
Abstract
Background
Chronic mucocutaneous candidiasis (CMC) is the most common clinical symptom of singer transducer and signal transducer and activator of transcription 1 (STAT1) gain-of-function (GOF) mutations. Bronchiectasis is a chronic lung disease that is characterized by permanent bronchiectasis, causing cough, expectoration, and even haemoptysis. The underlying pathogeny is not yet clear. Immunoglobulin (Ig) A is derived from memory B cells and correlates with immune-related diseases. STAT1 is closely associated with signal transmission and immune regulation.
Case presentation
We report a 17-year-old male patient carrying a GOF mutation in STAT1. The variant led to CMC, bronchiectasis, and elevated serum IgA levels, as well as stunting. Whole-exome sequencing (WES) revealed a c.986C>G (p.P329R) heterozygous mutation in the STAT1 gene.
Conclusion
Further Sanger sequencing analysis of STAT1 in the patient and his parents showed that the patient harboured a de novo mutation.
Similar content being viewed by others

Background
The STAT family of proteins are latent cytoplasmic transcription factors. When the proteins are translocated to the nucleus and bind to the corresponding cytokine or growth factor receptor, they are phosphorylated and activated by kinases, interferon, inflammatory factors, epidermal growth factor (EGF) and colony-stimulating factors (CFS) [1,2,3]. For example, STAT3 is activated by tyrosine phosphorylation in response to EGF and IL-6 [3]; Stat2 becomes phosphorylated after IFN-α treatment [4], and IFN-γ induces tyrosine phosphorylation of STAT91 [5]. The Janus kinase (JAK) family tyrosine phosphorylate STAT family proteins. Activated STAT proteins are translocated to the nucleus to promote transcription, forming the classic JAK-STAT pathway, which is involved in many important biological signal transmissions [6]. There are many members of the STAT family, 7 of which have been clearly defined: STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b and STAT6 [7, 8]. Among them, STAT1 was the first identified, and it is activated by IFN-α/β [6, 9], which plays an important role in autoimmune diseases [10], such as asthma [11], rheumatoid arthritis [12], and inflammatory bowel disease [13]. Mutations in STAT1 can affect the immune function mediated by IFN-γ and IFN-α/β, thereby affecting host immune defence capacity and increasing susceptibility to mycobacteria, fungi and viruses [14]. STAT1 is both a target of loss-of function (LOF) and GOF mutations [15]. Since STAT1 GOF mutations were first reported in 2011 [16], they have been identified in a growing number of patients and have attracted increasing attention.
GOF mutations in STAT1 are frequently enriched in the coiled-coil.
domain or DNA-binding domain [17], with a variety of clinical manifestations, including CMC infection, autoimmune diseases, and infection leading to early death [18]. Here, we report a case of pathogenic STAT1 GOF mutation in a young male in China with severe, recurrent and persistent pulmonary bacterial infections and aphthous stomatitis since childhood and who then developed bronchiectasis and increased IgA. The patient has undergone many repeated examinations in the past 10 years to confirm the diagnosis. However, genetic analysis was not performed until at age 17, revealing a novel STAT1 GOF mutation (c.986C> G). We propose that patients with unexplained chronic aphthous stomatitis, pulmonary bacterial infections, bronchiectasis and an increase in immunoglobulin IgA may carry STAT1 GOF mutations.
Case presentation
This case report was approved by Wuhan University Zhongnan Hospital. The 17-year-old patient has been slower than his peers in development and repeatedly experienced pulmonary bacterial infections and aphthous stomatitis since childhood. He became infected with varicella-zoster virus (VZV) and developed lung abscess when he was 10 years old. He was diagnosed with bronchiectasis and elevation of IgA (6.43 g/L) for the first time at the age of 15 years (Fig. 1a). The patient was treated with antiviral and antibiotics treatments discontinuous, and the treatments were interrupted when the symptoms were relieved. At the age of 17, he was 160 cm tall and weighed 40 kg, which was significantly shorter than a male of the same age. He revisited our hospital due to repeated coughing and the symptoms progressed with the treatment of oral antibiotics at home.

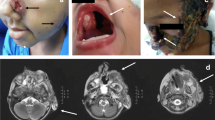
Severe bronchiectasis and bone marrow puncture in the 17-year-old male patient. a Bilateral bronchiectasis and bronchiolitis on chest computed tomography scan in the 16-year-old. b Cell morphous was observed by microscope of bone marrow puncture. c Bilateral bronchiectasis and bronchiolitis on chest computed tomography scan in the 17-year-old. d Bilateral bronchiectasis and bronchiolitis on chest PETCT
Further tests were conducted to determine the underlying pathogenesis, including quantitative serum immunoglobulin immunofixation electrophoresis and autoimmune disease-related antibody assays. We found no abnormality when detecting antibodies related to autoimmune diseases, with no abnormal bands by immunofixation electrophoresis. Therefore, we can rule out the common autoimmune diseases such as rheumatism, systemic lupus erythematosus and Sjogren’s syndrome. Levels of serum IgG (11.70 g/L) and IgM (1.00 g/L) were normal, but those of IgA (6.91 g/L) were elevated. We performed a bone marrow aspiration examination to rule out blood system diseases (Fig. 1b). The patient had repeated coughing; he was diagnosed with bronchiectasis many years ago, but the aetiology was not determined. Therefore, we reperformed chest CT, and the results indicated bronchiectasis (Fig. 1c), which was consistent with his previous diagnosis. At the same time, we performed whole-body positron emission tomography (PET) CT. Despite the presence of bronchiectasis, there were no signs of malignancy (Fig. 1d).
Since the examinations could not clarify the original cause of the patient’s condition and the patient has been ill since childhood, the possibility of genetically related disease was considered, and we further performed WES at the Institute of Hematology, Hematology Hospital, Chinese Academy of Medical Sciences with the consent of the patient’s parents. The report identified a missense variant (NM_007315.3, Exon11, c.986C>G, p.P329R) of the STAT1 gene (chr2:191856005) (Fig. 2a). The inheritance of this mutation type is heterozygous autosomal dominant (AD). The mutation occurs in the STAT DNA-binding domain. According to the American Society of Medical Genetics and Genomics (ACMG) guidelines, the c.986C>G mutation of the STAT1 gene is a suspected pathogenic mutation. The mutation was not found in Human Exon Database (ExAC), Reference Population Thousand Genomes (1000G), or Population Genome Mutation Frequency Database (gnom AD). This mutation occurs at the same position as a mutation that has been determined to be pathogenic, but it involves a different amino acid missense mutation. Based on functional prediction through SIFT (http://sift.jcvi.org/) and Polyphen (http://genetics.bwh.harvard.edu/pph2/), the mutant protein is harmful, leading to immunodeficiency 31C. The disease mainly manifests with repeated respiratory infections, immunodeficiency, immune disorders, recurrent infections, progressive lymphocytopenia, progressive memory B cell reduction, and progressive immunoglobulin reduction. Some patients have hypothyroidism. The disease characteristics are basically consistent with the phenotype of this case, but the level of IgA in this case was elevated. To clarify the origin of the patient’s STAT1 mutation, we performed Sanger sequencing of STAT1 in his parents. As his parents did not carry the variant (NM_007315.3, Exon 11, c.986C>G, p.P329R), this was a de novo mutation (Fig. 2b).

Pathogenic STAT1 variant identified in the 17-year-old male patient. a Integrative Genomics Viewer snapshot of the STAT1 pathogenic variant. (NM_007315.3, Exon11, c.986C>G, p.P329R) identified by whole-exome sequencing. b Validation of the STAT1 variant by Sanger sequencing. Chromatograms show the heterozygous variant in the proband (patient), indicated by the arrow, and the normal sequence in the unaffected parents
Discussion and conclusion
With STAT1 GOF mutations, the ability of the protein to respond to IFN-α/β/γ and IL-27 is enhanced, thereby suppressing the differentiation of T cells to IL-17-secreting T cells and decreasing IL-17 secretion. At the same time, STAT1 GOF mutation impairs the pathway by which IL-6, IL-21 and IL-23 induce the differentiation of IL-17 secretory T cells through STAT3, resulting in reduced IL-17 secretion and easily inducing CMC [16, 19]. The most common cause of CMC is a GOF STAT1 variant, with over half of these patients developing CMC [20]. A clinical case study reported in 2016 from France showed that 98% of patients with this type of mutation were infected with CMC [18]. Regarding our case, the patient had repeated aphthous stomatitis since childhood. The heterozygous c.986C>G (p.P329R) variant in STAT1 identified in our patient has not been reported before. This case report describes a new autosomal dominant GOF pathogenic variant in STAT1 that causes CMC.
CMC infection is the most common clinical symptom in patients with STAT1 GOF mutations, and bronchiectasis is common in these patients. In the study of Julie Toubiana’s group, 21% of the patients with STAT1 GOF mutation had bronchiectasis [18]. Impairment of class-switched memory B cells is involved in the pathogenesis of bronchiectasis [21, 22], and studies have reported a reduction in class-switched B cells in patients with STAT1 GOF mutation [23, 24]. Breuer and colleagues proposed that the main cause of bronchiectasis in patients with STAT1 GOF mutant is the reduction of memory B cells, but the links between STAT1 mutation and B cell dysfunction need further study [25]. In general, the level of IgA in patients with STAT1 GOF mutation and bronchiectasis should be decreased, though it was increased in our case. Bronchiectasis may be induced by repeat pulmonary bacterial infections [26], which might have been the pathogenesis of bronchiectasis in our patient.
Patients with STAT1 GOF mutation often experience bacterial infections and invasive fungal infections. Moreover, many patients have autoimmune manifestations, including hypothyroidism, type 1 diabetes, blood cytopenia, and systemic lupus erythaematosus. Invasive infections, cerebral aneurysms, and even cancers can occur. At the same time, many patients have normal results in initial immunologic investigations, such as lymphocyte subpopulations (T, B and NK cells) and immunoglobulin isotypes [18]. Therefore, it is very difficult to determine a patient’s diagnosis based on clinical symptoms or laboratory tests. Accordingly, genetic testing is currently the fastest approach to diagnosing such diseases. The patient in our case had repeated oral fungal and pulmonary bacterial infections since childhood as well as symptoms of immune dysfunction such as bronchiectasis and elevated levels of lgA, consistent with the clinical manifestations of STAT1 GOF mutation. We used WES to identify that the cause of the patient’s disease is STAT1 GOF mutation, and the results clarified that the patient’s variant was not inherited from his parents. This patient has also been stunted since childhood. At 17 years old, he weighed 40 kg, was 160 cm tall, and had a BMI of 15.625 kg/m2. The average BMI range of healthy young men is 18.5–23.9 kg/ m2. However, the development of his parents is within the range of normal adults, excluding the developmental retardation caused by genetic factors. Then we suspected this STAT1 variant might cause developmental retardation which is worth consideration and further research. This report describes the first patient carrying a pathogenic variant of the STAT1 gene causing CMC infection, bronchiectasis, elevated serum IgA levels and stunting. It is worth noting that WES can serve as an effective detection method to help quickly identify pathogenesis in the presence of diverse unexplainable symptoms.
Availability of data and materials
The necessary clinical data of the patient are presented in the case report.
There is no dataset to be shared in public repositories.
Abbreviations
- CMC:
-
Chronic mucocutaneous candidiasis
- STAT1:
-
Singer transducer and activator of transcription 1
- GOF:
-
Gain-of-function
- Ig A:
-
Immunoglobulin A
- WES:
-
Whole exome sequencing
- EGF:
-
Epidermal growth factor
- CFS:
-
Colony-stimulating factors
- AK:
-
Janus Kinase
- VZV:
-
Varicella-zoster virus
- AD:
-
Autosomal dominant
- PET:
-
Positron emission tomography
References
Sadowski HB, Shuai K, Darnell JE Jr. A common nuclear signal transduction pathway activated by growth factor and cytokine receptors. Science. 1993;261(5291):1739–44.
Wegenka UM, Lutticken C, Buschmann J. The interleukin-6-activated acute-phase response factor is antigenically and functionally related to members of the signal transducer and activator of transcription (STAT) family. Mol Cell Biol. 1994;14(6):3186–96.
Zhong Z, Wen Z Jr. JED: Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264(5155):95–8.
Schindler C, Shuai K, Prezioso VR, Darnell JE Jr. Interferon-dependent tyrosine phosphorylationof a latent cytoplasmic transcription factor. Science. 1992;257(5071):809–13.
Shuai K, Schindler C, Prezioso VR, Darnell JE Jr. Activation of transcription by IFN-y: tyrosine phosphorylation of a 91-kD-DNA binding protein. Science. 1992;258(5089):1808–12.
Darnell JE Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–21.
Darnell JE Jr. STATs and gene regulation. Science. 1997;277(5332):1630–5.
Vera J, Rateitschak K, Lange F, Kossow C, Wolkenhauer O, Jaster R. Systems biology of JAK-STAT signalling in human malignancies. Prog Biophys Mol Biol. 2011;106(2):426–34.
Dale TCIA, Kerr IM, Stark GR. Rapid activation by interferon alpha of a latent DNA-binding protein present in the cytoplasm of untreated cells. Proc Natl Acad Sci U S A. 1989;86(4):1203–7.
Chapgier A, Boisson-Dupuis S, Jouanguy E, et al. Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease. PLoS Genet. 2006;2(8):e131.
Sampath D, Look DC, Holtzman MJ. Constitutive activation of an epithelial signal transducer and activator of transcription (STAT) pathway in asthma. J Clin Invest. 1999;103(9):1353–61.
Yokota A, Narazaki M, Shima Y, et al. Preferential and persistent activation of the STAT1 pathway in rheumatoid synovial fluid cells. J Rheumatol. 2001;28(9):1952–9.
PR SS, Hampe J, Nikolaus S, Groessner B, Schottelius A, Kühbacher T, Hämling J, Fölsch UR, Seegert D. Activation of signal transducer and activator of transcription (STAT) 1 in human chronicinflammatory bowel disease. Gut. 2002;51(3):379–85.
Sampaio EP, Hsu AP, Pechacek J, et al. Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations and disseminated coccidioidomycosis and histoplasmosis. J Allergy Clin Immunol. 2013;131(6):1624–34 e1617.
Consonni F, Dotta L, Todaro F, et al. Signal transducer and activator of transcription gain-of-function primary immunodeficiency/.immunodysregulation disorders. Curr Opin Pediatr. 2017;29(6):711–7.
Liu L, Okada S, Kong XF, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. 2011;208(8):1635–48.
Lee JS, An Y, Yoon CJ, et al. Germline gain-of-function mutation of STAT1 rescued by somatic mosaicism in immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like disorder. J Allergy Clin Immunol. 2020;145(3):1017–21.
Toubiana J, Okada S, Hiller J, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. 2016;127(25):3154–64.
Van de Veerdonk FL, Plantinga TS, Hoischen A, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med. 2011;365(1):54–61.
Haake K, Wustefeld T, Merkert S, Luttge D, et al. Human STAT1 gain-of-function iPSC line from a patient suffering from chronic mucocutaneous candidiasis. Stem Cell Res. 2020;43:101713.
Alachkar H, Taubenheim N, Haeney MR, et al. Memory switched B cell percentage and not serum immunoglobulin concentration is associated with clinical complications in children and adults with specific antibody deficiency and common variable immunodeficiency. Clin Immunol. 2006;120(3):310–8.
Yong PL, Orange JS, Sullivan KE. Pediatric common variable immunodeficiency: immunologic and phenotypic associations with switched memory B cells. Pediatr Allergy Immunol Official Publ Eur Soc Pediatr Allergy Immunol. 2010;21(5):852–8.
Uzel G, Sampaio EP, Lawrence MG, Hsu AP, Hackett M, et al. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like. J Allergy Clin Immunol. 2013;131(6):1611e1623.
Frans G, Moens L, Schaballie H, et al. Gain-of-function mutations in signal transducer and activator of transcription 1 (STAT1): chronic mucocutaneous candidiasis accompanied by enamel defects and delayed dental shedding. J Allergy Clin Immunol. 2014;134(5):1209–13 e1206.
Breuer O, Daum H, Cohen-Cymberknoh M, et al. Autosomal dominant gain of function STAT1 mutation and severe bronchiectasis. Respir Med. 2017;126:39–45.
Olbrich P, Freeman AF. STAT1 and STAT3 mutations: important lessons for clinical immunologists. Expert Rev Clin Immunol. 2018;14(12):1029–41.
Acknowledgements
Not Applicable.
Funding
No fund was received in the preparation and submission of this case.
report.
Author information
Authors and Affiliations
Contributions
JW designed the plan of the work. YY and HS acquired the clinical data and analyzed the clinical features. YY and FX wrote the manuscript. FX and JW substantively revised it. JW interpreted the laboratory findings and revised the final article. All four authors participated in writing the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
None.
Consent for publication
Written informed consent was obtained from the parents of the patient for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yu, Y., Xu, F., Shen, H. et al. Chronic Candida infection, bronchiectasis, immunoglobulin abnormalities, and stunting: a case report of a natural mutation of STAT1 (c.986C>G) in an adolescent male. BMC Infect Dis 21, 38 (2021). https://doi.org/10.1186/s12879-020-05734-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-020-05734-9