
Abstract
Background
Favipiravir possesses high utility for treating patients with COVID-19. However, research examining the efficacy and safety of favipiravir for patients with COVID-19 is limited.
Methods
We conducted a systematic review of published studies reporting the efficacy of favipiravir against COVID-19. Two investigators independently searched PubMed, the Cochrane Database of Systematic Reviews, MedRxiv, and ClinicalTrials.gov (inception to September 2020) to identify eligible studies. A meta-analysis was performed to measure viral clearance and clinical improvement as the primary outcomes.
Results
Among 11 eligible studies, 5 included a comparator group. Comparing to the comparator group, the favipiravir group exhibited significantly better viral clearance on day 7 after the initiation of treatment (odds ratio [OR] = 2.49, 95% confidence interval [CI] = 1.19–5.22), whereas no difference was noted on day 14 (OR = 2.19, 95% CI = 0.69–6.95). Although clinical improvement was significantly better in the favipiravir group on both days 7 and 14, the improvement was better on day 14 (OR = 3.03, 95% CI = 1.17–7.80) than on day 7 (OR = 1.60, 95% CI = 1.03–2.49). The estimated proportions of patients with viral clearance in the favipiravir arm on days 7 and 14 were 65.42 and 88.9%, respectively, versus 43.42 and 78.79%, respectively, in the comparator group. The estimated proportions of patients with clinical improvement on days 7 and 14 in the favipiravir group were 54.33 and 84.63%, respectively, compared with 34.40 and 65.77%, respectively, in the comparator group.
Conclusions
Favipiravir induces viral clearance by 7 days and contributes to clinical improvement within 14 days. The results indicated that favipiravir has strong possibility for treating COVID-19, especially in patients with mild-to-moderate illness. Additional well-designed studies, including examinations of the dose and duration of treatment, are crucial for reaching definitive conclusions.
Similar content being viewed by others

Background
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was first identified in Wuhan, Hubei Province, China [1], and it is the causative agent of the coronavirus disease 2019 (COVID-19) pandemic [2]. By the end of September, 2020, it brings the cumulative numbers to over 61.8 million cases and 1.4 million death globally since the start of the pandemic [3]. COVID-19 is caused by the novel virus, so that pathophysiology and effective treatment methods were unknown, and vaccine was not available at the first stage of the outbreak. Although the discovery of effective treatment methods were urgently required, the discovery of new antiviral agents against SARS-CoV-2 takes a long time and it is not straightforward task.
SARS CoV-2 is a positive strand RNA (+RNA) virus, and is a member of the coronaviridae family. SARS CoV-2 is a single-stranded RNA beta-coronavirus encoding an RNA-dependent RNA polymerase (RdRp) and proteases. Both RdRp and viral proteases are considered important targets for potential therapeutic agents. Favipiravir, previously known as T-705, is a prodrug of the purine nucleotide favipiravir ribofuranosyl-5′-triphosphate [4]. The active agent inhibits RNA polymerase, halting viral replication [4]. Favipiravir was approved in 2014 by the Japan Pharmaceuticals and Medical Devices Agency under the brand name AVIGAN® for the treatment of novel and re-emerging influenza virus infection [5]. Several studies described its effectiveness against other RNA viruses such as Ebola virus [6], as well as the effectiveness against rhinovirus and respiratory syncytial virus [7]. In vitro, the 50% effective concentration (EC50) of favipiravir against SARS-CoV-2 was 61.88 μM/L in Vero E6 cells [8]. Thus, favipiravir possess high potential for treating patients with COVID-19. However, research examining the efficacy and safety of favipiravir in patients with COVID-19 is limited.
The aim of the present study was to review systematically on the application of favipiravir for patients with COVID-19 to identify empirical evidence of its efficacy.
Methods
This systematic review and meta-analysis was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement and the statement by the Meta-analysis of Observational Studies in Epidemiology (MOOSE) group [9, 10].
Eligibility criteria and outcome measures
Studies fulfilling the following selection criteria were included in the meta-analysis: (1) study design and language: randomized clinical trials (RCTs), observational studies, and case series involving > 10 patients written in the English language; (2) population: patients with laboratory-confirmed SARS-CoV-2 infection who were hospitalized or treated in clinics; (3) intervention: administration of favipiravir; (4) comparison intervention: placebo, standard of care (SOC), remdesivir, lopinavir/ritonavir, other available antivirals, hydroxychloroquine (HQ), different dosages of favipiravir, combination therapy with favipiravir, or no comparator; (5) primary outcomes: viral clearance and clinical improvement including improvement of chest computer tomography (CT); and (6) secondary outcomes, any outcome variable. The exclusion criteria were as follows: (1) ≤10 patients in case series, (2) no reporting of outcome variables, and (3) insufficient or incomplete data.
Information sources and search strategy
Two investigators (T.M. and D.K.) independently searched for eligible studies in PubMed, the Cochrane Library, and MedRxiv from inception to September 12,020. We used the following key words: “novel coronavirus” OR “new coronavirus” OR “emerging coronavirus” OR “2019-nCoV” OR “COVID-19” OR “SARS-CoV-2” AND “favipiravir” OR “avigan” OR “T-705.” We searched the reference lists of all included studies, reviews, and clinical trial registries for ongoing trials investigating the efficacy or safety of favipiravir for patients with COVID-19. We also reviewed the reference lists of eligible studies using Google Scholar and performed a manual search to ensure that all appropriate studies were included.
Data extraction
Two reviewers (T.M. and D.K.) extracted the data independently. Articles retrieved in the search were stored in a citation manager (EndNote X9; Thomson Reuters, New York, NY, USA). After removing redundant articles, titles, abstracts, and then full-text articles were investigated. We extracted the following data: study design, observational period, study site, and inclusion/exclusion criteria of each study. Outcome variables were extracted into predesigned data collection forms. We verified the accuracy of data by comparing the collection forms of each investigator. Any discrepancies were resolved through discussion among the authors.
Risk of bias assessment
For clinical trials and before and after controlled trials, we assessed the risk of bias (“low risk,” “some concerns,” or “high risk”) in the overall effect of favipiravir on viral clearance and clinical improvements using version 2 of the Cochrane Risk of Bias Assessment Tool [11]. Risk of bias assessments were performed independently by two investigators (T.M. and D.K.), with disagreements resolved through discussion. We used the Grading of Recommendations Assessment and Evaluation approach [12] to assess the certainty of the evidence that favipiravir reduced the time to viral clearance and contributed to clinical improvement.
Data analysis
Throughout the meta-analysis, we estimated the odds ratios (ORs) or the proportions of patients for primary outcome variables with 95% confidence intervals (CIs) using a random-effects model (generic inverse variance method). To assess the proportions of the outcome variables among patients with COVID-19, the standard error was calculated using the Agresti–Coull method [13]. Heterogeneity among the original studies was evaluated using the I2 statistic [14]. Publication bias was examined using a funnel plot. For all analyses, significance levels were two-tailed, and p < 0.05 was considered significant. All statistical tests were performed using Review Manager (RevMan) ver. 5.3.5 (Cochrane Collaboration, Copenhagen, Denmark) [15].
Results
Study selection and characteristics
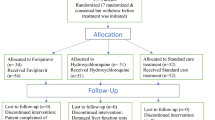
Of the 163 references screened, 11 studies were eligible (Fig. 1).

PRISMA flow diagram. N is the number of articles
Table 1 presents the characteristics of the included studies.
Among the 11 studies, three studies were RCTs [16, 18,19,20], one study was a non-randomized controlled study [17], one study was a before and after nonrandomized controlled study [21], and six studies were observational studies or case series [22,23,24,25,26]. Among the comparative studies, the comparators included umifenovir [17], baloxavir marboxil [18], standard of care (SOC) [19], lopinavir/ritonavir [21], and HQ alone or in combination with azithromycin [25]. An RCT that examined the early or late initiation of favipiravir treatment was the only study investigating asymptomatic or mildly ill patients [19]. Although the sample sizes were small, two case series assessed combination therapy involving favipiravir plus nafamostat mesylate [23] or methylprednisolone [22, 24] for patients with severe COVID-19.
The dose of favipiravir generally matched the standard dose for treating influenza infection, namely 1600 mg twice daily on the first day followed by 600 mg twice daily, but in some eligible studies, the dose was 1800 mg twice daily on the first day followed by 800 mg twice daily. One study examined the loading dose [22]. Among the 11 eligible studies, the duration of favipiravir therapy was primarily 14 days [18, 19, 21, 23,24,25].
Assessment of bias in studies comparing favipiravir with other antivirals or standard of care among patients with COVID-19
Data on viral clearance and clinical improvement were available for four trials [16,17,18,19] and the before and after controlled study [20]. We identified a high risk of bias, and the evidence was assessed at “low” for viral clearance and clinical improvement (Figs. S1–S2).
The major qualitative outcomes of each study are presented in Table 2.
Of the five trials with data on the primary outcomes, we estimated the ORs for the association between favipiravir treatment and viral clearance using meta-analysis (Fig. 2). The OR of viral clearance on day 7 was 0.40 (95% CI = 0.19–0.84) with statistical difference (p = 0.02) (Fig. 2a), whereas that on day 14 was 0.46 (95% CI = 0.14–1.45), with no significant difference (p = 0.18) (Fig. 2b) noted between the two points.

Forrest plots for viral clearance for patients with COVID-19 who were treated with favipiravir or a comparator for (a) 7 or (b) 14 days
We also estimated the proportion of patients with viral clearance in the favipiravir (Fig. 3) and comparative treatment arms (Fig. S3).

Proportions of patients with COVID-19 who achieved viral clearance on (a) 7 and (b) 14 days from the initiation of treatment
The estimated proportions of patients with COVID-19 who achieved viral clearance in the favipiravir arm on days 7 and 14 were 65.42 and 88.9% (Fig. 3), respectively, compared with 43.42 and 78.79%, respectively, for the comparator arm (Fig. S3).
Clinical improvement with favipiravir versus other antivirals or standard of care among patients with COVID-19
In terms of clinical improvement, the definition of clinical improvement varied among the studies as follows: continuous (> 72 h of recovery of body temperature, respiration rate, oxygen saturation and cough relief after treatment [16]); improvements on the seven-category ordinal scale, which references the National Early Warning Score or live discharge from the hospital, whichever came first [17]; and chest CT improvement [18, 20]. Despite the various definitions, favipiravir was associated with better clinical improvement than comparative therapy after 7 (OR = 1.60, 95% CI = 1.03–2.40, p = 0.04; Fig. 4a) and 14 days of treatment (OR = 3.03. 95% CI = 1.17–7.80, p = 0.02; Fig. 4b).

Forrest plots of clinical improvement for patients with COVID-19 treated with favipiravir for (a) 7 and (b) 14 days
The estimated proportions of patients with clinical improvement in the favipiravir group after 7 and 14 days were 54.33 and 84.63%, respectively (Fig. 5), compared with 34.40 and 65.77%, respectively, for the comparator group (Fig. S4).

Proportions of patients with COVID-19 who had clinical improvement until (a) 7 and (b) 14 days from the initiation of treatment
Secondary outcomes
Table 2 presents the various outcomes reported in each study. Among the comparative studies of favipiravir, most patients had moderate COVID-19, and death was observed in only two patients in the favipiravir group who had underlying diseases, such as diabetes mellitus, artificial hypertension, and obesity [18]. In two observational studies that examined the combination of favipiravir plus nafamostat mesylate [23] or methylprednisolone [24] in patients with severe COVID-19 reported one death each. The morality rates of the observational study that examined the loading dosage of favipiravir for patients including severe disease were 1.6% on day 14 and 4.8% on day 28 [22]. The median times to body temperature normalization in the favipiravir and standard of care arms in one study were 2 and 4 days, respectively [18], and those in patients with early and late favipiravir initiation were 2.1 and 3.2 days, respectively [19]. A study comparing HQ alone, HQ plus azithromycin, and favipiravir-containing regimens found that the length of hospital stay was shortest in the HQ alone group [25].
Adverse reactions of favipiravir
The reported adverse reactions of favipiravir in each study are presented in Table 3.
Concerning frequent adverse drug reactions, diarrhea or digestive tract reactions were reported in three studies, decreased albumin levels were reported in one study, and hyperuricemia was reported in one study. Serum uric acid levels were also determined in two studies. The number of adverse drug reactions was not significantly different among three trials (OR = 0.69, 95% CI = 0.13–3.52, p = 0.62; Fig. 6).

Forrest plots of the number of adverse events for patients with COVID-19 who were treated with favipiravir
Discussion
The present systematic review and meta-analysis using the limited available evidence revealed that favipiravir has high promise for treating patients with COVID-19. Among patients with moderate COVID-19, favipiravir accelerated viral clearance after 7 days of treatment. Favipiravir also contributed to clinical improvement, especially after 14 days of treatment. Drugs other than antiviral agents, such as nafamostat or methylprednisolone, can be used in combination with favipiravir for patients with moderate or severe COVID-19. However, we must await well-designed studies assessing effectiveness of favipiravir in patients with COVID-19, including examinations of the different doses and durations of therapy in patients with different levels of disease severity.
Favipiravir, which has displayed efficacy against many RNA viruses, acts by inhibiting RNA-dependent RNA polymerase, and it is one of several potential drugs that may be repurposed for treating COVID-19 [4, 7, 27]. In this study, although different comparators were used among the studies, the meta-analysis estimated that favipiravir was associated with a significantly higher likelihood of viral clearance on day 7. Contrarily, the proportions of viral clearance were not significantly different by day 14. The viral load of SARS-CoV-2 peaks around symptom onset or a few days thereafter, and the virus becomes undetectable within approximately 2 weeks [28]. In addition, among most eligible studies, the duration of favipiravir therapy was 14 days. The lack of a significant difference in the proportion of viral clearance at day 14 between the treatments may reflect the natural course of viral shedding. However, the emergence of patients and healthy viral carriers with early viral RNA clearance is a major concern for disease management and infection control measures. In fact, the study found that the median time to viral clearance among patients who were administered favipiravir on day 1 after onset was less than 12.8 days, compared with 17.8 days for patients who started treatment on day 6 [17]. Favipiravir is an oral drug; therefore, it is easy to administer to patients with asymptomatic or mild COVID-19. This result strengthened the importance of early favipiravir administration as well as the role of the drug in the management of early-stage or asymptomatic COVID-19.
Favipiravir contributed to significant clinical improvement by 14 days, but not 7 days, after treatment initiation. Contrarily, the time to body temperature normalization was approximately 2 days [18, 19]. The definition of clinical improvement differed among the studies; however, variables that defined clinical improvement included the respiration rate, oxygen saturation, cough relief, and chest CT improvement. These clinical signs and symptoms were affected by lung injury or pneumonia. Under the various clinical manifestations of COVID-19, ranging from an asymptomatic disease course to the clinical symptoms of acute respiratory distress syndrome and severe pneumonia, the lungs, which have extremely slow cell turnover, are the primary organs affected by SARS-CoV-2 [29]. Most patients in the present study had mild-to-moderate COVID-19, and favipiravir treatment may have contributed to lung recovery within 14 days from the initiation of treatment. A study of patients with asymptomatic or mild COVID-19 comparing early and late favipiravir initiation revealed a significant difference in the duration of hospitalization [19]. The result demonstrated the necessity of early favipiravir initiation even for patients with asymptomatic or mild COVID-19 before pneumonia develops or lung damage worsens [30]. In addition, the standard dosage of favipiravir for influenza was 1600 mg twice daily on the first day followed by 600 mg twice daily for a total of 5 days [5]. Most of the eligible studies followed the standard regimen, and the treatment duration was generally 14 days. However, some studies increased the dose to 1800 mg twice daily on the first day followed by 800 mg twice daily [28]. The losing variations are likely attributable to the lower favipiravir EC50 described against influenza than against Ebola and SARS-CoV-2 [31, 32]. Various dosing regimens have been proposed based on the type of infection indication [33]; a loading dose of 2400–3000 mg every 12 h (two doses) has been considered for the treatment of COVID-19, followed by a maintenance dose of 1200–1800 mg every 12 h [31, 32]. Rattanaumpawan et al. examined the loading dose of favipiravir and concluded that a low loading dose (≤45 mg/kg/day) was a poor prognosis factor for early clinical improvement. Doses at the higher end of the dosing range should be considered for the optimal treatment of COVID-19.
A review article demonstrated that favipiravir had a tolerable safety profile in terms of total and serious adverse effects compared with other drugs used for short-term treatment [33]. This is the compatible with the present systematic review and hyperuricemia was observed in 84.1% of patients with asymptomatic or mild COVID-19 patients in one study [19]. Although there is limited clinical experience with favipiravir for COVID-19 treatment, the present study demonstrated that serious adverse events induced by favipiravir were not observed. In addition, due to a risk of teratogenicity and embryotoxicity, the Ministry of Health, Labor and Welfare in Japan has therefore only granted conditional marketing approval for its production and clinical use for influenza virus infection [7]. This safety information will play a critical role of favipiravir in COVID-19 patients, especially pregnant women.
The present study had some limitations. First, only some of the studies included a comparator arm. In addition, although favipiravir treatment in most studies was followed by standard care for influenza, the dose and duration were not same among the trials. However, we included the studies that had no comparator arm and different dose and duration into the assessments for the proportions of viral clearance and clinical improvement if the outcome variable were presented. Second, the observation points of the primary outcomes were not strictly 7 and 14 days after treatment initiation in all studies. Third, the definition of clinical improvement differed among the studies. Despite these limitations, in an effort to expand the role of favipiravir in the clinical management of COVID-19, especially for patients with asymptomatic and mild-to-moderate disease, it was crucial to quickly examine the efficacy and safety of favipiravir.
Conclusions
Our study revealed that favipiravir can promote viral clearance within 7 days and clinical improvement within 14 days, especially in patients with mild-to-moderate COVID-19. The early initiation of treatment with favipiravir can contribute to positive outcomes for COVID-19. This systematic review and meta-analysis demonstrated the high potentiality for the use of favipiravir for COVID-19. In particular, since favipiravir is the oral form, it is easy to administer for asymptomatic or mildly ill patients with COVID-19. However, there is an urgent need for additional evidence, especially trials assessing different doses and durations of therapy and patients with different levels of disease severity.
Availability of data and materials
All relevant data are included within the paper and the Supporting Information file.
Abbreviations
- COVID-19:
-
Novel coronavirus disease
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus
- RdRp:
-
RNA-dependent RNA polymerase
- RCT:
-
Randomized clinical trials
- SOC:
-
Standard of care
- CT:
-
Computer tomography
- LPV/RTV:
-
Lopinavir/ritonavir
- HQ:
-
Hydroxychloroquine
- AZ:
-
Azithromycin
- SD:
-
Standard deviation
- IQR:
-
Interquartile range
- NA:
-
Not available
- OR:
-
Odds ratio
- HR:
-
Hazard ratio
- DIC:
-
Disseminated intravascular coagulation
- ICU:
-
Intensive care unit
- SpO2 :
-
Blood oxygen saturation
- MV:
-
Mechanical ventilation
- ECMO:
-
Extracorporeal membrane oxygenation
- NEWS2:
-
National Early Warning Score
- RBC:
-
Red blood cell
References
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan. China Lancet. 2020;395(10223):497–506. https://doi.org/10.1016/S0140-6736(20)30183-5.
World Health Organization. WHO Director-General’s opening remarks at the media briefing on COVID-19 - 11 March 2020. Available at https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19%2D%2D-11-march-2020 (Accessed April 5, 2020).
World Health Organization. Weekly epidemiological update-1 December 2020. Available at https://www.who.int/publications/m/item/weekly-epidemiological-update%2D%2D-1-december-2020 (Accessed December 5, 2020).
Furuta Y, Komeno T, Nakamura T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93(7):449–63. https://doi.org/10.2183/pjab.93.027.
FUJIFILM Toyama Chemical Co., Ltd. Drug Interview form. Avigan® http://fftc.fujifilm.co.jp/med/abigan/pack/pdf/abigan_if_01.pdf. Accessed 5 Apr 2020.
Lee JS, Adhikari NKJ, Kwon HY, Teo K, Siemieniuk R, Lamontagne F, et al. Anti-Ebola therapy for patients with Ebola virus disease: a systematic review. BMC Infect Dis. 2019;19(1):376. https://doi.org/10.1186/s12879-019-3980-9.
Shiraki K, Daikoku T. Favipiravir, an anti-influenza drug against life-threatening RNA virus infections. Pharmacol Ther. 2020;209:107512. https://doi.org/10.1016/j.pharmthera.2020.107512.
Eloy P, Solas C, Touret F, Mentré F, Malvy D, de Lamballerie X, et al. Dose rationale for Favipiravir use in patients infected with SARS-CoV-2. Clin Pharmacol Ther. 2020;108(2):188. https://doi.org/10.1002/cpt.1877.
Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gøtzsche PC, Ioannidis JP, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ. 2009;339(jul21 1):b2700. https://doi.org/10.1136/bmj.b2700.
Stroup DF, Berlin JA, Morton SC, Olkin I, Williamson GD, Rennie D, et al. Meta-analysis of observational studies in epidemiology: a proposal for reporting. Meta-analysis of observational studies in epidemiology (MOOSE) group. JAMA. 2000;283(15):2008–12. https://doi.org/10.1001/jama.283.15.2008.
Sterne JAC, Savović J, Page MJ, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ. 2019;366:l4898.
Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck-Ytter Y, Alonso-Coello P, et al. GRADE Working Group. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ. 2008;336(7650):924–6. https://doi.org/10.1136/bmj.39489.470347.
Agresti A, Coull BA. Approximate is better than “exact” for interval estimation of binomial proportions. Am Stat. 1998;52:119–26.
Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327(7414):557–60. https://doi.org/10.1136/bmj.327.7414.557.
Review Manager (RevMan) [Computer program] Version 5.3 Copenhagen: The Nordic Cochrane Center, The Cochrane Collaboration, 2014.
Chen C, Zhang Y, Huang J, Yin P, Cheng Z, Wu J, et al. Favipiravir versus Arbidol for COVID-19: A Randomized Clinical Trial. medRxiv 2020.03.17.20037432. https://doi.org/10.1101/2020.03.17.20037432.
Yan Lou, Lin Liu, Yunqing Qiu. Clinical Outcomes and Plasma Concentrations of Baloxavir Marboxil and Favipiravir in COVID-19 Patients: an Exploratory Randomized, Controlled Trial. medRxiv 2020.04.29.20085761; doi: https://doi.org/10.1101/2020.04.29.20085761.
Ivashchenko AA, Dmitriev KA, Vostokova NV, Azarova VN, Blinow AA, Egorova AN, et al. AVIFAVIR for Treatment of Patients with Moderate COVID-19: Interim Results of a Phase II/III Multicenter Randomized Clinical Trial. medRxiv 2020.07.26.20154724. https://doi.org/10.1101/2020.07.26.20154724.
Doi Y, Hibino M, Hase R, Yamamoto M, Kasamatsu Y, Hirose M, et al. A prospective, randomized, open-label trial of early versus late favipiravir in hospitalized patients with COVID-19. Antimicrob Agents Chemother. 2020 AAC.01897–20. Epub ahead of print. https://doi.org/10.1128/AAC.01897-20.
Cai Q, Yang M, Liu D, Chen J, Shu D, Xia J, et al. Experimental treatment with Favipiravir for COVID-19: an open-label control study. Engineering (Beijing). 2020;6(10):1192–8. https://doi.org/10.1016/j.eng.2020.03.007.
Rattanaumpawan P, Jirajariyavej S, Lerdlamyong K, Palavutitotai N, Saiyarin J. Real-world Experience with Favipiravir for Treatment of COVID-19 in Thailand: Results from a Multi-center Observational Study. medRxiv. 2020.06.24.20133249. https://doi.org/10.1101/2020.06.24.20133249.
Yamamura H, Matsuura H, Nakagawa J, Fukuoka H, Domi H, Chujoh S. Effect of favipiravir and an anti-inflammatory strategy for COVID-19. Crit Care. 2020;413(1):413. https://doi.org/10.1186/s13054-020-03137-5.
Doi K, Ikeda M, Hayase N, Moriya K, Morimura N, COVID-UTH Study Group. Nafamostat mesylate treatment in combination with favipiravir for patients critically ill with Covid-19: a case series. Crit Care. 2020;24(1):392. https://doi.org/10.1186/s13054-020-03078-z.
Murohashi K, Hagiwara E, Kitayama T, Yamaya T, Higa K, Sato Y, et al. Outcome of early-stage combination treatment with favipiravir and methylprednisolone for severe COVID-19 pneumonia: A report of 11 cases. Respir Investig. 2020:S2212–5345(20)30117–9. https://doi.org/10.1016/j.resinv.2020.08.001.
Çalik BaŞaran NC, Uyaroğlu OA, Telli Dizman G, Özişik L, Şahin TK, Taş Z, et al. Outcome of non-critical COVID-19 patients with early hospitalization and early antiviral treatment outside the ICU. Turk J Med Sci. 2020. https://doi.org/10.3906/sag-2006-173 Epub ahead of print.
Yaylaci S, Dheir H, Şenocak D, Genc AB, Kocayigit H, Çekiç D, et al. The effects of favipiravir on hematological parameters of covıd-19 patients. Rev Assoc Med Bras. 2020;66 Suppl 2(Suppl 2):65–70. https://doi.org/10.1590/1806-9282.66.S2.65.
Furuta Y, Takahashi K, Shiraki K, Sakamoto K, Smee DF, Barnard DL, et al. T-705 (favipiravir) and related compounds: novel broad-spectrum inhibitors of RNA viral infections. Antivir Res. 2009;82(3):95–102. https://doi.org/10.1016/j.antiviral.2009.02.198.
Xi X, Xu Y, Jiang L, Li A, Duan J, Du B, et al. Hospitalized adult patients with 2009 influenza A(H1N1) in Beijing, China: risk factors for hospital mortality. BMC Infect Dis. 2010;10:–256. https://doi.org/10.1186/1471-2334-10-256.
Walsh KA, Jordan K, Clyne B, Rohde D, Drummond L, Byrne P, et al. SARS-CoV-2 detection, viral load and infectivity over the course of an infection. J Infect. 2020;81(3):357–71. https://doi.org/10.1016/j.jinf.2020.06.067 Epub 2020 Jun 29. PMID: 32615199; PMCID: PMC7323671.
Klimczak A. Perspectives on mesenchymal stem/progenitor cells and their derivates as potential therapies for lung damage caused by COVID-19. World J Stem Cells. 2020;12(9):1013–22. https://doi.org/10.4252/wjsc.v12.i9.1013.
Sissoko D, Laouenan C, Folkesson E, M'Lebing AB, Beavogui AH, Baize S, et al. JIKI Study Group. Experimental Treatment with Favipiravir for Ebola Virus Disease (the JIKI Trial): A Historically Controlled, Single-Arm Proof-of-Concept Trial in Guinea. PLoS Med. 2016;13(3):e1001967. https://doi.org/10.1371/journal.pmed.1001967.
Mentré F, Taburet A-M, Guedj J, Anglaret X, Keïta S, Lamballerie X, et al. Dose regimen of favipiravir for Ebola virus disease. Lancet Infect Dis. 2015;15(2):150–1. https://doi.org/10.1016/S1473-3099(14)71047-3.
Sanders JM, Monogue ML, Jodlowski TZ, Cutrell JB. Pharmacologic treatments for coronavirus disease 2019 (COVID-19): a review. JAMA. 2020;323(18):1824–36. https://doi.org/10.1001/jama.2020.6019.
Acknowledgements
Authors thank to Serika Nakamura for her general assistance.
Funding
The study was supported by a grant from Japan Science and Technology (JST), JST-Mirai Program (#20345310). The funders had no role in the design, methods, participant recruitment, data collection, analysis, or preparation of the paper.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: TM and KK. Performed the experiments: TM, DK, HA, KK. Analysed the data: TM. Interpreted the study results: TM, DK, HA, KK. Supervision: KK. Wrote the first draft of the manuscript: TM. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The institutional review board and patient consent were not required because of the review nature of this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Manabe, T., Kambayashi, D., Akatsu, H. et al. Favipiravir for the treatment of patients with COVID-19: a systematic review and meta-analysis. BMC Infect Dis 21, 489 (2021). https://doi.org/10.1186/s12879-021-06164-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-021-06164-x