
Abstract
Background
Urinary and faecal metabolic profiling have been extensively studied in gastrointestinal diseases as potential diagnostic markers, and to enhance our understanding of the intestinal microbiome in the pathogenesis these conditions. The impact of bowel cleansing on the microbiome has been investigated in several studies, but limited to just one study on the faecal metabolome.
Aim
To compare the effects of bowel cleansing on the composition of the faecal microbiome, and the urine and faecal metabolome.
Methods
Urine and faecal samples were obtained from eleven patients undergoing colonoscopy at baseline, and then at day 3 and week 6 after colonoscopy. 16S rRNA gene sequencing was used to analyse changes in the microbiome, and metabonomic analysis was performed using proton nuclear magnetic resonance (1H NMR) spectroscopy.
Results
Microbiomic analysis demonstrated a reduction in alpha diversity (Shannon index) between samples taken at baseline and three days following bowel cleansing (p = 0.002), and there was no significant difference between samples at baseline and six weeks post colonoscopy. Targeted and non-targeted analysis of urinary and faecal bacterial associated metabolites showed no significant impact following bowel cleansing.
Conclusions
Bowel cleansing causes a temporary disturbance in bacterial alpha diversity measured in faeces, but no significant changes in the faecal and urine metabolic profiles, suggesting that overall the faecal microbiome and its associated metabolome is resistant to the effects of an induced osmotic diarrhoea.
Similar content being viewed by others


Synopsis
The intestinal microbiome has been shown to be impacted by bowel cleansing, but the impact on the related metabolome has been much less explored. This study demonstrated a temporary disturbance in faecal microbiome following bowel lavage, but the associated urinary and faecal metabolome remained stable.
Background
Investigation of the composition and functionality of the gut microbiome is of key interest for a range of gastro-intestinal (GI) diseases [1]. However, there is no standardised best practice regarding choice of sample type (stool or mucosal biopsies), or sample collection methodology and/or processing. It is also recognised that a wide range of external factors may influence the results of analysis. One such major influence is medication, including bowel purgatives such as polyethylene glycol (PEG) solutions, which are given as bowel cleansing prior to colonoscopy [2, 3]; this being of particular relevance when investigating GI disease.
PEG solutions cause a profound osmotic diarrhoea with a high volume lavage rapidly passing through the gastro-intestinal tract, which in turn alters the luminal contents including the microbiota [4, 5]. Effects of bowel cleansing on the intestinal microbiome have been studied [4,5,6,7,8,9,10], both to assess whether it can directly cause dysbiosis, and to assess how these vary in health and disease [5].
Results have been inconsistent across different published studies; some have shown a significant reduction in bacterial load [8] and alpha diversity [4, 7, 10] when examining the faecal and colonic mucosal microbiota post bowel cleansing, but this has not been universally demonstrated [5, 6, 9]. In those studies that demonstrate a dysbiosis however, it appears that the composition recovers quickly [5, 8, 10]—likely within 14 days—but the exact timing of this restoration is unclear. Understanding the longevity of these microbial changes may be important in determining study protocols and interpreting results. Of further interest is determining if there are functional metabolic changes (measured in the metabolome) associated with these microbiome perturbations, beyond simply measuring composition. Only one study to date [5] has assessed the metabolic effects of bowel cleansing, using faecal metabonomic analysis.
While faecal metabolic data have been extensively researched in GI conditions [11,12,13,14,15,16], sample collection can be unappealing for patients, and therefore be more difficult to obtain. Urine is a more readily acquired biosample, and has been shown to demonstrate gut host-microbiota metabolic changes in GI pathology including inflammatory bowel disease (IBD) [17,18,19,20,21] and colorectal cancer [22,23,24,25]. No studies have yet been published assessing the impact of bowel cleansing on bacterial associated urinary metabolites or their relationship to the faecal microbiome or metabolome.
This study aimed to compare the effects of bowel cleansing on the faecal microbiome and metabolome at baseline, day 3 and week 6 post colonoscopy.
Methods
Experimental design and subjects
This study had ethical approval from Imperial College Healthcare NHS Trust Research and Ethics Committee (Ref: 13/LO/1867). Eleven subjects were recruited from gastroenterology clinics at St. Mary’s Hospital in London who were due to undergo a colonoscopy. Nine participants with no previous history of gastrointestinal disorder were being investigated for a new change in bowel habit and/or diarrhoea; two participants with known ulcerative colitis were being assessed for the activity of inflammatory bowel disease. Subjects were excluded if they had received antibiotics, further purgatives, acid suppressing or immunosuppressive medication within 2 months of sample collection. Written informed consent was obtained from all participants. Detailed dietary and lifestyle data was taken from each subject. A low residue diet containing items such as refined carbohydrates (e.g. white rice), clear soups and white meats, and excluding items high in fibre (e.g. wholemeal bread, fruits with skins) was consumed from two days before the procedure. No solid food was eaten for the final 24 h before the colonoscopy. Participants were given Moviprep® two doses (each dose dissolved in 1 L of water) as a bowel cleanse according to manufacturer guidance.
Sample collection
32 urine samples and 30 faecal samples were collected from 11 subjects at 3 different time points around their colonoscopy (Additional file 1: Table S1). 9 of these subjects were male, and the mean age was 41 years. The mean BMI was 23.4 kg/m2. Two subjects did not provide faecal samples at the last time point, and one of these subjects also did not provide a urine sample at the last time point—however all recruited subjects were included in the final analysis. Baseline (t0) samples were collected 3 days prior to the procedure, and before a low residue diet or bowel purgatives were commenced. Further samples were collected 3 days post procedure (t1), and 6 weeks post procedure (t2). Samples were collected in sterile polypropylene containers and stored in a − 80 °C freezer once received from the subject. MoviPrep® (Macrogol 3350, Sodium sulphate anhydrous, Sodium chloride, Potassium chloride, Ascorbic acid and Sodium ascorbate) was used as bowel preparation in all cases.
Bacterial DNA extraction and 16S rRNA gene sequencing
DNA extraction was performed using the PowerLyzer PowerSoil DNA Isolation Kit (Mo Bio, Carlsbad, CA, USA). 250 mg of faeces from each sample was used for extraction, and the manufacturer’s instructions were followed. An in-house additional bead beating step [26] was included at speed 8 for 3 min using a Bullet Blender Storm (Chembio Ltd, St Albans, UK). The extracted DNA was then stored at − 80 °C. Illumina’s 16S Metagenomic Sequencing Library Preparation Protocol [27] was used to prepare the sample libraries; the V1-V2 regions of the 16S rRNA gene were amplified using previously-reported primers [28]. These libraries were quantified using the NEBNext Library Quant Kit for Illumina (New England Biolabs, Hitchin, UK). An Illumina MiSeq platform (Illumina Inc., Saffron Walden, UK) was used to perform the sequencing using the MiSeq Reagent Kit v3 (Illumina) and paired-end 300 bp chemistry.
16S rRNA sequencing data was then analysed using the Mothur package (version 2017a, The Mathworks, Inc.; Natwick, MA) following the MiSeq SOP Pipeline [29]. Sequence alignment was performed using the Silva bacterial database, and the Wang method using the ribosomal database project (RDP) database was used for classification of sequences [30]. Operational taxonomic unit (OTU) taxonomies (from phylum to genus) were established using the RDP MultiClassifier Script. The lowest number of reads in a sequenced sample was > 10,000, and subsampling was performed at 10,000 reads per sample. Calculations were performed within Mothur for alpha diversity (Shannon diversity index, H’), and Wilcoxon matched-pairs signed rank test using GraphPad Prism statistical analysis software programme version 8.0.2 was used to assess for statistical significance between time points. Beta diversity was assessed using the non-metric multidimensional scaling (NMDS) plot, and PERMANOVA p-values were generated using the UniFrac weighted distance matrix generated from Mothur, and analysed using the Vegan library within the R statistical package (version 3.3.3) [31]. The Statistical Analysis of Metagenomic Profiles software (STAMP) package [32] was used to assess for statistically significant differences in bacterial composition in the subjects at different time points using White’s non-parametric t-test with Benjamini–Hochberg false discovery rate (FDR).
Metabolomic analysis
Sample preparation
Preparation of urine samples for metabolomic analysis was performed in accordance to standardized protocols. In brief, 540 µL of defrosted urine sample was added to 60 µL of buffer (1.5 M KH2PO4/D2O, 2 mM NaN3 and 0.1% 3-(trimethyl-silyl)propionic acid-d4) (TSP), and centrifuged. 550 µL of the resulting supernatant was transferred into 5 mm diameter NMR tubes. Faecal water was extracted from whole faecal samples by mixing 500 mg of crude stool with two volumes of phosphate buffer saline (PBS) solution, and then vortexing at 2000 Hz for 15 min. These were then centrifuged at 9500 rpm for 20 min at 4 °C, and 600 μl. 400µL of supernatant was transferred to a new Eppendorf, where 100µL of buffer and 250ul D2O were added and the samples centrifuged again. 600µL was transferred to a 5 mm diameter NMR tube. Samples were loaded onto a refrigerated SampleJet robot (Bruker Corporation, Germany) and maintained at 4 °C until analysis. Pooled quality control (QC) samples were also generated for both sample types.
Sample analysis
Metabolomic analysis was undertaken using proton nuclear magnetic resonance (1H NMR) spectroscopy. All experiments were carried out using an Avance 600 MHz NMR spectrometer (Bruker Biospin), following a standardised protocol [33, 34]. In brief, one dimensional NOESY experiments were carried out on urine and faecal water samples. Spectra were obtained for both sample types at a constant temperature of 300 K. Spectra consisted of 96 K data points, with a spectral width of 20 ppm centred at 4.75 ppm, and pulse width of approximately ~ 13 µs. A double pre-saturation technique was carried out to attenuate the water resonance, harnessing the relaxation delay (4 s), and NOESY mixing time.
Data analysis for metabolomic samples
Following acquisition, all spectra were phased, calibrated, (using TSP) and baseline corrected automatically in Topspin (version 3.2, Bruker Biospin Ltd.). Spectral data was imported into MATLAB (version 2017a, The Mathworks, Inc.; Natwick, MA) using in-house scripts before spectral regions containing redundant information were removed. These included peaks corresponding to water and TSP, at 4.6–4.85 ppm and − 0.2 to 0.2 ppm respectively. Prior to modelling, all data were aligned using an in-house automatic alignment function. Spectral data was then normalized using a probabilistic quotient approach.
Multivariate statistical analysis was used to investigate differences between study groups. This was performed using SIMCA (version 15, Umetrics, Sweden). Principal components analysis (PCA) was carried out using univariate scaling to allow for the identification of any outliers and clustering based on principal components.
For targeted analysis, peak integral values for selected metabolites were obtained using an in-house Matlab script [26]. Using the integral values, a univariate statistical approach was used to compare the relative amount of metabolites of interest between the three time points. GraphPad Prism statistical analysis software programme version 8.0.2 was used to perform a Wilcoxon matched-pairs signed rank test between time points t0 and t1, t0 and t2, and t1 and t2. A Bonferroni calculation was used to correct for multiple comparisons. Urine metabolites [17, 19,20,21,22,23,24,25, 35, 36] and faecal metabolites [11,12,13, 37, 38] were selected for targeted analysis if they are produced by intestinal bacterial metabolism or host-bacterial co-metabolism, and in previous studies have been shown to be important in GI disease.
Results
Faecal 16 s RNA sequencing
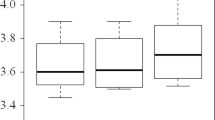
31 faecal samples were collected, which included 11 samples at baseline (t0), 11 at three days post colonoscopy (t1), and 9 at six weeks post procedure (t2). Analysis of the alpha diversity between baseline (t0) and 3 days post procedure (t1) showed that bowel preparation caused a significant decrease (p = 0.002) in the Shannon index of the bacteria present (Fig. 1). There was no significant change between baseline and 6 weeks post bowel cleansing (t0 and t2).

Faecal microbiota alpha diversity as measured by the Shannon index at baseline (t0), 3 days after bowel preparation (t1), and 6 weeks after bowel preparation (t2)
Compositional changes between samples (beta diversity) were analysed using non-metric multidimensional scaling (NMDS) plots measuring weighted Unifrac distances—see Fig. 2. There were no statistically significant differences when comparing samples across different time points permutational multivariate analysis of variance (PERMANOVA).

NMDS plot of microbial composition of subjects at differing time points. t0 = baseline, t1 = 3 days post colonoscopy, and t2 = 6 weeks post colonoscopy. Lines and labels have been added to figure to clarify subjects and time points
Taxanomic analysis at phylum, class, order, family, and genus levels was performed using the STAMP software package, and there was no significant difference in composition of bacteria between samples taken at baseline, 3 days post colonoscopy, and 6 weeks post procedure—see Fig. 3.

Percentage bar graph showing the relative proportion of bacterial phyla in each sample. Samples are grouped within subjects and in order of collection time; at baseline (t0), 3 days after bowel preparation (t1), and 6 weeks after bowel preparation (t2)
Urine metabonomic analysis
Metabonomic analysis was performed on 32 urine samples which were collected at the same time as the faecal samples. This included 11 baseline samples (t0), 11 collected at 3 days post procedure (t1), and 10 collected at 6 weeks post procedure (t2). Detailed lifestyle and dietary data obtained from the study subjects showed no significant differences between time points—see Additional file 1: Table S2.
Unsupervised multivariate analysis using principal component analysis (PCA) of the urine samples at each time point was performed—see Fig. 4. In 9 out of the 11 subjects, there was intra-subject clustering of samples despite the use of bowel cleansing.

Principal component analysis (PCA) scores plot of urine metabolic profiles of 11 subjects, with samples at baseline, 3 days post procedure, and 6 weeks post procedure. Samples from the same subject are plotted in the same colour, with participant number as shown in the key; sampling time points are also shown for each participant, i.e. baseline (t0), 3 days after bowel preparation (t1), and 6 weeks after bowel preparation (t2). This figure shows intra-subject clustering in samples from 9 out of 10 subjects over the 3 time points
Targeted analysis of 20 urine metabolites revealed that alanine excretion was higher at 3 days and 6 weeks post bowel cleansing in comparison to baseline (p value 0.005, and 0.020 respectively) but these differences were not significant after correcting for multiple comparisons—see Fig. 5 and Additional file 1: Table S3.

Bar plot graphs showing median relative quantity of each urine metabolite at three time points; at baseline (t0), 3 days after bowel preparation (t1), and 6 weeks after bowel preparation (t2)
Faecal metabonomic analysis
Metabonomic analysis was performed on ten faecal samples at baseline (t0), and ten samples 3 days post colonoscopy (t1). Unsupervised multivariate analysis of faecal water metabolites was performed using Principal Component Analysis—see Additional file 1: Figure S1. This showed that in 9 out of 10 subjects, there was intra-subject clustering of samples despite the use of bowel cleansing. Targeted analysis was performed on 10 metabolites. Butyrate was higher at 3 days in comparison to baseline (p value = 0.027) but this difference was not significant after correcting for multiple comparisons—see Additional file 1: Table S4.
Discussion
This is a novel study combining urinary and faecal metabolic data with microbiomic changes to assess the effect of bowel cleansing used pre-colonoscopy.
Stool analysis showed a reduction in microbiome ecological indices, with a significant decrease in the Shannon index (p = 0.002) following bowel preparation. In a similar study, Gorkiewicz et al. [4] demonstrated a reduction in richness following three days of PEG administration, and this change trended towards recovery but remained significantly lower than baseline one week after bowel cleansing. A trend towards reduced Shannon index was also shown by Shobar et al. [10] within one week post bowel cleansing. However, four other studies did not show a change in Shannon index following bowel cleansing [5, 6, 8, 9].
Inter-subject variability was more marked than intra-subject variability in all but three patients following bowel preparation, and there were no statistically significant changes in beta diversity between samples across the different time points. Similar beta diversity analysis published by Nagata et al. [5], using principal coordinated analysis (PCoA) plot of weighted UniFrac distances, also showed individuals who had received bowel cleansing clustered together, rather than clustering at time points. A study by Shobar et al. [10] also did not show a statistically significant difference in weighted Unifrac distances in faecal samples pre- and post-bowel cleansing, but did show a difference in unweighted distances, potentially suggesting that rarer species were affected more by bowel cleansing.
Taxonomic composition analysis showed no statistically significant changes from phylum to genus level following bowel cleansing. Negata et al. [5] showed no changes in the relative abundance at phyla level in eight patients receiving bowel cleansing, and in genera with a > 1% relative abundance. A larger study by O'Brien et al. [9] showed no consistent findings in taxonomic composition in 15 patients undergoing bowel lavage. Another study by Shobar et al. [10] showed a reduction in Bacteroidetes in ten healthy controls and eight IBD patients receiving bowel preparation, along with changes at other taxonomic levels including a reduction in the Clostridiales order. A reduction in Firmicutes and an increase in Proteobacteria was reported in a study by Drago et al. [6] at phyla level, who also showed a reduction in the Clostridia class. Discrepancies between previous studies [4,5,6, 8,9,10, 39] assessing the impact of bowel cleansing on the faecal microbiota are likely in part due to the differing study designs, time point analysis, analytical techniques and generally small sample sizes.
The effects of bowel cleansing on intestinal bacterial-associated urine metabolites was investigated in this study, measuring selected targeted metabolites which have been previously demonstrated to alter in GI diseases including IBD and colorectal cancer [17, 19,20,21,22,23, 25, 35, 36]. These included formate, hippurate, p-cresol sulphate, and alanine [17, 21]. Formate, produced both endogenously and from intestinal fermentation, has been associated with the Enterobacteriaceae phylum, and particularly Escherichia coli [40, 41]. Hippurate, a product of host and commensal co-metabolism of dietary aromatic compounds, has been shown in several studies to have reduced excretion in IBD [17, 19,20,21], and to have a positive association with Clostridia species [42]. Bacterial fermentation of tyrosine in the colon produces p-cresol sulphate, and its production has also been associated with Clostridia species [43]. Beta-alanine, an isomer of alanine, is a non-protein amino acid obtained from dietary muscle protein and additionally sourced from intestinal Escherichia coli [44]. Trimethylamine N-oxide (TMAO) has been associated with the development of colorectal cancer [16, 45]. TMAO is reduced to trimethylamine (TMA) by predominantly Enterobacteriaceae in the gut [46].
There were no significant changes in any of the urinary metabolites that were measured following bowel cleansing (with the exception of alanine, although this was not significant when corrected for multiple comparisons). Unsupervised multivariate analysis showed that in nine out of eleven patients, subject clustering was greater than time point clustering irrespective of bowel cleansing, suggesting stability of bacterial associated urinary metabolites despite alterations in the intestinal faecal microbiome. Untargeted multivariate analysis of faecal metabolites showed that at day 3 post bowel cleansing subject clustering was still present in 9 out of 10 subjects, and targeted univariate analysis showed higher butyrate excretion at day 3, although statistical significance for this change in butyrate was lost once corrected for multiple comparisons. A recent study by Nagata et al. [5] showed an immediate faecal metabolic perturbation in the first catch samples following bowel purgative ingestion, but subject clustering and changes in specific metabolites were restored at day 14 (the next sampling point in this study).
A stable metabolome despite a transient change in one aspect of the intestinal microbiome composition (alpha diversity) may suggest functional redundancy, where despite loss of specific bacterial species, the overall metabolic processes are preserved by cross-compensation by other bacteria [47]. These results suggest that in patients undergoing bowel preparation for colonoscopy, PEG may potentially confound results of microbiome studies, but appears less likely to impact studies of microbiomic function.
Several limitations were present in this study, including a relatively small number of subjects—although this was a similar cohort size to previous studies assessing the effects of bowel cleansing. Although all the colonoscopy examinations showed no significant active pathology, all subjects were recruited from gastroenterology clinic and reporting gastrointestinal symptoms, and so there was a mixture of subjects with quiescent ulcerative colitis and irritable bowel syndrome within the study subjects. More than 80% of the study participants were male, meaning that our results might not be reflective of a cohort with a lower proportion of male subjects. The week 6 sample cohort was incomplete, with two subjects not giving faecal samples, and one subject not giving a urine sample. Moreover, faecal metabolomics was not performed at week 6 owing to a lack of faecal material available for the analysis. The initial post-intervention sample time point was taken three days after bowel cleansing, and so more immediate perturbations in metabolites and the faecal microbiome may have been missed. NMR spectroscopy was used as the analytical platform, which gives a good overall qualitative and relative quantitative assessment of metabolites, but is less sensitive than mass spectrometry [48].
In conclusion, this study shows that bowel cleansing causes a temporary disturbance in bacterial alpha diversity measured in faeces, but no significant changes in the urine and faecal metabolome. This suggests overall the faecal microbiome and its associated metabolome is resistant to the effects of an induced osmotic diarrhoea.
Availability of data and materials
The datasets generated during and/or analysed during the current study are not publicly available at this time but are available from the corresponding author on reasonable request.
Abbreviations
- 1H NMR:
-
Proton nuclear magnetic resonance
- BMI:
-
Body mass index
- CD:
-
Crohn’s disease
- GI:
-
Gastrointestinal
- NMDS:
-
Non-metric multidimensional scaling
- PEG:
-
Polyethylene glycol
- t0:
-
Time point 0—baseline
- t1:
-
Time point 1—3 days post colonoscopy
- t2:
-
Time point 2—6 weeks post colonoscopy
- UC:
-
Ulcerative colitis
References
McIlroy J, Ianiro G, Mukhopadhya I, Hansen R, Hold GL. Review article: the gut microbiome in inflammatory bowel disease-avenues for microbial management. Aliment Pharmacol Ther. 2018;47(1):26–42.
Clark RE, Godfrey JD, Choudhary A, Ashraf I, Matteson ML, Bechtold ML. Low-volume polyethylene glycol and bisacodyl for bowel preparation prior to colonoscopy: a meta-analysis. Ann Gastroenterol. 2013;26(4):319–24.
Parra-Blanco A, Ruiz A, Alvarez-Lobos M, Amorós A, Gana JC, Ibáñez P, et al. Achieving the best bowel preparation for colonoscopy. World J Gastroenterol. 2014;20(47):17709–26.
Gorkiewicz G, Thallinger GG, Trajanoski S, Lackner S, Stocker G, Hinterleitner T, et al. Alterations in the colonic microbiota in response to osmotic diarrhea. PLoS ONE. 2013;8(2):e55817.
Nagata N, Tohya M, Fukuda S, Suda W, Nishijima S, Takeuchi F, et al. Effects of bowel preparation on the human gut microbiome and metabolome. Sci Rep. 2019;9(1):4042.
Drago L, Toscano M, De Grandi R, Casini V, Pace F. Persisting changes of intestinal microbiota after bowel lavage and colonoscopy. Eur J Gastroenterol Hepatol. 2016;28(5):532–7.
Harrell L, Wang Y, Antonopoulos D, Young V, Lichtenstein L, Huang Y, et al. Standard colonic lavage alters the natural state of mucosal-associated microbiota in the human colon. PLoS ONE. 2012;7(2):e32545.
Jalanka J, Salonen A, Salojarvi J, Ritari J, Immonen O, Marciani L, et al. Effects of bowel cleansing on the intestinal microbiota. Gut. 2015;64(10):1562–8.
O’Brien CL, Allison GE, Grimpen F, Pavli P. Impact of colonoscopy bowel preparation on intestinal microbiota. PLoS ONE. 2013;8(5):e62815.
Shobar RM, Velineni S, Keshavarzian A, Swanson G, DeMeo MT, Melson JE, et al. The effects of bowel preparation on microbiota-related metrics differ in health and in inflammatory bowel disease and for the mucosal and luminal microbiota compartments. Clin Transl Gastroenterol. 2016;7:e143.
Bjerrum JT, Wang Y, Hao F, Coskun M, Ludwig C, Gunther U, et al. Metabonomics of human fecal extracts characterize ulcerative colitis, Crohn’s disease and healthy individuals. Metabol Off J Metabol Soc. 2015;11:122–33.
Le Gall G, Noor SO, Ridgway K, Scovell L, Jamieson C, Johnson IT, et al. Metabolomics of fecal extracts detects altered metabolic activity of gut microbiota in ulcerative colitis and irritable bowel syndrome. J Proteome Res. 2011;10(9):4208–18.
Santoru ML, Piras C, Murgia A, Palmas V, Camboni T, Liggi S, et al. Cross sectional evaluation of the gut-microbiome metabolome axis in an Italian cohort of IBD patients. Sci Rep. 2017;7(1):9523.
Scott AJ, Alexander JL, Merrifield CA, Cunningham D, Jobin C, Brown R, et al. International Cancer Microbiome Consortium consensus statement on the role of the human microbiome in carcinogenesis. Gut. 2019;68(9):1624–32.
Weir TL, Manter DK, Sheflin AM, Barnett BA, Heuberger AL, Ryan EP. Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PLoS ONE. 2013;8(8):e70803.
Xu R, Wang Q, Li L. A genome-wide systems analysis reveals strong link between colorectal cancer and trimethylamine N-oxide (TMAO), a gut microbial metabolite of dietary meat and fat. BMC Genomics. 2015;16(Suppl 7):S4.
Dawiskiba T, Deja S, Mulak A, Ząbek A, Jawień E, Pawełka D, et al. Serum and urine metabolomic fingerprinting in diagnostics of inflammatory bowel diseases. World J Gastroenterol. 2014;20(1):163–74.
De Preter V. Metabolomics in the clinical diagnosis of inflammatory bowel disease. Dig Dis. 2015;33(Suppl 1):2–10.
Schicho R, Shaykhutdinov R, Ngo J, Nazyrova A, Schneider C, Panaccione R, et al. Quantitative metabolomic profiling of serum, plasma, and urine by (1)H NMR spectroscopy discriminates between patients with inflammatory bowel disease and healthy individuals. J Proteome Res. 2012;11(6):3344–57.
Stephens NS, Siffledeen J, Su X, Murdoch TB, Fedorak RN, Slupsky CM. Urinary NMR metabolomic profiles discriminate inflammatory bowel disease from healthy. J Crohns Colitis. 2013;7(2):e42–8.
Williams HR, Cox IJ, Walker DG, North BV, Patel VM, Marshall SE, et al. Characterization of inflammatory bowel disease with urinary metabolic profiling. Am J Gastroenterol. 2009;104(6):1435–44.
Chen JL, Fan J, Yan LS, Guo HQ, Xiong JJ, Ren Y, et al. Urine metabolite profiling of human colorectal cancer by capillary electrophoresis mass spectrometry based on MRB. Gastroenterol Res Pract. 2012;2012:125890.
Cheng Y, Xie G, Chen T, Qiu Y, Zou X, Zheng M, et al. Distinct urinary metabolic profile of human colorectal cancer. J Proteome Res. 2012;11(2):1354–63.
Kim ER, Kwon HN, Nam H, Kim JJ, Park S, Kim YH. Urine-NMR metabolomics for screening of advanced colorectal adenoma and early stage colorectal cancer. Sci Rep. 2019;9(1):4786.
Qiu Y, Cai G, Su M, Chen T, Liu Y, Xu Y, et al. Urinary metabonomic study on colorectal cancer. J Proteome Res. 2010;9(3):1627–34.
Mullish BH, Pechlivanis A, Barker GF, Thursz MR, Marchesi JR, McDonald JAK. Functional microbiomics: evaluation of gut microbiota-bile acid metabolism interactions in health and disease. Methods. 2018;149:49–58.
Illumina. 16S Metagenomic Sequencing Library Preparation: Illumina. 2017. https://support.illumina.com/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf.
Mullish BH, Pechlivanis A, Barker GF, Thursz MR, Marchesi JR, McDonald JAK. Functional microbiomics: evaluation of gut microbiota-bile acid metabolism interactions in health and disease. Methods (San Diego, Calif). 2018;149:49–58.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–41.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41(Database issue):D590–6.
R. The R Project for Statistical Computing 2017. https://www.rproject.org.
Parks D, Beiko R. STAMP: statistical analysis of metagenomic profiles. 2013 [
Dona AC, Jimenez B, Schafer H, Humpfer E, Spraul M, Lewis MR, et al. Precision high-throughput proton NMR spectroscopy of human urine, serum, and plasma for large-scale metabolic phenotyping. Anal Chem. 2014;86(19):9887–94.
Morris LS, Marchesi JR. Assessing the impact of long term frozen storage of faecal samples on protein concentration and protease activity. J Microbiol Methods. 2016;123:31–8.
Chen MX, Wang S-Y, Kuo C-H, Tsai IL. Metabolome analysis for investigating host-gut microbiota interactions. J Formos Med Assoc. 2019;118:S10–22.
De Preter V, Verbeke K. Metabolomics as a diagnostic tool in gastroenterology. World J Gastrointest Pharmacol Therap. 2013;4(4):97–107.
Jansson J, Willing B, Lucio M, Fekete A, Dicksved J, Halfvarson J, et al. Metabolomics reveals metabolic biomarkers of Crohn’s disease. PLoS ONE. 2009;4(7):e6386.
Marchesi JR, Holmes E, Khan F, Kochhar S, Scanlan P, Shanahan F, et al. Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J Proteome Res. 2007;6(2):546–51.
Nishimoto Y, Mizutani S, Nakajima T, Hosoda F, Watanabe H, Saito Y, et al. High stability of faecal microbiome composition in guanidine thiocyanate solution at room temperature and robustness during colonoscopy. Gut. 2016;65(9):1574–5.
Case GL, Benevenga NJ. Significance of formate as an intermediate in the oxidation of the methionine, S-methyl-L-cysteine and sarcosine methyl carbons to CO2 in the rat. J Nutr. 1977;107(9):1665–76.
Leonhartsberger S, Korsa I, Bock A. The molecular biology of formate metabolism in enterobacteria. J Mol Microbiol Biotechnol. 2002;4(3):269–76.
Li M, Wang B, Zhang M, Rantalainen M, Wang S, Zhou H, et al. Symbiotic gut microbes modulate human metabolic phenotypes. Proc Natl Acad Sci U S A. 2008;105(6):2117–22.
Bone E, Tamm A, Hill M. The production of urinary phenols by gut bacteria and their possible role in the causation of large bowel cancer. Am J Clin Nutr. 1976;29(12):1448–54.
Eaton K, Howard M, Hunnisett A. Urinary beta-alanine excretion is a marker of abnormal as well as normal gut fermentation. J Nutr Med. 2009;14:121–7.
Guertin KA, Li XS, Graubard BI, Albanes D, Weinstein SJ, Goedert JJ, et al. Serum trimethylamine N-oxide, carnitine, choline, and betaine in relation to colorectal cancer risk in the alpha tocopherol, beta carotene cancer prevention study. Cancer Epidemiol Biomark Prevent Publ Am Assoc Cancer Res Cospons Am Soc Prevent Oncol. 2017;26(6):945–52.
Hoyles L, Jiménez-Pranteda ML, Chilloux J, Brial F, Myridakis A, Aranias T, et al. Metabolic retroconversion of trimethylamine N-oxide and the gut microbiota. Microbiome. 2018;6(1):73.
Moya A, Ferrer M. Functional redundancy-induced stability of gut microbiota subjected to disturbance. Trends Microbiol. 2016;24(5):402–13.
Emwas AH. The strengths and weaknesses of NMR spectroscopy and mass spectrometry with particular focus on metabolomics research. Methods Mol Biol (Clifton, NJ). 2015;1277:161–93.
Acknowledgements
Nil.
Funding
The Department of Metabolism, Digestion and Reproduction at Imperial College London receives funding from the National Institute of Health Research (NIHR) Biomedical Research Centre (BRC) based at Imperial College Healthcare NHS Trust and Imperial College London. BHM and JLA are the recipients of NIHR Academic Clinical Lectureships. JLA receives funding for his Clinical Lectureship from Imperial College London and The Freed Foundation. None of the funders for this work provided any direct role in the design of the study, sample collection, sample analysis, data interpretation, writing the manuscript, or any other aspects of the study.
Author information
Authors and Affiliations
Contributions
TRO and HRTW conceptualised and supervised the study. STRP conceptualised study, recruited subjects, collected and processed the samples and analysed the data. KIG assisted in data analysis and producing results figures. LWLC assisted in sample collection, data analysis and processing. BHM, JLA, LCH, JAKM and JRM assisted in data analysis. All co-authors assisted in manuscript revision. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study had ethical approval from Imperial College Healthcare NHS Trust Research and Ethics Committee (Ref: 13/LO/1867).
Consent for publication
Full written informed consent obtained from study subjects at time of recruitment.
Competing interests
The authors of this study have no competing interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary Tables 1 to 4, and Supplementary Figure 1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Powles, S.T.R., Gallagher, K.I., Chong, L.W.L. et al. Effects of bowel preparation on intestinal bacterial associated urine and faecal metabolites and the associated faecal microbiome. BMC Gastroenterol 22, 240 (2022). https://doi.org/10.1186/s12876-022-02301-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12876-022-02301-1