
Abstract
Background
Gut pathological microbial imbalance or dysbiosis is closely associated with colorectal cancer. Although there are observable differences in molecular and clinical characteristics between patients with right- and left-sided colon cancer, differences in their gut microbiomes have not been thoroughly investigated. Furthermore, subsequent changes in microbiota status after partial colectomy remain unknown. We examined the human gut microbiota composition to determine its relationship with colon cancer and partial colon resection according to location.
Methods
Stool samples from forty-one subjects (10 in the control group, 10 in the right-sided colon cancer [RCC] group, 6 in the sigmoid colon cancer [SCC] group, 9 in the right colon resection [RCR] group and 6 in the sigmoid colon resection [SCR] group) were collected, and DNA was extracted. After terminal restriction fragment length polymorphism (T-RFLP) analysis, the samples were subjected to 16S rRNA gene amplicon sequencing, and the metabolic function of the microbiota was predicted using PICRUSt2.
Results
T-RFLP analysis showed a reduced ratio of clostridial cluster XIVa in the SCC patients and clostridial cluster IX in the RCC patients, although these changes were not evident in the RCR or SCR patients. 16S rRNA gene amplicon sequencing demonstrated that the diversity of the gut microbiota in the RCC group was higher than that in the control group, and the diversity in the SCR group was significantly higher than that in the RCR group. Principal coordinate analysis (PCoA) revealed significant differences according to the group. Analyses of the microbiota revealed that Firmicutes was significantly dominant in the RCC group and that the SCC group had a higher abundance of Verrucomicrobia. At the genus level, linear discriminant analysis effect size (LEfSe) revealed several bacteria, such as Ruminococcaceae, Streptococcaceae, Clostridiaceae, Gemellaceae, and Desulfovibrio, in the RCC group and several oral microbiomes in the SCC group. Metabolic function prediction revealed that cholesterol transport- and metabolism-related enzymes were specifically upregulated in the RCC group and that cobalamin metabolism-related enzymes were downregulated in the SCC group.
Conclusion
Gut microbial properties differ between RCC and SCC patients and between right hemicolectomy and sigmoidectomy patients and may contribute to clinical manifestations.
Similar content being viewed by others


Introduction
There is emerging evidence that gut pathological microbial imbalance or dysbiosis occurs in patients with colorectal cancer [1,2,3,4,5]. Changes in the intestinal microbiome can promote chronic inflammatory conditions and the production of carcinogenic molecules, subsequently increasing the risk of colorectal cancer. Specific alterations of the microbiome are observed during different stages of colorectal cancer [6,7,8]. These bacteria include Fusobacterium, Peptostreptococcus, Porphyromonas, Prevotella, Parvimonas, Bacteroides, Gemella and the oral microbiome. Their metabolism or immune modulation may directly or indirectly affect the colonic mucosal cells, causing colonic carcinogenesis [2,3,4,5]. However, how gut microbial dysbiosis may be involved in cancer pathogenesis remains undescribed.
In general, colorectal cancer pathogenesis depends on the location of the tumor. The proximal colon (right side) and distal colon (left side) exhibit different molecular characteristics and histologies. Right-sided tumors generally demonstrate flat histology associated with mutations in the DNA mismatch repair pathway. In contrast, left-sided tumors demonstrate epolypoid-like morphology with chromosomal instability-related mutations [9,10,11]. These findings raise new insights that the colorectal cancer-related gut microbiome may differ among tumor locations. However, few studies have analyzed microbiome changes between right- and left-sided colon cancers [12, 13].
However, a nationwide study suggested that the risk of diabetes mellitus (DM) is high in colectomy patients, especially in patients who received resection of the left part of the colon and the sigmoid colon, whereas resection of the rectum was not associated with a risk of DM [14]. These results suggest that the left colon may play a role in the regulation of glucose homeostasis. In addition, patients with left hemicolectomy were at higher risk of cerebrovascular disease [15]. These results indicate that different colectomy procedures may influence metabolic diseases after colectomy.
It is known that dysbiosis and alterations in gut microbial composition are linked to the development of metabolic diseases, including DM [16, 17]. In previous reports, patients with DM had enriched Clostridium clostridioforme and Lactobacillus species but low Roseburia, a major butyrate producer. The ratio of Bacteroidetes to Firmicutes demonstrated a significantly positive correlation with reduced glucose tolerance, which suggests that the gut microbiome may play an important role in the development of DM [18,19,20].
From this evidence, we suppose that the change in gut microbiota after colectomy may be involved in the regulation of multiple metabolic, signaling, and inflammatory pathways that are related to general physiological conditions. However, the gut microbiota after different sites of colectomy has not been well elucidated.
Therefore, we first investigated the gut microbiota in patients with right-sided colon cancer (RCC) and sigmoid colon cancer (SCC) to elucidate the different microbiota that may be responsible for colon carcinogenesis between different sites. We also analyzed the gut microbiota in patients with right colon resection (RCR) and with sigmoid colon resection (SCR) to reveal long-term changes in the microbiota after different colon resections. Finally, we aimed to provide new insights into the microbiota status of colon cancer and subsequent colectomy at different sites.
Methods
Human subjects
Subjects who were under 77 years of age and had undergone colonoscopy at the Mie Prefectural General Medical Center, Yokkaichi, Japan, between 2017 and 2020 were enrolled in the study. To evaluate differences in gut microbiota via terminal restriction fragment length polymorphism (T-RFLP) analysis, the subjects were classified into five groups: (i) control subjects who had no history of bowel disease; (ii) patients who had recently been diagnosed with colon cancer from the cecum to the transverse colon and who were awaiting surgery (the RCC group); (iii) patients who had recently been diagnosed with sigmoid colon cancer and were awaiting sigmoid colectomy (the SCC group); (iv) patients with a diagnosis of colorectal cancer undergoing right colon resection (the RCR group); and (v) patients with a diagnosis of sigmoid colon cancer undergoing sigmoid colon resection (the SCR group). We excluded all participants with a current use of antibiotics, history of current chronic bowel or liver disease, history of chemotherapy, and regular use of immunomodulators or probiotics. Patient assignments are shown in Table 1. Stool samples were collected from each participant, and fecal samples were stored at 4 °C until analysis.
DNA extraction
DNA extraction was performed using a previously described method [7]. In brief, an aliquot of the suspension of fecal samples was homogenized with zirconia beads in a 2.0-ml screw cap tube by a FastPrep 24 Instrument (MP Biomedicals, Santa Ana, CA, USA). We extracted DNA from each sample using an automatic nucleic acid extractor (Precision System Science, Chiba, Japan). We used MagDEA 200 (GC; Precision System Science) as the reagent for automated nucleic acid extraction.
T-RFLP
T-RFLP analysis of the microbial community structure in feces was performed by TechnoSuruga Laboratory Co., Ltd. (Shizuoka, Japan). We performed amplification of the bacterial 16S rRNA gene (16S rDNA), restriction enzyme digestion, size fractionation of fluorescently labeled terminal restriction fragments (T-RFs) and T-RFLP data analysis by a previously described method by Nagashima et al. [21, 22]. Briefly, the 5′ HEX-labeled 516 f and 1516 r primers were used for amplification of the bacterial 16S rRNA. Then, the PCR products were digested with 10 U of BslI (New England BioLabs, Ipswich, MA, USA). The resultant DNA fragments, namely, fluorescently labeled T-RFs, were analyzed by an ABI PRISM 3130xl genetic analyzer, and their length and peak area were determined using the genotype software GeneMapper (Applied Biosystems). T-RFs were divided into 29 operational taxonomic units (OTUs). The OTUs were quantified as the percentage of individual OTUs per total OTU area, which was expressed as the percentage of the area under the curve (%AUC). The bacteria were predicted for each classification unit, and the corresponding OTUs were identified according to reference Human Fecal Microbiota T-RFLP profiling (https://www.tecsrg.co.jp/t-rflp/t_rflp_hito_OTU.html).
To evaluate differences in gut microbiota composition at the species level, samples from 10 control subjects and 31 patients with carcinoma were selected for 16S rRNA gene amplicon sequencing; StatView-J 5.0 was used to match control and patient samples based on age, sex, BMI, smoking, alcohol, diabetes, hypertension, total cholesterol, triglyceride, HDL cholesterol, and LDL cholesterol.
Illumina library generation
Next-generation sequencing (NGS) analysis of the microbial community structure in the feces was performed using a MiSeq (Illumina, San Diego, CA, USA), as previously described by Takahashi et al. [23]. The V3-V4 regions of bacterial and archaeal 16S rRNA were amplified using the Pro341F/Pro805R primers and the dual-index method [23]. Barcoded amplicons were paired-end sequenced on a 2 × 284-bp cycler using the MiSeq system together with MiSeq Reagent Kit version 3 (600 cycles).
Quality filtering and amplicon sequencing analysis
Paired-end sequencing reads were merged using the fastq-join program with default settings. Only combined reads with a quality value score of ≥ 20 for more than 99% of the sequence were extracted using FASTX-Toolkit. The chimeric sequences were deleted with USEARCH ver. 6.1 [24]. Identification from sequence analyses of sequence reads was performed manually using the Ribosomal Database Project (RDP) Multiclassifier tool ver. 2.11, which is available from the RDP website (http://rdp.cme.msu.edu/classifier/). Bacterial and archaeal species identification from sequences was performed using Metagenome@KIN 2.2.1 analysis software (World Fusion, Japan) and the TechnoSuruga Lab Microbial Identification database DB-BA ver. 13.0 (TechnoSuruga Laboratory, Japan) with homology ≥ 97% [25]. Principal coordinate analysis (PCoA) was performed using Metagenome@KIN software (World Fusion Co., Ltd., Tokyo, Japan) based on data from bacterial genera with a 97% similarity cutoff with the Apollon DB-BA database ver. 13.0 (TechnoSuruga Laboratory).
Alpha and beta diversity analysis
The joined amplicon sequence reads were processed through QIIME 2 ver. 2020.6. Quality filtering and chimeric sequences were filtered with the default option, and representative sequences were created using the DADA2 denoise-single plugin ver. 2017.6.0. The taxonomy of representative sequences was assigned using the Greengenes database ver. 13.8 by training a naive Bayes classifier using the q2-feature classifier plugin. The sampling depth for alpha and beta diversity was 28,216, which was the minimum number of read counts among samples.
Alpha diversity indices (Chao1, Shannon and Simpson) were calculated using the alpha rarefaction plugin. The statistical significance of the Chao1, Shannon and Simpson indices among the groups was assessed by the Kruskal–Wallis test using the alpha-group-significance plugin.
Beta diversity was analyzed using weighted UniFrac, unweighted UniFrac and Bray–Curtis distances using a core-metrics-phylogenetic plugin. The Emperor tool was used to visualize the PCoA plots. The statistical significance of the similarity of bacterial communities among groups was assessed with the ANOSIM test using the beta-group-significance plugin.
Predictive functionality analysis
Predictive functionality analysis of bacterial 16S rDNA communities was performed using PICRUSt ver. 2.3.0-b. The analyzed representative sequences by QIIME 2 and the reference sequence of the Integrated Microbial Genomes database (IMG) were aligned using HMMER ver. 3.3. Phylogenetic placement analyses were applied using EPA-NG ver. 0.3.3 and GAPPA ver. 0.6.0. 16S rRNA gene copies were normalized using the caster package of R software. The gene families were predicted based on the Clusters of Orthologous Genes (COG) and Enzyme Classification (EC) databases. Biological pathways were reconstructed based on predicted gene families using MinPath ver. 1.4 [26].
Linear discriminant analysis (LDA)
To determine potential bacteria that differ in abundance between the groups, a linear discriminant analysis effect size (LEfSe) analysis in multilevel species was used. LEfSe can identify taxa with significantly normalized relative abundances and performs a linear discriminant analysis (LDA) to determine the effect size of each taxon through the website http://huttenhower.sph.harvard.edu/galaxy [27]. Taxa with an effect size greater than 2.0 (with P < 0.05) were considered significant.
Metabolic function prediction of the microbiota by PICRUSt2.
The mean (standard deviation [SD]) and median (interquartile range [IQR]) were used to describe normally and nonnormally distributed data, respectively. Numbers and percentages were calculated for categorical variables. The data were analyzed using the Kruskal–Wallis test or the Mann–Whitney test (two-sided) for continuous variables and Fisher’s exact test for categorical variables using StatView-J 5.0. P values < 0.05 were considered significant.
Results
Clinical patient characteristics
The demographic and clinical characteristics of the subjects are shown in Table 1. Ten healthy controls and 31 patients with cancer (breakdown: RCC, 10 patients; SCC, 6 patients; RCR, 9 patients; and SCR, 6 patients) were enrolled. The average postoperative observation period was 37 months (minimum 13 months, maximum 90 months). Blood tests showed no significant differences in total cholesterol, triglycerides, HDLs, or LDLs in any group, including the control group. The mean age and BMI of the healthy subjects were lower than those of the cancer patients. Two diabetic patients were found in the RCR group and one in the SCR group.
Differences in microbiota between control and cancer patients by T-RFLP analysis
The results of T-RFLP are shown in Table 2. Lactobacillales were more abundant in all the groups than in the control group. Bifidobacterium tended to increase in the RCR and SCR groups, while Prevotella tended to decrease in the RCR and SCR groups. In the Clostridium cluster, a decrease in Clostridium cluster XIVa increased in the SCC group (P < 0.05). In addition, Clostridium cluster IX decreased in the RCC group (P < 0.05). The postoperative RCR and SCR groups of both Clostridium cluster IX and XIVa were not different from those of the control group.
Differences in species richness and diversity between control and cancer subjects
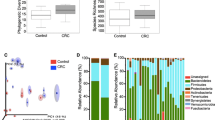
Because characteristic changes in the gut microbiota were observed in each group, 16S rRNA gene amplicon sequencing was performed to analyze the microbiota in more detail. First, we examined diversity. The Chao1 index was applied to assess the influence and diversity of the microbiota in the feces between the control group and cancer patients. Figure 1 shows the alpha diversity between each group, including the control group. Chao1 showed a predominant increase in diversity in the RCC group but not in the SCC group; the RCR group was less diverse than the RCC group (P < 0.05). In addition, the SCR group was more diverse than the RCR group (P < 0.01). Similar results were obtained with Shannon and Simpson analyses.

Richness and diversity analysis of 16S rRNA gene amplicon sequences obtained from fecal samples. The Chao1 index was used to evaluate microbial richness and diversity in fecal samples between patients in the healthy control and cancer groups (*P < 0.05, **P < 0.01)
PCoA was then performed to visually assess diversity. Figure 2 shows the beta diversity between each cancer group, including the control group. In Bray–Curtis ANOVA, there was a significant and clear cluster difference between the control group and the RCC group (P < 0.01), the control group and the SCR group (P < 0.05), and the RCC group and the RCR group (P < 0.01). There was no significant difference between the RCR and SCR groups, but the composition tended to be different (P = 0.095). Similar results were observed with weighted UniFrac and unweighted UniFrac with other analysis methods. In the unweighted UniFrac analysis, the P value for the comparison of the RCR and SCR groups was 0.001.

Beta diversity was assessed by the Bray–Curtis test. In (A), the red dots indicate the control group, the blue dots indicate the RCC group, and the yellow dots indicate the SCC group. In (B), the green dots indicate the RCR group, and the purple dots indicate the SCR group. The beta diversity represents the degree of difference in diversity between two samples. The Bray–Curtis distance was computed via the 16S rRNA gene amplicon sequence data. (P values: P < 0.01 for the control group and RCC group; P < 0.05 for the control group and SCR group; and P < 0.01 for the RCC group and RCR group. P = 0.095 for the RCR group and SCR group)
16S rRNA gene amplicon sequencing in each systematic classification
Figure 3 shows the relative proportions of gut microbiota at the phylum and class classification levels in each group. As shown in Fig. 3A, Firmicutes was more abundant in the RCC group than in the other groups, including the control group, and the SCC group had more Verrucomicrobia. Bacteroidetes decreased in the SCR group compared to the control group. As shown in Fig. 3B, Bacilli was more abundant in the RCC group, and Verrucomicrobia was more abundant in the SCC group than in all the other groups, including the control group. Clostridia increased in the postoperative SCR group compared to the SCC group. Fusobacteriia was not significantly different between groups, including the control group, even at the class level.

Phylum (A)- and class (B)-level classifications of bacteria identified in individual fecal samples of the control group and each cancer group. Each bar represents the percent contribution of phylum- and class-level profiles. The phylum and class represented by the different colors are shown below the figure
Comparison of bacterial classifications between control and cancer patients
Next, a linear discriminant analysis (LDA) effect size (LEfSe) approach was performed to clarify the characteristic genus-level bacteria in each group, as shown in Fig. 4. LEfSe can determine the taxonomic units most likely to explain differences between classes by coupling standard tests for statistical significance with additional tests encoding biological consistency and effect relevance (LDA score > 2.0, P < 0.05). An increase in the abundance of thirty-one genera was detected in the RCC group. Among them, Ruminococcaceae, Gemella, Streptococcaceae, Peptococcaceae, and Desulfovibrio have been suggested to be associated with carcinogenesis. On the other hand, eleven genera whose abundance decreased were identified. Only four bacteria were identified in the SCC group. Among them, Porphyromonas and Parvimonas ranked high among indigenous bacteria in the oral cavity. Only Butyricicoccus decreased in abundance. In postoperative patients, the number of bacteria decreased in the RCR group compared to the RCC group. Among the reduced bacteria, Gemella abundance decreased. In the SCR group, a large number of bacterial species were identified compared to those in the RCR group. In particular, more bacterial species belonging to the Ruminococcaceae family and Clostridiales order were identified. Fusobacteriaceae was also identified among those bacteria whose abundance was reduced.


LEfSe comparing the control and bacterial classifications of tumor-related microorganisms. The histograms of LDA scores for differentially abundant bacterial groups are shown in red for the control group and green for the cancer patient group. The control group and RCC group analyses are designated (A); the control group and SCC group analyses are designated (B); the control group and RCR group analyses are designated (C); and the control group and SCR group analyses are designated (D). Each analysis was performed at the genus level, but when the genera were not clear, the next level of hierarchy was used
Metabolic function prediction of the microbiota by PICRUSt2
Finally, predictive functionality analysis using PICRUSt2 was performed to clarify the functions of the genes of the microbiota. Tables 3 and 4 show the top 10 genes with the lowest P values among those whose expression increased and decreased in each group. The results revealed many enzymes involved in glucose metabolism, such as glutamyl aminopeptidase and maltose 6`-phosphate phosphatase. Cholesterol-related enzymes were found, especially in the RCC group. However, a decrease in enzymes related to short-chain fatty acids (SCFAs) was observed in the RCC group. The SCC group showed a decrease in vitamin-related enzymes. Moreover, enzymes related to glucose metabolism were found more frequently in the RCR and SCR groups.
Discussion
In this study, we clearly found that the composition of the gut microbiota differs between right- and left-sided colon cancer patients and after curative colectomy using T-RFLP analysis and 16S rRNA gene amplicon sequencing. These results demonstrated the specific change in gut microbiota in each group, suggesting that gut microbiota are closely associated with the development of colon cancer and physiological conditions after partial colectomy.
T-RFLP analysis showed a reduced ratio of clostridial cluster XIVa in the SCC patients and clostridial cluster IX in the RCC patients, although these changes were not seen in RCR and SCR patients. Clostridial cluster XIVa includes most butyrate producers that belong to the Firmicutes phylum in the human colon and clostridial cluster IX propionate-producing bacteria [28,29,30]. SCFAs such as butyrate or propionate are known to have an important role in preserving gut barrier functions and exerting immunomodulatory and anti-inflammatory properties [29, 31]. The antiproliferative, apoptotic and differentiating properties of the various SCFAs are linked to the degree of induced histone hyperacetylation [32]. Fewer SCFAs in feces are observed in patients with IBD or colorectal cancer than in normal patients [33]. Our study indicates that different types of main SCFA-producing bacteria are lower in colon cancer patients and that those of clostridial cluster IX are lower in the RCC group and those of clostridial cluster XIVa are lower in the SCC group. However, this type of change was not observed in patients after colectomy. Therefore, we speculate that low SCFA-producing bacteria may be related to colon carcinogenesis and are high-risk markers of colon cancer.
We further analyzed the gut microbiota in patients with RCC and SCC and with curative colectomy by 16S rRNA gene amplicon sequencing with QIIME 2, which is a next-generation microbiome bioinformatics platform. We found that the diversity of the gut microbiota in the RCC group was higher than that in the control group, but that in the RCR group was at the same level as that in the control group. In contrast, diversity in the SCR group was significantly higher than that in the RCR group. We also found that gut microbial genus composition using principal component analysis of the log-transformed relative abundances showed a separation between the control group and RCC group and between the RCR group and SCR group. A previous study suggested that patients with colon cancer have a less diverse microbiome than healthy individuals [34]. However, other reports found higher richness in the microbiomes of patients with colon cancer than controls, partly by the expansion of species derived from the oral cavity. A study of microbiota after curative colon surgery suggested that the right hemicolectomy group showed a tendency to decrease in terms of richness and diversity at the genus level [20]. These results are consistent with our current data. The diversity of the gut microbiome is defined as the number and relative abundance distribution of distinct types of microbiomes in the gut. Previous studies have suggested that dysbiosis of the gut microbiome is becoming increasingly recognized for its influence on host immunity and may influence the good response to a variety of cancer therapies, such as chemotherapy, radiotherapy, and immunotherapy [35]. From this point of view, it is very interesting that diversity and richness differed in patients with both pre- and postoperative colorectal cancer according to the tumor location, and high diversity in the SCC group may be associated with favorable outcomes in the SCR group.
Analysis of microbiota at the class level revealed that Clostridia and Bacilli belonging to Firmicutes were significantly dominant in the RCC group compared to the control group. The SCC group had a higher abundance of Verrucomicrobiae belonging to Verrucomicrobia. These characteristics were not observed in patients in the postcolectomy group. In particular, Firmicutes was more abundant and Verrucomicrobia was lower in the SCR group than in the RCR group. This tendency was consistent with the findings of a previous long-term study after curative colectomy [20]. These results indicated that there may be specific profiles of the gut bacterial population at the phylum and class taxonomic levels related to colon cancer, and this alteration was different between the locations of cancer. Furthermore, colectomy may also differentially lead to new gut microbiota compositions depending on the removal site.
Next, we performed discriminant analysis by using the LEfSe approach, which was applied to show the key taxa responsible for the difference between several groups and identified several gut microbes mainly at the genus and family levels. Among these bacteria, especially Ruminococcaceae, Streptococcaceae, Clostridiaceae, Gemellaceae, and Desulfovibrio, which are seen in the RCC group, these species have already been reported to constitute colon cancer-associated microbiomes [2,3,4, 36]. However, the exact mechanism by which these bacteria affect the development and progression of colon cancer has not been fully elucidated. In the SCC group, Porphynomonas, Parvimonas, Peptoniphilus, and Peptococcaceae were identified. These species are gram-positive anaerobic cocci mainly located among oral bacteria and influence mucosal gene expression, which might contribute to the development of colon cancers [3, 4, 37]. In addition, Butyricicoccus, which produces butyrate, was less abundant in the SCC group. Butyrate is an essential metabolite in the human colon and is the preferred energy source because colon epithelial cells have immunomodulatory and anti-inflammatory properties [31]. Some cross-sectional studies reported that, compared to control individuals, patients with colorectal cancer had a lower abundance of butyrate-producing species and lower fecal levels of butyrate, which is consistent with our current data.
Our data clearly suggest that the tumor microbiota between right- and left-sided colorectal cancer patients shows differential microbial diversity and bacterial taxa at several levels, meaning that the RCC and SCC groups may clearly exhibit different specific microbiome compositions.
Genetically, right-sided tumors are commonly associated with microsatellite instability and are highly immunogenic, presenting with BRAF mutations, whereas left-sided tumors show chromosomal instability with mutations in KRAS, APC, PIK3CA, and p53 [9, 11]. In fact, Fusobacterium nucleatum signatures in proximal colorectal cancer tissue are correlated with shorter patient survival and molecular alterations such as hypermutation with microsatellite instability and BRAF mutations [3, 38]. Clinically, patients with right-sided tumors present with a worse prognosis than those with left-sided tumors [4, 39].
Therefore, there is a possibility that the gut microbiome affects the development and progression of colon cancer differently according to tumor location.
Although there are a few studies regarding the differences in the tumor microbiota between right- and left-sided colon cancer, compositional alterations in the microbiota are not restricted to cancerous tissue and differ between distal and proximal cancers. [13]. Another report of analysis of on- and off-tumor microbiota suggests that the right and left colon show distinctive bacterial populations; however, the presence of a colonic tumor leads to a more consistent microbiota between locations [13]. Therefore, when we investigate the gut microbiota of colon cancers using feces, we should consider that the findings might be influenced not only by tumor-associated microbiota but also by the surrounding nontumor microbiota.
Because the number of patients enrolled in this study was small, we could not demonstrate clinical characteristics, such as DM, in postcolectomy patients. However, a previous study reported that right hemicolectomy patients rather than left anterior resection patients had higher serum fasting glucose levels than controls, implying that the proximal colon may play an important role in glucose control [20]. In contrast, a large study demonstrated an increased risk of clinically recorded type 2 diabetes among patients who had undergone total and partial colectomy, with the risk being elevated only among individuals who had the left part of their colon removed [14]. Therefore, the gut microbiota has recently been shown to play an important role in the development of metabolic diseases, including obesity and metabolic syndrome [16, 25].
In our current study, the RCR and SCR groups showed clear alterations in microbiome composition. At the family level, Gemellaceae, the members of which modulate immunological reactions, was less abundant in the RCR group than in the other groups. In contrast to the RCR group, in the SCR group, a variety of gut bacteria belonging to the Firmicutes phylum were abundant, but the abundance of Fusobacteriaceae was low. A previous study reported that the ratio of Firmicutes to Bacteroidetes was significantly lower in patients with DM [20]. However, our study showed a high Firmicutes-to-Bacteroidetes ratio in the SCR group. Further studies are needed to determine how gut microbiome composition is related to clinical manifestations in patients with colectomy.
Finally, we performed metabolic function prediction of the microbiota by PICRUSt2 to clarify the functional enzyme spectrum in each group. We revealed the differential expression of several genomes related to glucose metabolism in each group. A previous meta-analysis of metagenomic studies identified the microbiome function of gluconeogenesis and the putrefaction and fermentation pathways as being associated with colorectal cancer [40]. Furthermore, we revealed that cholesterol transport- and metabolism-related enzymes were specifically upregulated in the RCC group and that cobalamin metabolism-related enzymes were downregulated in the SCC group. These results supported previous reports in which cholesterol may inhibit the proliferative capacity of certain human colonic adenocarcinomas [41, 42] and plasma vitamin B12 concentrations are associated with the risk of colorectal cancer [41, 43]. Furthermore, some SCFA-related enzymes were identified in both the RCC and SCC groups. However, these specific characteristics were not observed in the postoperative groups. These results support the idea that pathological microbial dysbiosis is responsible for the gut of patients with colorectal cancer. Future shotgun metagenomic studies of the intestinal mucosa-associated microbiome will be important to further refine the list of colorectal cancer-associated gut microbes.
Several limitations associated with the present study warrant mentioning. First, this was a single-center study, and the sample size was relatively small, which may limit the generalizability of the results. Second, this was a cross-sectional study, and the mean age or background of the subjects was somewhat different, which may affect the gut microbiota among the groups. Third, our study analyzed the gut microbiota in subjects who did not consume controlled diets, which may also influence the results.
In conclusion, we clearly found that the composition of the gut microbiota dramatically differs between right and sigmoid colon cancer patients and between right hemicolectomy and sigmoidectomy patients. Our findings support the hypothesis of tumor location-specific microbiota. We found that high richness and diversity were associated with the RCC and SCR groups and the different gut microbiota compositions in each group. It is difficult to determine whether the gut microbiota could influence the pathophysiological condition of patients or whether the gut condition could alter the microbiota. However, in assessing the gut microbiota in patients with colon cancer, we should consider tumor location.
We hope that the results herein can provide useful information for using gut microbes as biomarkers to assess the location and progression of colon cancer or lead to interventional targets to control the development of this disease.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available in the DDBJ Sequenced Read Archive, [https://www.ddbj.nig.ac.jp/index-e.html], under the accession number DRA013452.
Abbreviations
- BMI:
-
Body mass index
- T-RFLP:
-
Terminal restriction fragment length polymorphism
- SCFA:
-
Short-chain fatty acid
References
Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol. 2012;10(8):575–82.
O’Keefe SJ. Diet, microorganisms and their metabolites, and colon cancer. Nat Rev Gastroenterol Hepatol. 2016;13(12):691–706.
Song M, Chan AT, Sun J. Influence of the gut microbiome, diet, and environment on risk of colorectal cancer. Gastroenterology. 2020;158(2):322–40.
Ternes D, Karta J, Tsenkova M, Wilmes P, Haan S, Letellier E. Microbiome in colorectal cancer: how to get from meta-omics to mechanism? Trends Microbiol. 2020;28(5):401–23.
Wang F, Song M, Lu X, Zhu X, Deng J. Gut microbes in gastrointestinal cancers. Semin Cancer Biol. 2021. https://doi.org/10.1016/j.semcancer.2021.03.037.
Shen XJ, Rawls JF, Randall T, Burcal L, Mpande CN, Jenkins N, Jovov B, Abdo Z, Sandler RS, Keku TO. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes. 2010;1(3):138–47.
Kasai C, Sugimoto K, Moritani I, Tanaka J, Oya Y, Inoue H, Tameda M, Shiraki K, Ito M, Takei Y, Takase K. Comparison of human gut microbiota in control subjects and patients with colorectal carcinoma in adenoma: terminal restriction fragment length polymorphism and next-generation sequencing analyses. Oncol Rep. 2016;35(1):325–33.
Sheng Q, Du H, Cheng X, Cheng X, Tang Y, Pan L, Wang Q, Lin J. Characteristics of fecal gut microbiota in patients with colorectal cancer at different stages and different sites. Oncol Lett. 2019;18(5):4834–44.
Yamauchi M, Morikawa T, Kuchiba A, Imamura Y, Qian ZR, Nishihara R, Liao X, Waldron L, Hoshida Y, Huttenhower C, Chan AT, Giovannucci E, Fuchs C, Ogino S. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut. 2012;61(6):847–54.
Baran B, Mert Ozupek N, Yerli Tetik N, Acar E, Bekcioglu O, Baskin Y. Difference between left-sided and right-sided colorectal cancer: a focused review of literature. Gastroenterol Res. 2018;11(4):264–73.
De Renzi G, Gaballo G, Gazzaniga P, Nicolazzo C. Molecular biomarkers according to primary tumor location in colorectal cancer: current standard and new insights. Oncology. 2021;99(3):135–43.
Flemer B, Lynch DB, Brown JM, Jeffery IB, Ryan FJ, Claesson MJ, O’Riordain M, Shanahan F, O’Toole PW. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut. 2017;66(4):633–43.
Phipps O, Quraishi MN, Dickson EA, Steed H, Kumar A, Acheson AG, Beggs AD, Brookes MJ, Al-Hassi HO. Differences in the on- and off-tumor microbiota between right- and left-sided colorectal cancer. Microorganisms. 2021;9(5):1108.
Jensen AB, Sørensen TI, Pedersen O, Jess T, Brunak S, Allin KH. Increase in clinically recorded type 2 diabetes after colectomy. Elife. 2018;7: e37420.
Jensen AB, Ajslev TA, Brunak S, Sørensen TI. Long-term risk of cardiovascular and cerebrovascular disease after removal of the colonic microbiota by colectomy: a cohort study based on the Danish National Patient Register from 1996 to 2014. BMJ Open. 2015;5(12): e008702.
Ussar S, Griffin NW, Bezy O, Fujisaka S, Vienberg S, Softic S, Deng L, Bry L, Gordon JI, Kahn CR. Interactions between gut microbiota, host genetics and diet modulate the predisposition to obesity and metabolic syndrome. Cell Metab. 2015;22(3):516–30.
Zhang S, Cai Y, Meng C, Ding X, Huang J, Luo X, Cao Y, Gao F, Zou M. The role of the microbiome in diabetes mellitus. Diabetes Res Clin Pract. 2021;172: 108645.
Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett. 2009;294(1):1–8.
Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, Al-Soud WA, Sørensen SJ, Hansen LH, Jakobsen M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE. 2010;5(2): e9085.
Lin XH, Jiang JK, Luo JC, Lin CC, Ting PH, Yang UC, Lan YT, Huang YH, Hou MC, Lee FY. The long term microbiota and metabolic status in patients with colorectal cancer after curative colon surgery. PLoS ONE. 2019;14(6): e0218436.
Nagashima K, Hisada T, Sato M, Mochizuki J. Application of new primer-enzyme combinations to terminal restriction fragment length polymorphism profiling of bacterial populations in human feces. Appl Environ Microbiol. 2003;69(2):1251–62.
Nagashima K, Mochizuki J, Hisada T, Suzuki S, Shimomura K. Phylogenetic analysis of 16S ribosomal RNA gene sequences from human fecal microbiota and improved utility of terminal restriction fragment length polymorphism profiling. Biosci Microflora. 2006;25(3):99–107.
Takahashi S, Tomita J, Nishioka K, Hisada T, Nishijima M. Development of a prokaryotic universal primer for simultaneous analysis of Bacteria and Archaea using next-generation sequencing. PLoS ONE. 2014;9(8): e105592.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–6.
Kasai C, Sugimoto K, Moritani I, Tanaka J, Oya Y, et al. Comparison of the gut microbiota composition between obese and non-obese individuals in a Japanese population, as analyzed by terminal restriction fragment length polymorphism and next-generation sequencing. BMC Gastroenterol. 2015;15:100.
Ye Y, Doak TG. A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes. PLoS Comput Biol. 2009;5(8): e1000465.
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60.
Van den Abbeele P, Grootaert C, Marzorati M, Possemiers S, Verstraete W, Gérard P, Rabot S, Bruneau A, El Aidy S, Derrien M, Zoetendal E, Kleerebezem M, Smidt H, Van de Wiele T. Microbial community development in a dynamic gut model is reproducible, colon region specific, and selective for Bacteroidetes and Clostridium cluster IX. Appl Environ Microbiol. 2010;76(15):5237–46.
Van den Abbeele P, Belzer C, Goossens M, Kleerebezem M, De Vos WM, Thas O, De Weirdt R, Kerckhof FM, Van de Wiele T. Butyrate-producing Clostridium cluster XIVa species specifically colonize mucins in an in vitro gut model. ISME J. 2013;7(5):949–61.
Oya M, Tokunaga T, Tadano Y, Ogawa H, Fujii S, Murakami W, Tamai K, Ikomi F, Morimoto Y. The composition of the human fecal microbiota might be significantly associated with fecal SCFA levels under hyperbaric conditions. Biosci Microbiota Food Health. 2021;40(4):168–75.
Rivière A, Selak M, Lantin D, Leroy F, De Vuyst L. Bifidobacteria and butyrate-producing colon bacteria: importance and strategies for their stimulation in the human gut. Front Microbiol. 2016;7:979.
Hinnebusch BF, Meng S, Wu JT, Archer SY, Hodin RA. The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. J Nutr. 2002;132(5):1012–7.
Xu K, Jiang B. Analysis of mucosa-associated microbiota in colorectal cancer. Med Sci Monit. 2017;23:4422–30.
Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, Shi J, Goedert JJ, Hayes RB, Yang L. Human gut microbiome and risk for colorectal cancer. J Natl Cancer Inst. 2013;105(24):1907–11.
McQuade JL, Daniel CR, Helmink BA, Wargo JA. Modulating the microbiome to improve therapeutic response in cancer. Lancet Oncol. 2019;20(2):e77–91.
Allali I, Boukhatem N, Bouguenouch L, Hardi H, Boudouaya HA, Cadenas MB, Ouldim K, Amzazi S, Azcarate-Peril MA, Ghazal H. Gut microbiome of Moroccan colorectal cancer patients. Med Microbiol Immunol. 2018;207(3–4):211–25.
Drewes JL, White JR, Dejea CM, Fathi P, Iyadorai T, Vadivelu J, Roslani AC, Wick EC, Mongodin EF, Loke MF, Thulasi K, Gan HM, Goh KL, Chong HY, Kumar S, Wanyiri JW, Sears CL. High-resolution bacterial 16S rRNA gene profile meta-analysis and biofilm status reveal common colorectal cancer consortia. NPJ Biofilms Microbiomes. 2017;3:34.
Yu J, Chen Y, Fu X, Zhou X, Peng Y, Shi L, Chen T, Wu Y. Invasive Fusobacterium nucleatum may play a role in the carcinogenesis of proximal colon cancer through the serrated neoplasia pathway. Int J Cancer. 2016;139(6):1318–26.
Nakagawa-Senda H, Hori M, Matsuda T, Ito H. Prognostic impact of tumor location in colon cancer: the Monitoring of Cancer Incidence in Japan (MCIJ) project. BMC Cancer. 2019;19(1):431.
Thomas AM, Manghi P, Asnicar F, Pasolli E, Armanini F, Zolfo M, Beghini F, Manara S, Karcher N, Pozzi C, Gandini S, Serrano D, Tarallo S, Francavilla A, Gallo G, Trompetto M, Ferrero G, Mizutani S, Shiroma H, Shiba S, Shibata T, Yachida S, Yamada T, Wirbel J, Schrotz-King P, Ulrich CM, Brenner H, Arumugam M, Bork P, Zeller G, Cordero F, Dias-Neto E, Setubal JC, Tett A, Pardini B, Rescigno M, Waldron L, Naccarati A, Segata N. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat Med. 2019;25(4):667–78.
Wang C, Li P, Xuan J, Zhu C, Liu J, Shan L, Du Q, Ren Y, Ye J. Cholesterol enhances colorectal cancer progression via ROS elevation and MAPK signaling pathway activation. Cell Physiol Biochem. 2017;42(2):729–42.
Broitman SA, Cerda S, Wilkinson J 4th. Cholesterol metabolism and colon cancer. Prog Food Nutr Sci. 1993;17(1):1–40.
Dahlin AM, Van Guelpen B, Hultdin J, Johansson I, Hallmans G, Palmqvist R. Plasma vitamin B12 concentrations and the risk of colorectal cancer: a nested case-referent study. Int J Cancer. 2008;122(9):2057–61.
Acknowledgements
The authors express their appreciation to the staff at the TechnoSuruga Laboratory Co., Ltd. (Sizuoka, Japan) for their technical assistance.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Conceived and designed the study: DS, KS. Recruited the subjects: DS, HM, SF, MK, YS, YN, YN, YK, IM, YY, HI, EO, YM, KD, KT, HW, and KS. Wrote the paper: DS and KS. Supervised the study: HN and KS. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All patients received an explanation of the procedures and possible risks associated with the study, and they gave their written informed consent to participate. This study was performed in accordance with the Declaration of Helsinki and was approved by our institutional ethics committee (authorization number 2016-17, Mie Prefectural General Medical Center, Yokkaichi, Japan).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Suga, D., Mizutani, H., Fukui, S. et al. The gut microbiota composition in patients with right- and left-sided colorectal cancer and after curative colectomy, as analyzed by 16S rRNA gene amplicon sequencing. BMC Gastroenterol 22, 313 (2022). https://doi.org/10.1186/s12876-022-02382-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12876-022-02382-y