
Abstract
Background
Febrile Urinary Tract Infection (FUTI) is frequently treated initially with intravenous antibiotics, followed by oral antibiotics guided by clinical response and bacterial susceptibility patterns. Due to increasing infection rates with multiresistant Enterobacteriaceae, antibiotic options for stepdown treatment decline and patients more frequently require continued intravenous antibiotic treatment for FUTI. Fosfomycin is an antibiotic with high bactericidal activity against Escherichia coli and current resistance rates are low in most countries. Oral Fosfomycin-Trometamol 3000 mg (FT) reaches appropriate antibiotic concentrations in urine and blood and is considered safe. As such, it is a potential alternative for stepdown treatment.
Methods
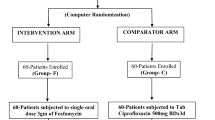
The FORECAST study (Fosfomycin Randomized controlled trial for E.coli urinary tract infections as Alternative Stepdown Treatment) is a randomized, double-blind, double-dummy, non-inferiority trial in which 240 patients will be randomly allocated to a stepdown treatment with FT or ciprofloxacin (standard of care) for FUTI, caused by Escherichia coli with in vitro susceptibility to both antibiotics. The study population consists of consenting female patients (≥18 years) with community acquired E. coli FUTI. After intravenous antibiotic treatment during at least 48 (but less than 120) hours, and if eligibility criteria for iv-oral switch are met, patients receive either FT (3 g every 24 h) or ciprofloxacin (500 mg every 12 h) for a total antibiotic duration of 10 days. The primary endpoint is clinical cure (resolution of symptoms) 6–10 days post-treatment. Secondary endpoints are microbiological cure 6–10 days post-treatment, clinical cure, mortality, ICU admittance, relapse, reinfection, readmission, additional antibiotic use for UTI, early study discontinuation, adverse events, days of hospitalization and days of absenteeism within 30–35 days post-treatment. The sample size is based on achieving non-inferiority on the primary endpoint, applying a non-inferiority margin of 10%, a two-sided p-value of < 0.05 and a power of 80%.
Discussion
The study aims to demonstrate non-inferiority of oral fosfomycin, compared to oral ciprofloxacin, in the stepdown treatment of E. coli FUTI.
Trial registration
Registered at the Nederlands trial register (Dutch trial register) on 4-10-2017. Trial registration number: NTR6449. Secondary ID (national authority): NL60186.041.17.
Similar content being viewed by others
Background
Urinary Tract Infections (UTI) are the most common bacterial infections requiring antibiotic treatment in the western world [1]. Complicated urinary tract infections (cUTI) are defined upon the presence of systemic symptoms or upon the susceptibility of the host for a complicated course [2]. Systemic symptoms are often fever, febrile UTI (FUTI), or other symptoms, reflecting the presence of tissue infection such as pyelonephritis, prostatitis or the urosepsis syndrome [2]. Guidelines recommend to treat FUTI with a 7–14 day course of antibiotics [2, 3] and in the majority of cases empiric intravenous antibiotics will be followed by oral stepdown therapy. The choice of stepdown treatment should be targeted to the susceptibility pattern of the causal uropathogen. Unfortunately, antibiotic options for stepdown treatment are becoming limited as the result of increasing antibiotic resistance [2].
Escherichia coli (E.coli) is the causative organism in around 70–75% of FUTI [4, 5]. In 2017, resistance rates of E. coli isolates of patients admitted to Dutch hospitals were 14% for ciprofloxacin (CPX), 23% for trimethoprim-sulfamethoxazole and 36% for amoxicillin-clavulanic acid with higher resistance rates in patients from urology departments [6]. Though this leaves fluoroquinolones still as one of the oral antibiotics to treat FUTI, one of the goals of antibiotic stewardship is to reduce fluoroquinolone use because of its selective properties for antibiotic resistance [2]. In the Netherlands, about 2–5% of patients hospitalized for community-acquired FUTI cannot be treated with oral antibiotics due to antibiotic resistance of the causal uropathogen [7]. This implies the need of prolonged intravenous antibiotic treatment, prolonged hospitalization, increased healthcare costs and higher risks of forthcoming complications.
Fosfomycin is an alternative antibiotic treatment for the stepdown treatment of FUTI [8, 9]. Fosfomycin is a phosphoenolpyruvate analogue and orally available as Fosfomycin-Trometamol (FT). It has been used extensively for uncomplicated cystitis in women, has a good safety profile [10] and possesses a suitable pharmacokinetic profile in healthy volunteers.
In the FORECAST trial (FOsfomycin Randomized controlled trial for E.coli urinary tract infections as Alternative Stepdown Treatment), we will investigate the efficacy of FT when used as stepdown therapy of FUTI in women.
Methods
The objective of this randomized controlled double-blind, double-dummy, non-inferiority, multicentre, investigator-initiated trial is to determine whether oral FT is non-inferior to oral CPX in the step-down treatment of E.coli FUTI in women for achieving clinical cure. The study will be performed in 15 Dutch hospitals; 4 academic centers and 11 large teaching hospitals.
Participants
Inclusion criteria
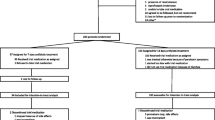
Competent women (≥18 years), that are hospitalized for the presumed diagnosis FUTI and receive appropriate empirical intravenous antibiotics for ≥48 - ≤120 h, who are judged to be eligible for an intravenous-oral switch by the attending physician, according to the Dutch guideline, that recommends to switch therapy until the patient has been afebrile for 24–48 h and symptoms have improved [2]. Urine (≥104 CFU/ml) or blood culture must reveal E.coli, susceptible to both CPX and fosfomycin. A patient is not eligible if non-E.coli-type Enterobacteriaceae are present in urine culture (≥103 CFU/ml) or blood culture.
FUTI is defined as UTI with at least one forthcoming systemic symptom or sign and one local symptom at presentation. Local symptoms are dysuria, urinary urgency, urinary frequency, suprapubic/pelvic discomfort, macroscopic hematuria, new urinary incontinence or worsening of pre-existing incontinence, lower abdominal pain, low back pain, flank pain, costo-vertebral angle pain or tenderness on physical examination. Systemic symptoms or signs are fever (≥38.0 C ͦ) or low temperature (< 36 C ͦ), rigors, delirium, hemodynamic instability as a result of sepsis requiring intravenous fluids or an increase in CRP (≥30 mg/L) or leucocytes (≥12*109/L). Local symptoms are not required in case the urine and blood culture yield are positive for a phenotypically identical matched E.coli and UTI is the presumed source of infection according to the treating physician.
Adequate empirical intravenous treatment may consist of amoxicillin+/−clavulanic acid, 2nd or 3rd generation cephalosporin, aminoglycoside, carbapenem, fluoroquinolones, trimethoprim-sulfamethoxazole or a combination and in vitro susceptibility of the causative E.coli to at least one of the used agents.
Exclusion criteria
Pregnancy or breastfeeding, glomerular filtration rate below 30 ml/min/1,73 m3 or renal replacement therapy, concomitant systemic antibacterial treatment, ascertained or presumptive hypersensitivity to (compounds of) the study products, participation in any trial with an investigational product involved in the 30 days before the screening visit, specific patient groups: patients with renal transplant, polycystic kidney disease, neutropenia (< 500 /μl), paraplegia, suspicion/presence of renal abscess, suspicion of septic metastatic foci/endocarditis, urostomy, ileal loops, long-term urinary catheter (placed ≥24 h before admission), e.g., double-J catheter, nephrostomy catheter, suprapubic catheter, specific contra-indications for CPX or fosfomycin: concurrent use of tizanidin, clozapin or theophylline, a history of tendon disease/disorder related to quinolone treatment, patients with known risk factors for prolongation of the QT interval, glucose-6-phosphate dehydrogenase deficiency, inadequate understanding of the study risks or its requirements or unwilling to plan a follow-up visit and every other laboratory result, clinical condition, disease or treatment that, in investigator’s opinion, make the subject non suitable for the study. Patients with partial obstruction of the urinary tract, ureteral stones or intermittent catheterization will not be excluded.
Setting
Potential subjects for inclusion will be identified through daily screening for E. coli in urine and blood cultures in the clinical microbiology laboratories of participating centres. The study investigator will assess potential subjects for eligibility with consultation of the attending physician. After identification, eligible patients will be informed and, after obtaining consent, will be randomized.
Randomization
The data manager of the University Medical Centre Utrecht constructed a randomization list, that connects the unique medication code with the active substance (1:1). Only the data manager and the pharmacist on duty, responsible for the deblinding process in case of emergency, have access to the randomization code. Constrained randomization will take place with permuted blocks, that contain a pre-specified number of treatment assignments in a random order. As we expect to include more than 200 participants in a two-arm trial, the probability of a significant imbalance is negligible [11]. To prevent imbalances between both study arms stratification will be performed per study centre, as each study centre handles their own local guideline for the empirical treatment of FUTI.
Treatment
Participants will be randomized for an intravenous-oral antibiotic switch to either FT 3000 mg every 24 h or CPX 500 mg every 12 h [1]. CPX is chosen for the control group, because most evidence exists for CPX as an oral treatment of FUTI [12, 13]. A double-dummy design is chosen as it is impossible to equalize fosfomycin granules to any form of CPX. An identical placebo will be used for both active substances. All medicines will be manufactured according to Good Manufacturing Principles by the pharmaceutical company Basic Pharma BV, the Netherlands. The duration of total antibiotic therapy is 10 days for all participants, of which 2–5 days empirical intravenous antibiotics and the remaining 5–8 days stepdown therapy with FT or CPX.
FT will be dosed 3 g every 24 h, which is based on a review of the scientific data and expert opinion on the treatment of FUTI and the pharmacokinetics of FT that was available at the moment the study was designed (May 2017). The use of FT every 24 h for more than a single dose was tolerated well in retrospective studies and according to personal off-label use [14]. We do not expect negative effects of accumulation as fosfomycin is registered for intravenous use in dosages as high as 24 g per 24 h [15]. A medication diary needs to be filled by the participant in order to improve medication adherence. All patients receive oral and written information on the trial, including possible risks of participating. Additionally, patients receive a medication folder with instructions for use, contra-indications, interactions and adverse events of both FT and CPX (probable, possible and seldom adverse events). For questions or concerns, participants could contact the local or principle investigator or an independent expert physician.
Data collection
In each hospital, authorized and qualified clinical investigators and research nurses will obtain informed consent for participation. Demographic, clinical and microbiological data will be collected and stored for at least 15 years. Data will be collected from questionnaires and supplemented with data from the electronic medical record, see Table 1.
All participating microbiological and clinical chemistry laboratories are accredited by the Dutch foundation for the promotion of the quality and the accreditation of laboratories in health care (ISO). Susceptibility to fosfomycin and CPX is tested using validated local susceptibility methods, following the European Committee on Antimicrobial Susceptibility (EUCAST) recommendations. The clinical (E. coli) isolates will be stored for additional susceptibility measurements, i.e. agar dilution for fosfomycin susceptibility, and molecular analysis.
The primary and secondary endpoints will be collected 6–10 days post-end-of-treatment during a visit and 30–35 days post-end-of-treatment by telephone. Data will be entered into the Case Report Form (CRF) pseudo-anonymously in each participating centre using an unique participant code. The data will be processed by the data management program ‘ResearchOnline’. All steps of the data process will be stored to be able to check validity and plausibility.
Endpoints
The primary endpoint is clinical cure (resolution of symptoms) 6–10 days post end-of-treatment. Definitions and criteria of primary and secondary endpoints are described in Table 2.
Ethics
This study has been set up in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines. The medical ethics committee of the University Medical Centre Utrecht approved the study protocol, followed by the Institutional Scientific Boards of each participating centre. Written informed consent is obtained prior to randomization. Structural protocol modifications, including amendments, will be communicated as soon as possible to the medical ethics committee, the trial register, and all involved investigators and participants.
Statistical analysis
Sample size calculation
We determined the sample size based on an assumed cure rate of 92.5% for both groups and a margin of inferiority of 10%, consistent with assumptions in similar UTI trials [5, 12, 13, 16]. Assuming that a difference of 10.0 percentage points or less is irrelevant and with the two-sided alpha set at 0.05, the sample size needed in the two groups is 109. The study has a 80% power to reject the null hypothesis that the clinical cure rate for the study arm (FT) is 10% lower than the cure rate for the standard of care (CPX). Equivalently, the likelihood is 80% that the two-sided 95.0% confidence interval for the difference in clinical cure rates will exclude a 10% difference in favour of standard care. In order to evaluate 109 patients in both treatment arms, taking into account 10% of lost participants for various reasons, 240 subjects will be enrolled in total.
Intention to treat and per-protocol analysis
Participants are evaluable if they are randomized and received at least one dose of the study drug, and all will be included in the intention to treat analysis. Analysis of all endpoints will be performed according to the intention-to-treat principle, on which data non-inferiority is based. A per-protocol analysis will be performed for the primary endpoint and the secondary endpoint ‘microbiological cure’ for patients that completed at least 80% of study medicines.
Statistical methods
For the primary endpoint, the difference between the study arm and standard of care (p < 0.05) will be calculated with a two-sided Z-score for proportions. Secondary outcomes will be analysed using the following tests: two-tailed Z-score for proportions (p < 0.05), t-test, chi-square, logistic regression, when appropriate.
Linear and logistic regression will be used to identify associations between patient, disease and treatment characteristics) with both study arms with regard to the primary and secondary endpoints. Clinical cure 6–10 and 30–35 days post-treatment in specific subgroups will be investigated in exploratory analyses, based on host or disease characteristics, e.g. age, BMI, Charlson index, use of immunosuppressive drugs, presence of host factors, presence of a short term indwelling catheter, creatinine clearance, diabetes mellitus, days of empirical intravenous antibiotics, class of empirical intravenous antibiotics, and concomitant bacteraemia.
Missing data
Attempts will be made to complete the data from all enrolled participants. Missing data will be tracked or retrieved by the coordinating team after consultation with the local team. Missing information will be extracted directly from the electronic patient file without disclosing patient identifiers. Multiple imputation will be used for missing data.
Safety
Our entry criteria are designated to reflect daily practice with regard to the safety of the participants. Candidates should be diagnosed with FUTI as the primary reason for hospitalization. All identified uropathogens must be E. coli susceptible to both fosfomycin and CPX. Patients should be treated with appropriate intravenous antibiotics for at least 48 h. The attending physician determines the eligibility for the intravenous-to-oral switch. Patients that require more than 5 days of intravenous antibiotics will be excluded as this is considered a complicated course. Our goal is to investigate a representative population by including as many patients as possible, and only exclude extremely vulnerable patients (e.g. non-competent patients, renal transplant recipients, pregnant women), patients that require an alternate antibiotic strategy (e.g. renal abscess, endocarditis) or patients for which other endpoints should be used to estimate efficacy (e.g. men). Adverse events should be actively reported in the medication diary by participants in order to measure tolerability. Furthermore, these will be checked actively with questionnaires at 6–10 days and 30–35 days post-end of treatment. De-blinding is possible 24/7 h in case severe harm could be potentially prevented, and after consultation of the coordinating investigator. Serious adverse events and Suspected unexpected serious adverse reactions (SUSARs) will be reported during follow-up. All participants are automatically insured by a subject and liability insurance.
The study will be monitored by qualified monitors from the University Medical Centre Utrecht. For each participating centre the following procedures will be checked after the first 5 inclusions and thereupon annually: at least 10% of signed Informed Consent forms, in- and exclusion criteria, defined variables including the primary endpoint, source data verification, missed SAE’s, or SUSAR’s.
An interim analysis will be performed by the Data and Safety Monitoring Board (DSMB) after inclusion of 50 and 100 participants. De-blinded data will be delivered by an independent statistician to the DSMB, which will function as an advisory board, i.e. it will provide non-binding advice to the principal investigator. After inclusion of 50 participants an interim analysis will be performed regarding adverse events, medication adherence and early study medicine discontinuation as a result of adverse events/intolerance. Based on these results, the DSMB could recommend to adjust the dosage of FT. After inclusion of 100 participants, an interim analysis will be performed regarding Serious Adverse Events (SAE’s), early study withdrawal and the primary endpoint. Based on these results, the DSMB could recommend to stop the study, in case the interim analysis shows a probability of finding non-inferiority at the final analysis of less than 5% based on the conditional power (futility) [17] or an unexpected high rate of possible related SAEs or early study withdrawals, in particular in the FT arm (safety).
Discussion
As a result of emerging antibiotic resistance among Enterobacteriaceae the options for oral antibiotic stepdown treatment of FUTI are becoming limited. Whereas new intravenous antibiotics are being developed and registered, little interest has been put in the development of oral applications in the past decade [4, 16, 18]. As a consequence, patients with FUTI caused by (or carrying) a multiresistant Enterobacteriaceae usually require full intravenous antibiotic regimens with a higher risk on subsequent complications, psychological burden and increased health care costs, partly due to the forthcoming extended length of hospital stay [19].
This has led to renewed interest in FT, a relatively infrequent used antibiotic, invented in 1969 [20]. FT could be a suitable choice for targeted stepdown treatment of E.coli FUTI. FT was non-inferior in comparison to carbapenems in observational studies considering cUTIs [8, 9]. This investigator-initiated randomized controlled trial aims to evaluate FT as stepdown treatment for E.coli FUTI in women.
The eligibility criteria of this study were defined such that results will be generalizable to the population being treated for FUTI in clinical practice. Consequently, patients with FUTI with a positive urine or blood culture revealing E.coli will be included if both local and systemic UTI signs or symptoms are present and the patient is treated with adequate intravenous antibiotics with the presumptive focus of infection being the urinary tract. The presence of fever is not required for enrolment, as we aimed to include certain vulnerable patient populations that sometimes do not develop fever (i.e. elderly, immunocompromised patients) during sepsis. In order to better fit our domain, minor changes were made in eligibility criteria after we recruited 15 participants [1]: As a systemic sign, we included an increase in CRP (≥30 mg/L) or leucocytes (≥12*109/L) [2]. Next, local symptoms are not required in case the urine and blood culture are positive for a phenotypically matched E.coli and UTI is the presumed source of infection according to the treating physician [3]. Short term urinary catheters, if placed at least within 24 h before admission, will be allowed, e.g. double-J catheter, nephrostomy catheter, suprapubic catheter [4]. Finally, a patient is not eligible if any non-E.coli-type Enterobacteriaceae are present in urine (≥103 CFU/ml) or blood culture.
As primary endpoint, clinical cure 6 to 10 days post-end of treatment was chosen, with, next to safety endpoints, microbiological cure at 6–10 days and clinical cure at 30–35 days post-end of treatment as secondary endpoints. For future perspectives, cost-effectiveness variables will be collected, i.e. length of hospital and intensive care stay and days of absenteeism, in order to perform a model-based cost-effectiveness analysis between oral FT as stepdown treatment versus a full intravenous antibiotic course (e.g. with carbapenems) for the treatment of multiresistant E.coli FUTI.
There were several aspects related to the spectrum of FT that guided the study design. First, it is not rational to use FT for the empirical treatment of FUTI. The pharmacokinetic-pharmacodynamics profile is presumably not suitable to treat FUTI empirically. One gift of 3 g (oral) FT results in a peak fosfomycin urinary concentration of 1.600 mg/L and concentrations above the Minimal Inhibitory Concentration (MIC) of most E.coli isolates (below 2-4 mg/L including Extended Spectrum Beta-Lactamase-ESBL) for about 48 h [21,22,23]. The same gift leads to a peak serum concentration of around 20 mg/L, declining below 4 mg/L within 8–12 h [21, 24]. Second, in our opinion, FT is investigated for its use as targeted stepdown treatment of E.coli pathogens only, as Klebsiella and Proteus spp. are less susceptible to fosfomycin with proportions of resistance to fosfomycin of 31 and 16%, respectively, in the Netherlands [25]. The prevalence of resistance to fosfomycin among E.coli isolates from urine samples has been stable for years (+ − 1%) in the Netherlands [25], and is even low among ESBL-producing Enterobacteriaceae [26]. Finally, determination of fosfomycin susceptibility in daily practice seems more reliable for E.coli than for non-E.coli Enterobacteriaceae [27]. Third, although FT has been used in men to treat prostatitis and FUTI, we decided not to include men as they require longer antibiotic treatment and because treatment failure expresses itself as relapses for an extended period of time [12]. Stratification for gender has been considered, but would, within the limits of perceived feasibility, reduce statistical power to draw meaningful conclusions about non-inferiority. Finally, a study that was published recently evaluated the tolerability and pharmacokinetics of using FT every 48 h for three doses versus every 24 h for 7 doses [24]. The ‘every 24 hours’ arm was associated with significantly less days of being free of diarrhoea, however no subject discontinued FT due to adverse events. The study was not placebo-controlled, and was performed in healthy volunteers, therefore it is unknown how this reflects on the participants in this study. For safety reasons, adverse events and adherence will be evaluated during the interim analyses after 50 participants. Currently, the FOREST study (http://www.clinicaltrial.gov, NCT02142751) compares the efficacy of intravenous fosfomycin to meropenem for bacteraemic ESBL-E.coli UTI. In this study, intravenous fosfomycin is followed by stepdown therapy with 3 g FT every 48 h [18].
In summary, the FORECAST study will determine the efficacy of fosfomycin-trometamol, compared to CPX, in the stepdown treatment of E.coli FUTI in women.
Abbreviations
- CPX:
-
Ciprofloxacin
- CRF:
-
Case Report Form
- DSMB:
-
Data and Safety Monitoring Board
- E.coli:
-
Escherichia coli
- ESBL:
-
Extended Spectrum Beta-Lactamase
- EUCAST:
-
European Committee on Antimicrobial Susceptibility
- FORECAST:
-
FOsfomycin Randomized controlled trial for E.coli Complicated urinary tract infections as Alternative Stepdown Treatment
- FT:
-
Fosfomycin-Trometamol
- FUTI:
-
Urinary Tract Infection with systemic symptoms or signs
- MIC:
-
Minimal Inhibitory Concentration
- SAE:
-
Serious Adverse Event
- SUSAR:
-
Suspected Unexpected Serious Adverse Reactions
- UTI:
-
Urinary Tract Infection
References
Flores-Mireles AL, Walker JN, Caparon M, Hultgren SJ. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat Rev Microbiol. 2015;13(5):269–84.
Stichting Werkgroep Antibioticabeleid. SWAB Guidelines for Antimicrobial Therapy of Complicated Urinary Tract Infections in Adults; 2013. p. 1–94. Available from: http://www.swab.nl/richtlijnen
Gupta K, Hooton TM, Naber KG, Wullt B, Colgan R, Miller LG, et al. International clinical practice guidelines for the treatment of acute uncomplicated cystitis and pyelonephritis in women: a 2010 update by the Infectious Diseases Society of America and the European Society for Microbiology and Infectious Diseases. Clin Infect Dis. 2011;52(5):103–20.
Wagenlehner FM, Sobel JD, Newell P, Armstrong J, Huang X, Stone G, et al. Ceftazidime-avibactam versus Doripenem for the treatment of complicated urinary tract infections, including acute pyelonephritis: RECAPTURE, a phase 3 randomized trial program. Clin Infect Dis. 2016;61(1):e5–12.
Naber KG, Llorens L, Kaniga K, Kotey P, Hedrich D, Redman R. Intravenous doripenem at 500 milligrams versus levofloxacin at 250 milligrams, with an option to switch to oral therapy, for treatment of complicated lower urinary tract infection and pyelonephritis. Antimicrob Agents Chemother. 2009;53(9):3782–92.
National institute of Public health and the Environment. Nethmap/Maran 2018 [Internet]. 2018. Available from: http://www.swab.nl/swab/cms3.nsf/viewdoc/20BCD3983B5C390AC12575850031D33D.
van der Starre WE, van Nieuwkoop C, Paltansing S, van’t Wout JW, Groeneveld GH, Becker MJ, et al. Risk factors for fluoroquinolone-resistant Escherichia coli in adults with community-onset febrile urinary tract infection. J Antimicrob Chemother. 2011;66(3):650–6.
Veve MP, Wagner JL, Kenney RM, Grunwald JL, Davis SL. Comparison of fosfomycin to ertapenem for outpatient or step-down therapy of extended-spectrum Beta-lactamase urinary tract infections. Int J Antimicrob Agents. 2016;48(1):56–60.
Senol S, Tasbakan M, Pullukcu H, Sipahi OR, Sipahi H, Yamazhan T, et al. Carbapenem versus Fosfomycin Tromethanol in the treatment of extended-Spectrum Beta-lactamase-producing Escherichia Coli -related complicated lower urinary tract infection. J Chemother. 2010;22(5):355–7.
Surber C. Product monograph of monurol. 2016;(version 4.0). Available from: http://www.paladin-labs.com/our_products/Monurol-Sachet-PM-En.pdf
Dettori J. The random allocation process: two things you need to know. Evid Based Spine Care J. 2010;1(3):7–9.
van Nieuwkoop C, van der Starre WE, Stalenhoef JE, van Aartrijk AM, van der Reijden TJK, Vollaard AM, et al. Treatment duration of febrile urinary tract infection: a pragmatic randomized, double-blind, placebo-controlled non-inferiority trial in men and women. BMC Med. 2017;15(1):70.
Sandberg T, Skoog G, Hermansson AB, Kahlmeter G, Kuylenstierna N, Lannergård A, et al. Ciprofloxacin for 7 days versus 14 days in women with acute pyelonephritis: a randomised, open-label and double-blind, placebo-controlled, non-inferiority trial. Lancet. 2012;380(9840):484–90.
Grayson ML, Macesic N, Trevillyan J, Ellis AG, Zeglinski PT, Hewitt NH, et al. Fosfomycin for treatment of prostatitis: new tricks for old dogs. Clin Infect Dis. 2015;61(7):1141–3.
Falagas ME, Kastoris AC, Kapaskelis AM, Karageorgopoulos DE. Fosfomycin for the treatment of multidrug-resistant, including extended-spectrum Beta-lactamase producing, Enterobacteriaceae infections: a systematic review. Lancet Infect Dis. 2010;10(1):43–50.
Wagenlehner FM, Umeh O, Steenbergen J, Yuan G, Darouiche RO. Ceftolozane-tazobactam compared with levofloxacin in the treatment of complicated urinary-tract infections, including pyelonephritis: a randomised, double-blind, phase 3 trial (ASPECT-cUTI). Lancet. 2015;385(9981):1949–56.
Bratton DJ, Williams HC, Kahan BC, Phillips PP, Nunn a J. When inferiority meets non-inferiority: implications for interim analyses. Clin Trials. 2012;9(5):605–9.
Rosso-Fernández C, Sojo-Dorado J, Barriga A, Lavín-Alconero L, Palacios Z, López-Hernández I, et al. Fosfomycin versus meropenem in bacteraemic urinary tract infections caused by extended-spectrum β-lactamase-producing Escherichia coli (FOREST): study protocol for an investigator-driven randomised controlled trial. BMJ Open. 2015;5(3):e007363.
Sevinc F, Prins JM, Koopmans R, Langendijk PNJ, Bossuyt PMM, Dankert J, Speelman P. Early switch from intravenous to oral antibiotics: guidelines and implementation in a large teaching hospital. J Antimicrob Chemother. 1999;43:601–6.
Hendlin D, Stapley EO, Jackson M, Wallick H, Miller AK, Wolf FJ, et al. Phosphonomycin, a new antibiotic produced by strains of streptomyces. Science. 1969;166(3901):122–3.
Bergan T, Thorsteinsson SB, Albini E. Pharmacokinetic profile of fosfomycin trometamol. Chemotherapy Jan;39(5):297–301.
Vardakas KZ, Legakis NJ, Triarides N, Falagas ME. Susceptibility of contemporary isolates to fosfomycin: a systematic review of the literature. Int J Antimicrob Agents. 2016;47(4):269–85.
Wijma RA, Koch BCP, van Gelder T, Mouton JW. High interindividual variability in urinary fosfomycin concentrations in healthy female volunteers. Clin Microbiol Infect. 2018;24(5):528–32.
Wenzler E, Ellis-Grosse EJ, Rodvold KA. Pharmacokinetics, Safety, and Tolerability of Single Dose Intravenous (ZTI-01) and Oral Fosfomycin in Healthy Volunteers. Antimicrob Agents Chemother [Internet]; 2017. p. AAC.00775-17. Available from: http://aac.asm.org/lookup/doi/10.1128/AAC.00775-17
The Dutch Foundation of the Working Party on Antibiotic Policy. NethMap/Maran 2017; Consumption of antimicrobial agents and antimicrobial resistance among medically important bacteria in the Netherlands [internet]. 2017. Available from: http://www.swab.nl/swab/cms3.nsf/viewdoc/20BCD3983B5C390AC12575850031D33D
Cho YH, Il JS, Chung HS, Yu HS, Hwang EC, Kim SO, et al. Antimicrobial susceptibilities of extended-spectrum beta-lactamase-producing Escherichia coli and Klebsiella pneumoniae in health care-associated urinary tract infection: focus on susceptibility to fosfomycin. Int Urol Nephrol. 2015;47(7):1059–66.
van den Bijllaardt W, Schijffelen MJ, Bosboom RW, Cohen Stuart J, Diederen B, Kampinga G, et al. Susceptibility of ESBL Escherichia coli and Klebsiella pneumoniae to fosfomycin in the Netherlands and comparison of several testing methods including Etest, MIC test strip, Vitek2, Phoenix and disc diffusion. J Antimicrob Chemother [Internet]. 2018;(July):2380–2387. Available from: https://academic.oup.com/jac/advance-article/doi/10.1093/jac/dky214/5047839
Acknowledgements
We are grateful to all patients and their relatives for their participation in this study. We thank the staff (physicians, nurses, medical microbiologists, pharmacists, secretaries and assistants) at all participating sites for their cooperation. We thank Henri van Werkhoven for his statistical advice, Miquel van Ekkelenkamp and Joost Wiersinga for their role as independent expert physicians, Judith Vlooswijk, Edwin Boel, Rob Willems and Fernanda Paganelli for their microbiological expertise, and Johan Mouton and Rixt Wijma for their pharmacokinetic expertise.
Funding
Investigator-initiated study, sponsor: University Medical Center Utrecht (M.J.M. Bonten). The authors declare that there are no other sources of funding.
Availability of data and materials
Data sharing not applicable to this article as no datasets were yet generated or analysed during the current study.
Author information
Authors and Affiliations
Contributions
TtD, SvM, MB, CvN, SG and AH made substantial contributions to conception and design of the study. TtD drafted the protocol and performed the sample size calculation. All authors contributed to the manuscript and revised it critically for intellectual content and approved the final version to be published. All authors agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The medical ethics committee of the University Medical Centre Utrecht approved the study protocol, followed by the Institutional Scientific Boards of the following Dutch participating centres:
-
Amphia Hospital, Breda
-
Maastricht University Medical Center, Maastricht
-
Academic Medical Center, Amsterdam
-
Haaglanden Medical Center, The Hague
-
Diakonessenhuis hospital, Utrecht
-
St. Catharina hospital, Eindhoven
-
Flevohospital, Almere
-
Hagahospital, The Hague
-
Leiden University Medical Centre, Leiden
-
Rijnstate hospital, Arnhem
-
Spaarne Gasthuis, Harlem
-
St. Antonius hospital, Nieuwegein
-
Tergooi hospital, Hilversum
-
University Medical Centre, Utrecht
-
Viecuri Medical Centre, North-Limburg
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
ten Doesschate, T., van Mens, S.P., van Nieuwkoop, C. et al. Oral fosfomycin versus ciprofloxacin in women with E.coli febrile urinary tract infection, a double-blind placebo-controlled randomized controlled non-inferiority trial (FORECAST). BMC Infect Dis 18, 626 (2018). https://doi.org/10.1186/s12879-018-3562-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-018-3562-2