciate MS. Parand for her assistance in submitting the article.
Funding
This research was funded by the Research Vice-Chancellor of Shiraz University of Medical Sciences.
Author information
Authors and Affiliations
Contributions
HM and MB contributed to the concept and design of the study. KK and MS did and interpreted laboratory tests. SH performed statistical analysis. HM gathered the data. MB wrote the initial draft. All the authors reviewed the drafts, provided critical comments, and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by the Ethics Committee of Shiraz University of Medical Sciences (IR.SUMS.REC.1399.1281). Also, all methods were carried out in accordance with Declaration of Helsinki.
A written informed consent form was signed by the parents or the legal guardians of the participants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mottaghipisheh, H., Kalantar, K., Amanati, A. et al. Comparison of the clinical features and outcome of children with hemophagocytic lymphohistiocytosis (HLH) secondary to visceral leishmaniasis and primary HLH: a single-center study. BMC Infect Dis 21, 732 (2021). https://doi.org/10.1186/s12879-021-06408-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-021-06408-w