
Abstract
Background
The incidence and severity of tuberculosis chemotherapy toxicity is poorly characterised. We used data available from patients in the REMoxTB trial to provide an assessment of the risks associated with the standard regimen and two experimental regimens containing moxifloxacin.
Methods
All grade 3 & 4 adverse events (AEs) and their relationship to treatment for patients who had taken at least one dose of therapy in the REMoxTB clinical trial were recorded. Univariable logistic regression was used to test the relationship of baseline characteristics to the incidence of grade 3 & 4 AEs and significant characteristics (p < 0.10) were incorporated into a multivariable model. The timing of AEs during therapy was analysed in standard therapy and the experimental arms. Logistic regression was used to investigate the relationship between AEs (total and related-only) and microbiological cure on treatment.
Results
In the standard therapy arm 57 (8.9%) of 639 patients experienced ≥1 related AEs with 80 of the total 113 related events (70.8%) occurring in the intensive phase of treatment. Both four-month experimental arms (“isoniazid arm” with moxifloxacin substituted for ethambutol & “ethambutol arm” with moxifloxacin substituted for isoniazid) had a lower total of related grade 3 & 4 AEs than standard therapy (63 & 65 vs 113 AEs). Female gender (adjOR 1.97, 95% CI 0.91–1.83) and HIV-positive status (adjOR 3.33, 95% CI 1.55–7.14) were significantly associated with experiencing ≥1 related AE (p < 0.05) on standard therapy. The most common adverse events on standard therapy related to hepatobiliary, musculoskeletal and metabolic disorders. Patients who experienced ≥1 related AE were more likely to fail treatment or relapse (adjOR 3.11, 95% CI 1.59–6.10, p < 0.001).
Conclusions
Most AEs considered related to standard therapy occurred in the intensive phase of treatment with female patients and HIV-positive patients demonstrating a significantly higher risk of AEs during treatment. Almost a tenth of standard therapy patients had a significant side effect, whereas both experimental arms recorded a lower incidence of toxicity. That patients with one or more AE are more likely to fail treatment suggests that treatment outcomes could be improved by identifying such patients through targeted monitoring.
Similar content being viewed by others


Background
Combination therapy for drug-susceptible tuberculosis (TB) using isoniazid (H), rifampicin (R), pyrazinamide (Z) and ethambutol (E) has been the standard of care for several decades. While there is recognised toxicity associated with some of the individual drugs (for example hepatotoxicity [1], peripheral neuropathy [2], and gastrointestinal upset [3]) there are few prospective studies of regimen-related toxicity [4,5,6,7]. Consequently, clinicians treating tuberculosis largely rely on retrospective or anecdotal evidence to guide their choices when patients experience adverse events.
Previous reports include female gender, alcohol use, human immunodeficiency virus (HIV) infection and certain ethnic groups [3,4,5, 8,9,10,11] as risk factors for experiencing adverse events during therapy. Accurate definition of the risk is confounded by the varying definitions of adverse events or drug-related toxicity, and uncertainty of the recording mechanism or its completeness. For example, ethambutol related impairment of visual acuity rates vary from 0.02% [12] and 9.4% [13] despite the importance of the complication and the simplicity of its measurement.
To overcome the difficulties of biased reporting we used the consistent adverse event reporting system used in the REMoxTB trial [14] to investigate drug related toxicity, as we believe that this is the most comprehensive source of safety data for standard tuberculosis therapy currently available. The aim of the paper was to accurately characterise the patients at greatest risk, the incidence and nature of the toxicity related to standard TB therapy, and to investigate the impact of toxicity on treatment outcomes. Additionally, the paper aimed to compare the incidence of toxicity for standard TB therapy and the two experimental arms in REMoxTB.
Methods
REMoxTB trial
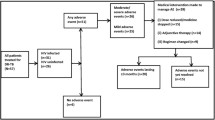
The REMoxTB trial [14] was a double-blind, placebo-controlled, randomised phase III trial to investigate two experimental moxifloxacin (M)-containing treatment regimens to treat pulmonary tuberculosis. There were 1931 patients randomised between 2007 and 2012 with 655 assigned to the “isoniazid arm” (2MHRZ/2MHR), 636 assigned to the “ethambutol arm” (2EMRZ/2MR), and 639 allocated to standard TB therapy as a control (2EHRZ/4HR). Patients were followed for 18 months after randomisation. We included all randomised patients in the REMoxTB trial who had received at least one dose of their allocated treatment.
Handling of safety data
Adverse events (AEs) were defined as any untoward medical occurrence in a patient administered the trial medication (with or without a causal relationship to the drugs) and were graded on a severity scale of 1 (least severe) to 4 (most severe) based on the Division of AIDS of the National Institute of Allergy and Infectious Diseases criteria [15]. The seriousness of an event (e.g. hospitalisation, life-threatening, death) irrespective of the severity was defined according to standard criteria. The local clinicians made the relatedness assessment for each event and those that were classified as “possibly”, “probably” or “definitely” related to drug therapy were considered to be “related” for the purposes of this analysis. Events that were assessed as either “unlikely related” or “not related” were considered to be “not related”. During the trial, sites were regularly monitored for data collection, and serious AEs (SAEs) were discussed in detail between the study medical monitor and the local clinician before being discussed in a safety board meeting with senior clinical specialists in the trial consortium to ensure quality control. Analyses and data handling were done using Stata statistical software version 14.1 (StataCorp, Texas).
Baseline characteristics in standard therapy
Baseline characteristics for all the patients assigned to standard therapy, and the characteristics for those patients who experienced one or more related or unrelated grade 3/4 AE and those who experienced one or more related grade 3/4 AE were tabulated. Univariable logistic regression was performed using each of the baseline characteristics for patients receiving standard therapy in the table against a binary outcome for experiencing one or more total or related grade 3/4 AE. Those variables with a p-value of < 0.10 were manually selected for inclusion in a multivariable model, with age, gender and baseline weight included regardless of univariable p value due to their clinical relevance. Random-effects multivariable logistic regression was used to test for associations between the selected variables and grade 3/4 AEs with trial centre used as the panel variable to account for any effect from the individual sites.
Adverse events in standard therapy over time
Incidence of grade 3/4 AEs and serious adverse events of any severity grade (SAEs) by treatment phase of standard therapy were categorised based on the grade 3/4 AE start date: intensive (weeks 0–8), continuation (weeks 9–26), and follow-up (week 27-month 18 after randomisation). MedDRA coding for System Organ Class and Preferred Term was used to identify the most common classes of adverse event. Patients were categorised based on the number of grade 3/4 AEs experienced in each phase of treatment. The mean number of SAEs per patient was calculated by dividing the total number of SAEs by the total number of patients with one or more SAE in each treatment phase. Patients who were withdrawn or died in the previous treatment phase were not included in later phases in order to present an accurate denominator for the number of patients at risk of an event in each of the three treatment phases. To illustrate the risk of grade 3/4 AE occurrence by time on standard treatment we constructed an Epanechnikov kernel smoothed hazard estimate with 95% confidence intervals and plotted the hazard function on the y-axis and the number of weeks from first dose on the x-axis.
Adverse events and treatment outcomes on standard therapy
The number of grade 3/4 AEs reported by each patient taking standard therapy was related to their microbiological outcome at 18 months after starting treatment. Patients were scheduled for 8 weekly visits followed by 8 visits until 18 months after randomisation, and early morning and spot sputum samples were to be collected (where possible) at each visit. Sputum culture results were available for both solid and liquid media in the trial database.
For this analysis, cure was defined as patients who were culture negative at either 18 months or when they were last reviewed in the trial with at least two consecutive negative cultures on both solid and liquid media prior to their final negative result. This outcome was based exclusively on recorded culture status and was independent of the patient’s outcome in the original publication [14]. Patients were grouped according to whether they had experienced ≥1 total or related grade 3/4 AE, and the proportions of cured patients in these groups were tabulated. The Chi square test was used to test for significance and binary logistic regression with cure as an outcome was used to test the association between experiencing ≥1 total or related grade 3/4 AE and odds of cure. Sex, age, baseline weight and HIV status were included in a multivariable logistic model.
Adverse events in all treatment arms
The incidence and classification of grade 3 or 4 adverse events (grade 3/4 AEs) and number of patients affected were calculated across the treatment arms according to the timing of the event: weeks 0–8 (EHRZ received on standard arm; MHRZ on isoniazid arm; EMRZ on ethambutol arm), weeks 9–17 (HR on standard arm; MHR on isoniazid arm; MR on ethambutol arm), weeks 18–26 (HR on standard arm; placebo on both isoniazid and ethambutol arms), and months 7–18 (no treatment administered for any arm and in trial follow-up). Grade 3/4 AEs were considered “clinically significant”. The proportion of patients with one or more grade 3/4 AE was compared across the treatment arms at each time window using the Chi square test. Time to first grade 3/4 AE in days after the first dose received was taken as the event of interest and Kaplan-Meier curves were constructed to illustrate the timing of grade 3/4 AEs in the three arms. The log rank test was used to compare the time to event in the standard therapy arm against the experimental arms individually.
Ethics approval and participant consent
The REMoxTB study was carried out with approval from the ethics board at University College London, and this included approval for the use of data and samples collected in other studies to improve the diagnosis and treatment of tuberculosis. All randomised patients agreed to any data and samples collected as part of the trial being used in further studies to improve the diagnosis and treatment of tuberculosis, as stated on the informed consent form for the study. All the research activities and data collection for the study was compliant with the Helsinki Declaration and the principles of Good Clinical Practice.
Results
Baseline characteristics for patients allocated to standard therapy
Of 639 patients taking standard therapy 57 (8.9%) experienced one or more grade 3/4 AEs judged to be related to their treatment, compared to 45 (6.9%) of 655 in the isoniazid and 40 (6.3%) of 636 in the ethambutol arm (p = 0.21, see Tables 1 and 5). Baseline weight as a categorical variable (OR 0.79, 95% CI 0.65–0.97), female sex (OR 1.60, 95% CI 1.06–2.39), and HIV infection (OR 3.45, 95% CI 1.86–6.42) were significantly associated with ≥1 grade 3/4 AE in univariable logistic regression. However, only HIV infection was significantly associated with experiencing any grade 3 or 4 AE in a multivariable model (adjOR 3.43, 95% CI 1.82–6.49). Female sex (adjOR 1.97, 95% CI 0.91–1.83) and HIV infection (adjOR 3.33, 95% CI 1.55–7.14) were significantly associated with grade 3/4 AEs considered related to standard therapy after being selected for inclusion in the multivariable model (both p values < 0.05, see Table 2).
Adverse events in standard therapy
Among the 113 related grade 3/4 AEs in the standard therapy group 80 (70.8%) were reported in the intensive phase of treatment (month 1 &2) as shown in Table 3 and illustrated in Fig. 1. Of the 57 patients who experienced ≥1 related grade 3/4 AE on treatment, 47 (82.5%) experienced an event in the intensive phase. The related adverse events most commonly reported were elevated liver enzymes (38 of 38 “hepatobiliary” events), arthralgia (15 of 22 “musculoskeletal” events), and diabetic complications (4 of 12 events attributed to “metabolism & nutrition”) (see Table 3). There was one case of deterioration in visual acuity reported in the standard arm (data not shown).

Hazard Curve for Related Grade 3 & 4 Adverse Events. Hazard Curve for Grade 3 or 4 Related Adverse Events According to Number of Weeks Since First Dose of Standard Therapy. Hazard function for the occurrence of a grade 3 or 4 related adverse event (with 95% confidence intervals) is plotted on the y axis, with the number of weeks following the first dose of standard tuberculosis therapy on the x axis. The rise in the hazard function after week 25 is accounted for by 2 events reported as “possibly” related to study drug
While the majority of SAEs were reported during therapy 10 of the 16 deaths in this treatment arm occurred after treatment was completed (see Table 3). The most common causes of death were trauma, suicide or unknown cause but presumed to be violent (8 of 16), and related to TB disease (3 of 16). Right heart failure, sepsis of unknown origin, and uncontrolled hypertension accounted for the remaining three deaths. None of the deaths in the standard therapy group were assessed as related to treatment.
Treatment outcomes for patients on standard therapy
Patients who had one or more total or related grade 3/4 AE were less likely to achieve microbiological cure compared to patients who did not experience a grade 3/4 AE (see Table 4). Of patients taking standard TB therapy, 21.1% (27 of 128) of patients with ≥1 grade 3/4 AE were not cured, compared to 9.2% (47 of 511) of patients who did not experience any grade 3/4 AE. Similarly, 26.3% (15 of 57) of patients who experienced ≥1 grade 3/4 AE considered related to treatment did not achieve cure compared to 10.1% (59 of 582) of patients who did not experience a related grade 3/4 AE (p value < 0.001).
Experiencing ≥1 related or unrelated grade 3/4 AE was significantly associated with not being cured of TB in a multivariable logistic regression model (adjOR 2.60, 95% CI 1.52–4.46, p < 0.001). A similar relationship was seen between ≥1 related-only grade 3/4 AE and an outcome of not cured (adjOR 3.11, 95% CI 1.59–6.10, p value < 0.001). The multivariable model included sex, age and baseline weight (clinical significance) and HIV status (due to earlier reported association with AE incidence).
Adverse events across all treatment arms over time
Most grade 3/4 AEs occurred during the intensive phase for all regimens (see Table 5) with 80 (73.4%), 51 (81.0%) and 44 (67.7%) related grade 3/4 AEs during the intensive phase in the standard, isoniazid, and ethambutol arms respectively. Both experimental arms had lower numbers of related grade 3/4 AEs (64 and 66 in the isoniazid and ethambutol arms vs 113 during standard therapy). There was a significant difference in the proportion of patients experiencing ≥1 related grade 3/4 AE in the intensive phase (p value 0.03) with the smallest proportion in the ethambutol arm (25 of 636 [3.9%], see Table 5). In all treatment arms the most common type of related grade 3/4 AEs were “hepatobiliary disorders” (40.7% in standard therapy, and 42.2% & 37.9% in isoniazid & ethambutol arms).
There was no difference in either the overall total or related total of grade 3/4 AEs between the three treatment arms during weeks 18–26 when patients in the experimental regimens were receiving placebo. The Kaplan Meier curves in Fig. 2 illustrate the majority of events occurring in the intensive phase followed by a plateau from approximately 9 weeks after starting treatment (log rank p = 0.19 for comparing standard therapy and isoniazid arm; p = 0.07 for standard therapy and ethambutol arm). The drop seen at 8 weeks of treatment in the number of patients at risk was driven by one site reporting 40 grade 3/4 AEs (30 considered related) from all treatment arms in a 30-week time window of the trial between May and December 2010 (the site reported a total of 146 grade 3/4 AEs). 36 of 40 (90%) of these events were reported in the intensive phase of the patient’s treatment.

Related Grade 3 or 4 Adverse Events By Treatment Arm. Kaplan Meier Curves for Time to First Event for Related Grade 3 or 4 Adverse Events in the Treatment Arms. The time to first event is plotted for all the patients at risk in the standard (blue), isoniazid (red) and ethambutol (green) arms. The y axis plots the proportion of the patients still at risk, and the risk table presents this numerically. The data was censored at 200 days after the first dose for all three arms, and there was no significant difference between the isoniazid arm (p = 0.19) or the ethambutol arm (p = 0.07) when compared to the standard therapy using the log rank test
Discussion
In a study encompassing a large number of drug-sensitive TB patients from across the world there is evidence that almost a tenth of patients experienced serious side effects due to their TB medication. The existing literature quotes a rate of approximately 5–20% for significant toxicity from standard TB therapy [5, 9, 16,17,18,19,20,21] and hepatotoxicity is the most frequently detected [1, 22, 23]. The liver enzyme profile on treatment for standard TB therapy and the experimental arms in REMoxTB has been described in more detail elsewhere [24]. As the exclusion criteria removed those with severe disease or concomitant diseases, our estimate must be considered a minimum [14], however the follow-up period was comparable to two other recent large tuberculosis trials [25, 26].
We observed that most of the AEs in standard therapy occurred in the intensive phase. The explanation for this is uncertain, and could involve a degree of survivorship bias or increased tolerance of side effects, but may relate to the presence of pyrazinamide in all three regimens. Pyrazinamide is a drug with a well-recognised toxicity profile [11], while ethambutol (the other drug only present for the intensive phase) has few reported side effects [27]. Additionally, hepatotoxicity and arthralgia were among the most common events and these are frequently reported side effects of pyrazinamide [28, 29]. The sterilising activity of pyrazinamide makes it an essential component of standard therapy [30], but there is still some uncertainty surrounding its ideal dosing [31] and there is evidence of a dose-response relationship with toxicity [32]. There is a pressing need to direct more research to optimise the most effective and least toxic dose alongside the other components of the standard regimen [33].
It is perhaps significant there was little difference in the number of related AEs in months 5 and 6 between those receiving active treatment and those on placebo. This could emphasise the importance of TB induced pathology on the presence and reporting of significant medical events. Reducing toxicity associated with medication is one of the factors driving the development of shorter treatment regimens for TB [34], however this finding suggests that concerns about toxicity may not be as important as previously thought. While the experimental arms were less toxic, it should be noted that they were also less effective. It was notable that both experimental regimens were less toxic and both of these reduced the bacterial load more quickly than standard regimen [14]. Whether there is a causal relationship between these observations is not known. This means, perhaps, that the motivation for shortening treatment needs to focus around patient acceptability and logistical benefits of few doses, visits to clinics and enhanced adherence.
We found female patients and HIV-positive patients to be at significantly higher risk of toxicity. Existing guidelines acknowledge the issues surrounding TB-HIV co-infection [35, 36], but these do not reference female gender as a risk factor for a more complicated treatment course (outside of pregnancy). It is unclear if reporting bias has played a role in AE recording for the trial, as there have been discrepancies noted between the genders in regards to healthcare-seeking behaviour previously [37, 38]. Nonetheless, clinicians should consider closer monitoring of both HIV-positive and female patients taking HRZE, especially in the intensive phase of treatment.
Those patients reporting one or more related grade 3/4 AE were more likely to fail to achieve sustained sputum culture negative status. This is an important observation and emphasises the need to detect toxicity early and manage it properly. The reasons for this difference in outcome is uncertain and would merit further investigation in prospective studies. It may be that better management of drug toxicity in tuberculosis treatment could deliver better outcomes.
It is notable that the majority of deaths occurred after completing treatment and were unrelated to trial medication, emphasising the importance of social context of TB infection. It may be that this is due to other underlying conditions that also contribute to a poor outcome or that experiencing toxicity reduces adherence to therapy. This relationship may explain why the cure rate with standard therapy for drug-sensitive disease can be as low as 80% in real-world settings [39].
This study is limited by innate reporting bias and reliance on a subjective assessment of severity in many cases (for example, pain scores). An example of this is the reporting activity at one site in the trial. After a trial pause this site reported almost one third of its total grade 3/4 AEs, and assessed 75% of them as being related to treatment in a 30 week period. Attributing causality to AEs has been shown to produce unreliable and subjective data [40] and caution has been advised when using trial data to evaluate drug safety profiles [41]. Given the proximity of the recent pause in trial recruitment it could be that there was concern over the safety of the experimental regimens and that in a double-blind trial this translated into a lower threshold to both report events and to attribute causality to the drugs.
While there is still merit in using AEs to investigate drug safety profiles, the often subjective nature of the reporting is a limitation. We are also aware of the potential dangers of drawing conclusions based on relatedness assessments for AEs [40, 42] and to this end have presented both total and related AEs in the analysis. Overall, the careful and consistent way in which data were recorded for this large number of patients does mean, however, that we are able to generate important observations and suggest future research.
In this paper we have shown that most adverse events occur in the intensive phase of treatment with female patients and those who are HIV positive constituting a demographic that should be closely monitored for toxicity. We have also found that those who experience clinically significant drug related-toxicity while taking standard TB therapy are at greater risk of failing treatment. From this we conclude that we need to improve our methods of detecting and managing patients experiencing toxicity, and that there is real need for novel drugs with more favourable toxicity profiles. Our data provide an evidence base to plan future research and to support improved treatment guidelines. Tuberculosis remains a global health threat, predominantly affecting a vulnerable and disadvantaged population, and this paper illustrates the need for clinicians to be quick to respond to side effects from treatment to ensure their patients have the best chance of achieving a cure.
References
Saukkonen JJ, Cohn DL, Jasmer RM, Schenker S, Jereb J a, Nolan CM, et al. an official ATS Statement : hepatotoxicity of Antituberculosis therapy. Am J Respir Crit Care Med. 2006;174:935–52.
Schaberg T, Rebhan K, Lode H. Risk factors for side-effects of isoniazid, rifampin and pyrazinamide in patients hospitalized for pulmonary tuberculosis. Eur Respir J. 1996;9:2026–30.
Yee D, Valiquette C, Pelletier M, Parisien I, Rocher I, Menzies D. Incidence of serious side effects from first-line Antituberculosis drugs among patients treated for active tuberculosis. Am J Respir Crit Care Med. 2003;167:1472–7.
Pepper DJ, Marais S, Wilkinson RJ, Bhaijee F, Maartens G, McIlleron H, et al. Clinical deterioration during antituberculosis treatment in Africa: incidence, causes and risk factors. BMC Infect Dis. 2010;10:83.
Lorent N, Sebatunzi O, Mukeshimana G, van den Ende J, Clerinx J. Incidence and risk factors of serious adverse events during antituberculous treatment in Rwanda: a prospective cohort study. PLoS One. 2011;6:1–8.
Zhang T, Du J, Yin X, Xue F, Liu Y, Li R, et al. Adverse events in treating smear-positive tuberculosis patients in China. Int J Environ Res Public Health. 2015;13:1–11.
Sekaggya-Wiltshire C, von Braun A, Scherrer AU, Manabe YC, Buzibye A, Muller D, et al. Anti-TB drug concentrations and drug-associated toxicities among TB/HIV-coinfected patients. J Antimicrob Chemother. 2017:1172–7.
Vasakova M. Challenges of antituberculosis treatment in patients with difficult clinical conditions. Clin Respir J. 2015;9:143–52.
Sadiq S, Khajuria V, Tandon VR, Mahajan A, Singh JB. Adverse drug reaction profile in patients on anti-tubercular treatment alone and in combination with highly active antiretroviral therapy. J Clin Diagnostic Res. 2015;9:FC01–4.
McIlleron H, Abdel-Rahman S, Dave JA, Blockman M, Owen A. Special populations and Pharmacogenetic issues in tuberculosis drug development and clinical research. J Infect Dis. 2015;211:S115–25.
Marra F, Marra CA, Bruchet N, Richardson K, Moadebi S, Elwood RK, et al. Adverse drug reactions associated with first-line anti-tuberculosis drug regimens. Int. J. Tuberc. Lung Dis. 2007;11:868–75.
Ezer N, Benedetti A, Darvish-Zargar M, Menzies D. Incidence of ethambutol-related visual impairment during treatment of active tuberculosis. Int J Tuberc Lung Dis. 2013;17:447–55.
Garg P, Garg R, Prasad R, Mishra AK. A prospective study of ocular toxicity in patients receiving ethambutol as a part of directly observed treatment strategy therapy. Lung India. 2015;32:16–9.
Gillespie SH, Crook AM, McHugh TD, Mendel CM, Meredith SK, Murray SR, et al. Four-month moxifloxacin-based regimens for drug-sensitive tuberculosis. N Engl J Med. 2014;371:1577–87.
AIDS UD of HD of. Table for grading the severity of adult and pediatric adverse events. DAIDS. 2017;Version 2.1.
Girling DJ. Adverse reactions to rifampicin in antituberculosis regimens. J Antimicrob Chemother England. 1977;3:115–32.
Addington WW. The side effects and interactions of antituberculosis drugs. Chest United States. 1979;76:782–4.
Chiba S, Tsuchiya K, Sakashita H, Ito E, Inase N. Rifampicin-induced acute kidney injury during the initial treatment for pulmonary tuberculosis: a case report and literature review. Intern Med. 2013;52:2457–60.
Chung-Delgado K, Revilla-Montag A, Guillen-Bravo S, Velez-Segovia E, Soria-Montoya A, Nuñez-Garbin A, et al. Factors associated with anti-tuberculosis medication adverse effects: a case-control study in Lima, Peru. PLoS One. 2011;6:1–5.
Devadatta S, Gangadharam PR, Andrews RH, Fox W, Ramakrishnan CV, Selkon JB, et al. Peripheral neuritis due to isoniazid. Bull World Health Organ. 1960;23:587–98.
Girling DJ. Adverse effects of antituberculosis drugs. Drugs New Zealand. 1982;23:56–74.
Devarbhavi H. Antituberculosis drug-induced liver injury: current perspective. Trop Gastroenterol. 2011;32:167–74.
Bright-Thomas RJ, Gondker AR, Morris J, Ormerod LP. Drug-related hepatitis in patients treated with standard anti-tuberculosis chemotherapy over a 30-year period. Int. J. Tuberc. Lung Dis. 2016;20:1621–4.
Tweed CD, Wills GH, Crook AM, Dawson R, Diacon AH, Louw CE, et al. Liver toxicity associated with tuberculosis chemotherapy in the REMoxTB study. BMC Med. 2018;16:46.
Jindani A, Harrison TS, Nunn AJ, Phillips PPJ, Churchyard GJ, Charalambous S, et al. High-dose Rifapentine with moxifloxacin for pulmonary tuberculosis. N Engl J Med. 2014;371:1599–608.
Gninafon M, Lo MB, Mthiyane T, Sc M, Kassa F, Diaye AN, et al. A four-month Gatifloxacin-containing regimen for treating tuberculosis. N Engl J Med. 2014;371:1588–98.
Sarkar S, Ganguly A, Sunwoo HH. Current overview of anti-tuberculosis Drugs : metabolism and toxicities. Mycobact Dis. 2016;6:1–6.
Chang KC, Leung CC, Yew WW, Lau TY, Tam CM. Hepatotoxicity of pyrazinamide: cohort and case-control analyses. Am J Respir Crit Care Med. 2008;177:1391–6.
Jenner PJ, Ellard GAA, Allan WGLG, Singh D, Girling DJ, Nunn AJ. Serum uric acid concentrations and arthralgia among patients treated with pyrazinamide-containing regimens in Hong Kong and Singapore. Tubercle Scotland. 1981;62:175–9.
Steele MA, Des Prez RM. The role of pyrazinamide in tuberculosis chemotherapy. Chest. 1988;94:845–50.
Sahota T, Della PO. Feasibility of a fixed-dose regimen of pyrazinamide and its impact. Antimicrob Agents Chemother. 2012;56:5442–9.
Ramakrishnan C V., Janardhanam B, Krishnamurthy D V., Stott H, Subbammal S, Tripathy SP. Toxicity of pyrazinamide, administered once weekly in high dosage, in tuberculous patients Bull World Health Organ 1968;39:775–779.
Boeree MJ, Diacon AH, Dawson R, Narunsky K, Bois J, Venter A, et al. A dose-ranging trial to optimize the dose of rifampin in the treatment of tuberculosis. Am J Respir Crit Care Med. 2015;191:1058–65.
Zumla AI, Gillespie SH, Hoelscher M, Philips PPJ, Cole ST, Abubakar I, et al. New antituberculosis drugs, regimens, and adjunct therapies : needs, advances, and future prospects. Lancet Infect Dis. 2014;14:327–40.
World Health Organization. Treatment of tuberculosis: guidelines 4th edition. Geneva; 2010.
Nahid P, Dorman SE, Alipanah N, Barry PM, Brozek JL, Cattamanchi A, et al. Official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America clinical practice guidelines: treatment of drug-susceptible tuberculosis. Clin Infect Dis. 2016;63:853–67.
Samal J. Health seeking behaviour among tuberculosis patients in India: a systematic review. J Clin Diagn Res. 2016;10:LE01–6.
Thompson AE, Anisimowicz Y, Miedema B, Hogg W, Wodchis WP, Aubrey-Bassler K. The influence of gender and other patient characteristics on health care-seeking behaviour: a QUALICOPC study. BMC Fam Pract. 2016;17:38.
World Health Organization. Global Tuberculosis Report. Geneva; 2016.
Hillman SL, Meyers J, Loprinzi CL, Limburg PJ, Mandrekar SJ. Statistical controversies in clinical research : value of adverse events relatedness to study treatment : analyses of data from randomized double-blind placebo-controlled clinical trials. Ann Oncol. 2017:1–8.
Hammad TA, Pinheiro SP, Neyarapally GA. Secondary use of randomized controlled trials to evaluate drug safety : a review of methodological considerations. Clin Trials. 2011;8:559–70.
Darssan D, Thompson MH, Pettitt AN. Incorporating adverse event relatedness into dose-finding clinical trial designs. Stat Med. 2014;33:1146–61.
Funding
Supported by the Global Alliance for TB Drug Development with support from the Bill and Melinda Gates Foundation, the European and Developing Countries Clinical Trials Partnership (grant IP.2007.32011.011), U.S. Agency for International Development, U.K. Department for International Development, Directorate General for International Cooperation of the Netherlands, Irish Aid, Australia Department of Foreign Affairs and Trade, and National Institutes of Health, AIDS Clinical Trials Group and by grants from the National Institute of Allergy and Infectious Diseases (NIAID) (UM1AI068634, UM1 AI068636, and UM1AI106701) and by NIAID grants to the University of KwaZulu Natal, South Africa, AIDS Clinical Trials Group (ACTG) site 31,422 (1U01AI069469); to the Perinatal HIV Research Unit, Chris Hani Baragwanath Hospital, South Africa, ACTG site 12,301 (1U01AI069453); and to the Durban International Clinical Trials Unit, South Africa, ACTG site 11,201 (1U01AI069426); Bayer Healthcare for the donation of moxifloxacin; and Sanofi for the donation of rifampin.
Availability of data and materials
The data that support the findings of this study are available from MRC CTU at UCL but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of MRC CTU at UCL.
Author information
Authors and Affiliations
Contributions
CDT, AC and SHGanalysed and interpreted the data for publication. CDT, AC, and SHG were responsible for drafting the manuscript. All other authors were involved in proposing further analysis, the interpretation of findings, revising the manuscript critically for intellectual content, and gave final approval prior to publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All patients provided written consent to any data and samples collected as part of the trial being used in further studies to improve the diagnosis and treatment of tuberculosis, as stated on the informed consent form for the study. Ethics approval was granted for REMoxTB by the ethics board at University College London, and at each of the study sites:
Kenya
-
KEMRI Scientific Steering Committee.
-
KEMRI Ethical Review Committee.
South Africa
-
Medicines Controll Council, Pretoria.
-
Pharma Ethics, Pretoria.
-
University of Cape Town Human Research Ethics Committee, Cape Town.
-
Biomedical Research Ethics Committee, Durban.
-
Wits Human Research Ethics Committee, Johannesburg.
Tanzania
-
Kilimanjaro Christian Medical College Research Ethics and Review Committee, Moshi.
-
Mbeya Ethics and Research Committee, Mbeya.
-
National Institute for Medical Research, Dar Es Salaam.
Zambia
-
University of Zambia Biomedical Research Ethics Committee, Lusaka.
China
-
Beijing Chest Hospital of Capital Medical University Ethics Committee, Beijing.
-
Shanghai Pulmonary Hospital Ethics Committee, Shanghai.
-
Tianjin CDC Biomedical Ethics Committee, Tianjin.
India
-
Biomedical Ethics Committee, New Dehli.
-
Institutional Ethics Committee, Mahatma Gandhi Medical College and Hospital, Jaipur.
Mexico
-
Comité de Investigación y Ética (División de Enseñanza, Investigación, Capacitación, Ética y Calidad) Hospital General de Occidente, Jalisco.
-
National Jewish Health, Denver USA.
Thailand
-
Ethics Committees on Researches Involving Human Subjects, Rajavithi Hospital, Bangkok.
-
The Khon Kaen University Ethics Committee for Human Research Faculty of Medicine, Khon Kaen University, Muang Khon Kaen.
-
Ethical Review Committee for Research in Human Subjects, Ministry of Public Health, Nonthraburi.
-
Ethical Review Committee of Chest Disease Institute, Department of Medical Services and Ministry of Public Health, Nonthraburi.
Malaysia
-
Medical Research & Ethics Committee, Ministry of Health Malaysia, Kuala Lumpur.
Consent for publication
Not applicable.
Competing interests
The authors’ declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tweed, C.D., Crook, A.M., Amukoye, E.I. et al. Toxicity associated with tuberculosis chemotherapy in the REMoxTB study. BMC Infect Dis 18, 317 (2018). https://doi.org/10.1186/s12879-018-3230-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-018-3230-6