
Abstract
Background
Hepatocellular carcinoma (HCC) was frequently considered as a kind of malignant tumor with a poor prognosis. Cyclin-dependent kinases (CDK) 4 was considered to be cell-cycle-related CDK gene. In this study, we explored the clinical significance of CDK4 in HCC patients.
Methods
Data of HCC patients were obtained from The Cancer Genome Atlas database (TCGA) and the Gene Expression Omnibus (GEO) database. Kaplan–Meier analysis and Cox regression model were performed to calculate median survival time (MST) and the hazard ration (HR), respectively. The joint-effect analysis and prognostic risk score model were constructed to demonstrate significance of prognosis-related genes. The differential expression of prognostic genes was further validated using reverse transcription-quantitative PCR (RT-qPCR) of 58 pairs of HCC samples.
Results
CDK1 and CDK4 were considered prognostic genes in TCGA and GSE14520 cohort. The result of joint-effect model indicated patients in CDK1 and CDK4 low expression groups had a better prognosis in TCGA (adjusted HR = 0.491; adjusted P = 0.003) and GSE14520 cohort (adjusted HR = 0.431; adjusted P = 0.002). Regarding Kaplan–Meier analysis, high expression of CDK1 and CDK4 was related to poor prognosis in both the TCGA (P < 0.001 and = 0.001 for CDK1 and CDK4, respectively) and the GSE14520 cohort (P = 0.006 and = 0.033 for CDK1 and CDK4, respectively). However, only CDK4 (P = 0.042) was validated in RT-qPCR experiment, while CDK1 (P = 0.075) was not.
Conclusion
HCC patients with high CDK4 expression have poor prognosis, and CDK4 could be a potential candidate diagnostic biomarker for HCC.
Similar content being viewed by others


Background
In 2020, liver cancer was known to rank sixth among diagnosed malignant cancers worldwide and the third leading cause of cancer death, ranking second in terms of cancer death for males [1]. 75–85% of primary liver cancer was HCC [1]. The main risk factors were chronic hepatitis B or C, aflatoxin B1 exposure, excessive alcohol intake and alcohol-related liver disease [1, 2]. Metabolic diseases such as type 2 diabetes (T2DM) and non-alcoholic fatty liver (NAFLD) are high-risk factors for HCC [3, 4]. NAFLD can progress to nonalcoholic steatohepatitis and then to HCC, and T2DM increases the risk of HCC by a factor of 3 through the PTEN/P13K/Akt and MAPK kinase molecular pathway [3,4,5]. Although there were many well-established diagnoses for HCC, including computed tomography, ultrasonography, serum tumor markers and magnetic resonance imaging [2], HCC patients are usually in advanced liver failure when they develop symptoms and are usually untreatable [6]. Therefore, patients can be diagnosed early to obtain longer overall survival and it is necessary to explore molecular biomarkers to provide early diagnosis and prognostic assessment for HCC patients.
The CDKs genes family play a vital role in cell division and modulating transcription [7]. A total of 21 genes in the CDKs gene family were divided into 11 subfamilies, of which CDK1 (CDK1, CDK2, CDK3), CDK4 (CDK4, CDK6) subfamily were considered to be cell-cycle-related subfamilies [7, 8]. Cell cycle regulators were frequently mutated in the tumor, including overexpression of CDKs [9]. There are many studies reporting the involvement of CDK1 and CDK4 subfamilies in the progression of multiple cancer [10,11,12,13,14,15,16,17,18,19,20]. However, the relationship between CDK1-4, 6 expression and the risk of HCC patients was rarely reported. Thus, this study aims to explore the role of CDK1-4, 6 expressions in HCC patients based on public cancer data.
Materials and methods
Data source
The mRNA expression and clinical information of HCC in the TCGA database were obtained from the University of California, Santa Cruz Xena (UCSC Xena, https://xenabrowser.net/datapages/) [21]. The GSE14520 dataset from the GEO database was analyzed. The platform of GSE14520 was GPL3921, which collected the mRNA expression levels of 225 HCC tissues and 220 matched liver tissues. The clinical information and mRNA gene expression matrix of HCC were downloaded from GSE14520 on the GEO website (https://www.ncbi.nlm.nih.gov/geo) [22, 23]. The limma package was used to process and normalize the raw data of the GSE14520 gene expression matrix in R platform. The gene expression and clinical data from Chinese HCC (CHCC) patients were accessed via the National Omics Data Encyclopedia (NODE) website (https://www.biosino.org/node/project/detail/OEP000321) [24].
Tissues processing and RT-qPCR experiments
In RT-qPCR experiment, a total of 58 pairs of tumor and adjacent normal liver tissues (> 3 cm margin from the tumor) of patients pathologically diagnosed as HCC in The First Affiliated Hospital of Guangxi Medical University were collected for further analysis. Inclusion criteria: Patients received no other non-surgical treatments before surgery and hospitalization time was from December 2016 to July 2018. HCC patients with follow-up time < 3 months were excluded. Small pieces of HCC and adjacent normal liver tissues were stored in RNAstore Reagent (Tiangen Biotech Co., Ltd.) at -80℃. According to the manufacturer’s instructions, RNA was extracted from tissues by the Trizol method and reversed into cDNA via PrimeScriptTMRT reagent kit (Takara Bio, Inc.). Primers GAPDH, CDK1, and CDK4 were purchased from TsingKe Biotech Co., Ltd. and their sequences (5'-3') were as follows: GAPDH, forward GTCAGCCGCATCTTCTTT, reverse CGCCCAATACGACCAAAT; CDK1, forward TTTCTTTCGCGCTCTAGCCA, reverse GGTAGATCCGCGCTAAAGGG; CDK4, forward AGCCAGAGAACATTCTGGTGACA, reverse TCGGCTTCAGAGTTTCCACAG. DEPC-treated water and FastStart Universal SYBR Green Master (ROX) were purchased from Sangon Biotech Co., Ltd., and Roche Diagnostics (Shanghai) Co., Ltd., respectively. The reaction cycle of PCR is as follows: hold the stage at 95 degrees for 35 s, then 40 cycles of PRC stage at 95 degrees for 5 s and 55 degrees for 34 s, and finally melt curve stage at 95 degrees for 15 s, 60 degrees for 1 min and 95 degrees for 15 s. The relative expression of mRNAs in tissues was calculated by the 2-∆∆Ct method.
Ethical approval
This study was approved by the Ethical Review Committee of the First Affiliated Hospital of Guangxi Medical University and informed consent of all HCC patients participating in this study was provided.
Bioinformatics analysis and correlation analysis
The Gene Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were used to study the potential biological functions and potential metabolic pathways of CDK1-4,6 by the clusterProfiler package in the R software [25]. The interactive gene‑gene networks and protein–protein interaction (PPI) networks were depicted by the GeneMANIA tool in the Cytoscape software v.3.6.1 [26, 27] and STRING (https://string-db.org) [28], respectively. Pearson’s correlation matrix was depicted correlations among CDK1-4, 6 genes by the ggcorrplot package in the R software.
Diagnostic values assessment and survival analysis
Receiver operating characteristic (ROC) curve was completed by GraphPad Prism v.8 to investigate the diagnostic value of CDK1-4, 6 genes with differential expression for HCC. Univariate Cox regression model was performed to identify the relationship between clinical information and prognosis of HCC patients. Clinical information statistically associated with prognosis in the univariate Cox regression model (P < 0.05) was selected as the adjusted factors for the multivariate Cox regression model. According to the median expression levels of CDK1-4, 6 of tumor tissues, patients were classified into high and low expression groups. The multivariate cox regression model and Kaplan‑Meier survival analysis were performed to explore the relationship between CDK1-4, 6 gene expression and overall survival (OS) in HCC patients. Only CDK1 and CDK4 were found to be statistically associated with OS in the multivariate cox regression model. Therefore, CDK1 and CDK4 were selected as prognosis-related CDK genes in HCC patients. Joint-effect analysis was performed to evaluate the combined effect of CDK1 and CDK4. In the TIMER2.0 (http://timer.comp-genomics.org/) [29] and Gene Expression Profiling Interactive Analysis (GEPIA, http://gepia.cancer-pku.cn/) [30] website, we queried the expression of CDK1 and CDK4 genes in different cancers and the relationship between the expression of CDK1 and CDK4 genes and TP53 gene mutation, a common mutation site in liver cancer. The prognostic value of CDK1 and CDK4 genes in liver cancers were obtained from the Kaplan–Meier Plotter website (https://kmplot.com/analysis/) [31].
Prognostic signature construction
To further explore the influence of CDK1 and CDK4 expression levels on the prognosis of HCC patients. The prognostic risk score model was established by including each the prognosis-related genes respectively weighted by their regression coefficients (β) from the multivariate Cox regression model. To assess the predictive value of the model, the area under the curve (AUC) of the time-dependent ROC curve was completed by the survivalROC package to indicate the predictive accuracy of the model for 1-, 2-, 3- and 5-year survival.
Gene set enrichment analysis
The gene set enrichment analysis (GSEA) was used to explore potential biological mechanisms that prognosis-related CDK genes may be involved in. [32, 33] The referenced gene sets derived from Molecular Signatures Database (MSigDB) of c2 (c2.all.v7.0. symbols) and c5 (c5.all.v7.0.symbols) [34]. C2 gene set contained two subsets of Chemical and genetic perturbations and Canonical pathways and c5 gene set derived from GO annotations [34]. The statistically significant results satisfied the following criteria: P < 0.05 and false discovery rate < 0.25.
Statistical analysis
T-test was used to assess differential expression of CDK1-4, 6 genes between HCC and matched normal tissues. Correlation among CDK1-4, 6 genes was assessed by Pearson’s correlation coefficients. MST was obtained by Kaplan–Meier survival analysis with the log-rank test. Association of CDK gene expression levels and clinical information with OS was assessed by HR and 95% confidence interval (CI) was calculated by Cox regression model. All statistical analyses were done by SPSS v.22.0 software (IBM Corporation, USA). Kaplan–Meier survival curve, scatter plots and ROC curve was depicted by GraphPad Prism v.8 software. Scatter plots, heat maps, histograms and matrix plots were depicted by the R platform (version 3.6.3). P < 0.05 was considered to be statistically significant in this study.
Results
The design of this study is displayed in the flow chart (Fig. 1).

Schematic diagram of the study design
Data source
TCGA cohort included 370 HCC tissues and 50 adjacent normal liver tissues and corresponding 370 patients’ prognostic information. In GSE14520 dataset, the majority of patients were Hepatitis B virus (HBV)-infected patients. To reduce confounding factors, 212 HCC tissues and 204 matched normal liver tissues from 212 HBV-infected HCC patients and the corresponding clinical information were retained in the GSE14520 cohort. In the CHCC cohort, gene expression was acquired in tumor and normal liver tissues from 159 Chinese HCC patients who underwent radical resection.
Bioinformatics analysis and correlation analysis
The results of GO analysis suggested that biological functions (Cellular component, Biological process, Molecular function) of CDK1-4, 6 were involved in regulation of cell cycle, serine/threonine protein kinase complex and protein serine/threonine kinase activity, etc. (Fig. 2A) The KEGG pathway analysis indicated pathways involved in CDK1-4, 6 were enriched cell cycle, p53 signaling pathway, cellular senescence and PI3K-Akt signaling pathway (Fig. 2B and Additional files 1, 2: Figure S1-2) [35,36,37]. The PPI networks suggested that CDK1-4, 6 proteins were associated with Cyclins (CCN) family proteins, CDC20, CDKN1A and CKS1B (Fig. 3A). Moreover, the gene-gene interaction network showed that nine CCN family number genes (CCNA1, CCNA2, CCNB1, CCNB2, CCND1, CCND2, CCND3, CCNE1 and CCNE2) and other genes (CDKN2A, RUNX1, CDKN1A, and so on) also were associated with CDK1-4, 6 (Fig. 3B). Pearson's correlation coefficient of CDK1-4, 6 was used to manifest the correlation among genes (Fig. 3C). The results suggested that CDK1-4, 6 genes had a certain correlation with each other in TCGA.

GO enrichment and KEGG pathway analysis of CDK1-4, 6. GO analysis (A); KEGG pathway analysis (B)

The protein–protein interaction networks among CDK1-4, 6 proteins with other proteins (A) and gene–gene interaction networks among CDK1-4, 6 genes with other genes (B). Matrix graphs of Pearson's correlations cofficient of CDK1-4, 6 gene expressions in the TCGA database (C). Note: *P < 0.05
Diagnostic values assessment
The expression of CDK1-4, 6 genes in different tissues was shown in the scatter plot (Fig. 4), and a total of 3 CDK genes (CDK1, CDK3 and CDK4) were found to be overexpressed in HCC tissues of TCGA cohort. Similarly, CDK1, CDK3, CDK4 and CDK6 were found to be overexpressed in HCC tissues of GSE14520 cohort. To evaluate the diagnostic value of CDK1-4, 6 genes expression, the ROC curve indicated that CDK1 (P < 0.001 and AUC = 0.965) and CDK4 (P < 0.001 and AUC = 0.834) had potential predictive value in TCGA cohort (Fig. 5A–E). Moreover, a total of 3 genes in GSE14520 cohort, CDK1 (P < 0.001 and AUC = 0.964), CDK3 (P < 0.001 and AUC = 0.836) and CDK4 (P < 0.001 and AUC = 0.926) suggested potential diagnostic value (Fig. 5F–I). In particular, CDK1 and CDK4 showed high accuracy in both TCGA and GSE14520 cohort (P < 0.001 and AUC > 0.800).

Scatter plot of expression level of CDK1-4, 6 genes between tumor tissue and adjacent normal liver tissues in TCGA cohort (A) and in GSE14520 cohort (B). Note: CDK2 was unavailable in GSE14520 of GEO database. NS: p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001

The ROC curves of CDK gens in distinguish HCC tumor tissue and adjacent normal tissues. The ROC curves of CDK1 (A), CDK2 (B), CDK3 (C), CDK4 (D), CDK6 (E) in TCGA cohort; the ROC curves of CDK1 (F), CDK3 (G), CDK4 (H), CDK6 (I) in GSE14520 cohort. Note: CDK2 was unavailable in GSE14520 of GEO database
Survival analysis
Kaplan‑Meier survival analysis (Fig. 6A–I) indicated that high expression of CDK1 and CDK4 had statistically significant worse prognosis in both TCGA cohort (Fig. 6A, D; P < 0.001 and = 0.001 for CDK1 and CDK4, respectively) and GSE14520 cohort (Fig. 6F, H; P = 0.006 and = 0.033 for CDK1 and CDK4, respectively).

The Kaplan–Meier survival curves for CDK gens in HCC. Overall survival curves were plotted for CDK1 (A), CDK2 (B), CDK3 (C), CDK4 (D) and CDK6 (E) in TCGA cohort; Overall survival curves were plotted for CDK1 (F), CDK3 (G), CDK4 (H) and CDK6 (I) in GSE14520 cohort. The Kaplan–Meier survival curves for joint-effects analysis of CDK1 and CDK4 in HCC of TCGA cohort (J), GSE14520 cohort (K) and CHCC cohort (L). Note: CDK2 was unavailable in GSE14520 of GEO database
The clinical information for the TCGA cohort of 370 HCC was presented in Table 1. Radical resection (P = 0.007) and III or IV TNM stage (P < 0.001) were statistically significant for OS. The clinical information for GSE14520 cohort of 212 HBV-infected HCC patients was presented in Table 2. Tumor size (P = 0.002), cirrhosis (P = 0.041), BCLC stage (P = 0.050, 0.004 and < 0.001 for A stage, B stage and C stage, respectively), serum AFP (P = 0.049) and TNM stage (P = 0.005 and < 0.001 for II stage and III or IV stage, respectively) were statistically significant for OS in HCC patients. Tumor size (P < 0.001), tumor thrombus (P = 0.005), preoperative AFP (P < 0.001) and BCLC stage (P = 0.014 and < 0.001 for B stage and C stage, respectively) were statistically associated with OS in the CHCC cohort (Additional file 10: Table S1). In TCGA, GSE14520 and CHCC cohort, the above clinical information statistically relevant to OS was considered as prognostic-related information for adjustion in the multivariate Cox regression model.
After adjusting tumor stage and radical resection, the multivariate Cox regression model of TCGA cohort (Table 3) suggested that high expression of CDK1 (adjusted HR = 1.541; adjusted P = 0.028) and CDK4 (adjusted HR = 1.721; adjusted P = 0.005) were statistically related to OS. In GSE14520 cohort (Table 4), tumor size, cirrhosis, and BCLC stage were considered as adjusted factors in the multivariate Cox regression model, which suggested that high expression of CDK1 (adjusted HR = 2.237; adjusted P < 0.001) and CDK4 (adjusted HR = 1.579; adjusted P = 0.044) were statistically related to OS in HBV-infected HCC patients.
In particular, gene expression of CDK1 and CDK4 were statistically related to OS in the multivariate Cox regression model of TCGA and GSE14520 cohort. Therefore, CDK1 and CDK4 were considered to be the genes associated with the prognosis of HCC patients for further joint-effect analysis. Patients in group D (MST = 2456 days, low CDK1 and CDK4 expression) had statistically better prognosis than patients in group A (MST = 899 days, high CDK1 and CDK4 expression) in TCGA cohort (Table 5 and Fig. 6J, adjusted HR = 0.491; adjusted P = 0.003). Similarly, patients in group d (low CDK1 and CDK4 expression) had statistically better prognosis than patients in group a (high CDK1 and CDK4 expression) in GSE14520 cohort (Table 5 and Fig. 6K, adjusted HR = 0.431; adjusted P = 0.002). The CHCC cohort, used as validation for the joint-effects analysis, showed similar results: patients in group IV (low CDK1 and CDK4 expression) had better survival (Additional file 11: Table S2 and Fig. 6L, adjusted HR = 0.287; adjusted P = 0.002).
To validate the prognostic value of CDK1 and CDK4 genes, we searched the value of CDK1 and CDK4 in HCC in multiple datasets. In the CHCC cohort, HCC patients with high expression of CDK1 (Additional file 3: Figure S3A; P < 0.001) and CDK4 (Additional file 3: Figure S3B; P < 0.001) had statistically worse prognosis. In TIMER2.0 results as shown in Additional files 4, 5, 6: Figure S4-6, CDK1 and CDK4 showed high expression in HCC and other cancers and were positively correlated with TP53 gene mutation, a common mutation site in HCC. As the results of the GEPIA website shown in Additional file 7: Figure S7, CDK1 and CDK4 were highly expressed in HCC than normal liver tissues and they also were highly expressed in other cancers, and expression of CDK1 and CDK4 was positively correlated with the stages of HCC. From the Kaplan–Meier Plotter website, we obtained the survival curves of CDK1 and CDK4, and the results showed that HCC patients with high expression of CDK1 and CDK4 had shorter OS (Fig. 7A, B), relapse-free survival (Fig. 7C, D), progression-free survival (Fig. 7E, F) and disease-free survival (Fig. 7G, H) than those with low expression, and these results were statistically significant.

Expression of CDK1 and CDK4 in HCC in Kaplan–Meier plotter tool. Overall survival curves were plotted for CDK1 (A) and CDK4 (B); Relapse free survival curves were plotted for CDK1 (C) and CDK4 (D); Progression free survival curves were plotted for CDK1 (E) and CDK4 (F); Disease free survival curves were plotted for CDK1 (G) and CDK4 (H)
Prognostic signature construction
Prognostic model was constructed to determine the combined predictive value of CDK1 and CDK4 expression. In TCGA cohort, the tumor stage and radical resection were as adjusted factors in the multivariate Cox regression model and regression coefficients (β) of CDK1 and CDK4 were calculated. Therefore, risk score = expression of CDK1 × 0.251 + expression of CDK4 × 0.444. HCC patients were divided into high-risk group (above the median risk score) and low-risk group (below the median risk score) for the calculation of the relationship between risk score and OS by cox regression model and the results were shown in Table 6 and Fig. 8A, B. Compared with the low-risk group (MST = 2456 days), the high-risk group (MST = 1149 days) showed statistically increased risk of death (adjusted HR = 1.643; adjusted P = 0.01) in HCC patients. The model’s predictive value was assessed by time-dependent ROC curves, which had AUC of 0.700, 0.691, 0.681 and 0.616 for the 1-year, 2-year, 3 year and 5-year ROC curves, respectively (Fig. 8C). Similarly, when β was calculated when tumor size, BCLC stage and cirrhosis as adjusted factors, risk score = expression of CDK1 × 0.792 + expression of CDK4 × 0.024 in GSE14520 cohort. The results suggested that the high-risk group suffer experience a worse prognosis (adjusted HR = 2.237; adjusted P < 0.001 Table 6 and Fig. 9A, B). The AUC was 0.533, 0.601, 0.601 and 0.642 for 1-year, 2-year, 3 year and 5-year ROC curves, respectively (Fig. 9C).

Prognostic risk score models of CDK1 and CDK4 genes in HCC patients of TCGA cohort. A Risk score from low to high, distribution of patient survival status and risk score and heat map of CDK1 and CDK4 genes; B Kaplan–Meier survival curves for low-risk and high-risk groups; C Time-dependent ROC analysis of the risk score predicts the HCC OS

Prognostic risk score models of CDK1 and CDK4 genes in HCC patients of GSE14520 cohort. A Risk score from low to high, distribution of patient survival status and risk score and heat map of CDK1 and CDK4 genes. B Kaplan–Meier survival curves for low-risk and high-risk groups. C Time-dependent ROC analysis of the risk score predicts the HBV-related HCC OS
GSEA
In the GSEA analysis, GEO and TCGA datasets were sorted according to expression of CDK1 and CDK4, respectively. In the TCGA cohort, the GSEA results suggested that high expression of CDK1 and CDK4 was correlated with cell cycle, liver cancer survival, cell cycle checkpoints, DNA replication and cell cycle G2 and M phase transition (Additional file 8: Figure S8A–L). In GSE14520 cohort, the GSEA results suggested high expression of CDK1 and CDK4 was correlated with cell cycle, liver cancer survival, DNA repair, regulation of TP53 activity and viral gene expression (Additional file 9: Figure S9A–L).
RT-qPCR experiment
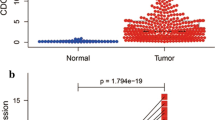
CDK1 and CDK4 were differentially expressed in the RT-qPCR experiment (Fig. 10A, B), respectively (P < 0.001), whereas only CDK4 was overexpressed in HCC tissues consistent with the results from TCGA and GEO cohort. ROC curve analysis suggested that CDK1 (Fig. 10C, AUC = 0.722, P < 0.001) and CDK4 (Fig. 10D, AUC = 0.744, P < 0.001) had statistically predictive value. According to median mRNA expression, patients were divided into high expression group and low expression group. The results of the CDK4 Kaplan–Meier analysis suggested high expression group had statistically worse prognosis (Fig. 10E, P = 0.042), but CDK1 was not statistically significant (Fig. 10F, P = 0.075).

Scatter plot of expression level of CDK1 (A) CDK4 (B) genes between tumor tissue and adjacent normal liver tissues in validation cohort. The ROC curves of CDK1 (C) and CDK4 (D) genes in distinguish HCC tumor tissue and adjacent normal tissues of validation cohort. Kaplan–Meier survival curves of CDK4 (E) and CDK1 (F) in HCC of validation cohort. Note: NS: p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
Discussion
CDK gene families were serine/threonine kinases and their main function was involved in cell cycle regulation, which required the specific cyclin subunits to provide domains essential [7]. The results of bioinformatics analysis suggested CDK1-4, 6 were involved in the regulation of cell cycle and related to the CCN gene and protein family, of which CCNB1, CDC20 and CCND1 were involved in the development of HCC in other reports [38,39,40,41]. In addition, KEGG pathway analysis suggested that CDK1-4, 6 were involved in the p53 signaling pathway and TI3K-Akt signaling pathway.
CDK genes mutations often occur in human tumors [9, 42]. It had been reported that CDK1 interacts with SOX2 to promote tumor initiation in human melanoma and colon [10], and patients with overexpression of CDK1 were reported to have poor prognosis in epithelial ovarian cancer [43], pancreatic ductal adenocarcinoma [11], lung adenocarcinoma [44] and might be a relevant prognostic biomarker. In addition, CDK1 was a promising biomarker for metastasis risk in colon cancer [45]. Moreover, lncRNA PVT1 promoted proliferation, migration and invasion of bladder cancer cells by increasing the expression of CDK1 which down-regulated miR-31 [46]. CDK4 had been reported to be related to a poor prognosis of osteosarcoma, triple-negative breast cancer, elderly lung cancer, and nasopharyngeal carcinoma [17, 18, 47, 48].
Our study suggested that CDK1 and CDK4 were highly expressed in HCC tissues compared to normal controls, and patients with high CDK1 and CDK4 expression had poor prognosis. The results of the joint-effects analysis suggested patients with CDK1 and CDK4 low expression had better prognosis. In the prognostic model, patients in the high-risk group had worse prognosis. The overexpression of CDK4 had been verified in the RT-qPCR experiment, but not CDK1. Although the clinical significance of CDK1 was not validated in RT-qPCR experiments, CDK1 is considered a prognostic factor for HCC in various cohorts and online databases, and its clinical significance in HCC needs to be further explored. Therefore, in this study, HCC patients with overexpression of CDK4 were considered to have poor prognosis, and CDK4 might serve as a potential prognostic biomarker of HCC. Previous reports suggested that high levels of CDK4 can cause hepatic steatosis, fibrosis, and hepatocellular carcinoma in non-alcoholic fatty liver mouse models and patients with fatty liver [49]. CDK1 had been found highly expressed in HCC tissues, and CDK1 mediated nuclear accumulation of apoptin and participated apoptosis in cancer [50]. Overexpression of CDK1 and CCNB1 can promote HCC cell proliferation and migration through the mitogen-activated protein/extracellular signal-regulated kinase (MEK/ERK) signaling pathway, and trials of MEK1/2 inhibitors for the treatment of HCC are currently underway [51, 52]. JIN et al.'s study suggested that LINC00346 affected p53 signaling pathway by regulating the expression of CDK1/CCNB1 and ultimately regulated apoptosis, invasion and cell cycle of HCC cells [53]. Wu et al. demonstrated that CDK1 inhibitor RO3306 can increase the antitumor effect of sorafenib in a PDX tumor model, and can provide a basis for personalized treatment for patients with CDK1-aberrant HCC [54]. Furthermore, Bollard et al.'s preclinical trials found that Palbociclib, a selective CDK4/6 inhibitor, can promote reversible cell cycle arrest to suppress growth of human liver cancer cell lines [55]. CDK4 expression had been reported to be associated with histopathologic grade and progression of HCC and can be used as a prognostic marker for HCC [56, 57].
GSEA results of the current study suggested that CDK1 and CDK4 are significantly related to liver cancer survival and some mechanisms that might be involved in cancer development: DNA repair, cell cycle, regulation of TP53 activity and viral gene expression. It is well known that the major functions of the CDK gene family are involved in cell cycle regulation, and mutations often occur in human tumor cells, of which the most common is CDK4 [9]. According to previous reports, CDK4/Cyclin D1 can phosphorylate the Ser249 of p53-RS, enhancing the binding of p53-RS and c-Myc, it can thereby activating the c-Myc transcription pathway, and promoting the growth of HCC cells [58]. Studies by Gan et al. showed that CDK1 protein interacts with iASPP protein to affect proliferation and apoptosis of colorectal cancer through p53 pathway [59]. The mechanisms of the CDK1 and p53 pathway in HCC needed further studies.
In our current study, ROC curves suggested that CDK1 and CDK4 were sensitive to diagnosis of HCC. At present, α-fetoprotein (AFP) is the serum tumor marker most commonly used for surveillance and early diagnosis of HCC [2]. However, in the retrospective case–control study, even with the most effective cutoff (10–20 ng/mL), the sensitivity was about 60% and the specificity was 80% [2]. Serum AFP > 400 ng/ml was considered to be of diagnostic efficiency, however, the possibility of false-negative results of AFP were high with early-stage HCC [60]. Other serum tumor markers of HCC included des-γ carboxyprothrombin, Golgi protein 73, glypican-3, Neprilysin and AFP-L3, which did not provide better accuracy [2, 60,61,62]. In recent years, some novel biomarkers of HCC had been discovered, including serum metabolite biomarker panel [63], gut microbiota [64] and serum miRNA (miR-193a-3p, miR-369-5p, miR-672.ect) [65]. In this study, the expression of CDK1 and CDK4 in HCC was statistically related to poor prognosis. Therefore, we believe that CDK1 and CDK4 might be biomarkers of HCC’s early diagnosis and prognosis prediction.
However, there were some limitations in this study. First, in the RT-qPCR experiment, not all results were consistent with the previous analysis, which resulted from low sample size and other potentially influencing factors. Second, lack of other factors may be involved in the progress of HCC, including smoking status, eating habits, region, drinking status and family history of liver cancer which could be used to further evaluate the relationship between CDK1-4,6 expression and HCC. Third, this study only explored the relationship between the mRNA expression level of the CDK family genes and HCC. Multi-omics analyses of other CDK genes such as protein and methylation need to be further explored.
In summary, our study showed that high mRNA expression of CDK4 was associated with a poor prognosis in HCC patients. CDK4 may showed as a potential prognostic biomarker of HCC.
Availability of data and materials
The datasets generated and analysed during the current study are available from University of California, Santa Cruz Xena, https://xenabrowser.net/datapages/; Gene Expression Omnibus, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE14520 and The National Omics Data Encyclopedia, https://www.biosino.org/node/project/detail/OEP000321.
Abbreviations
- HCC:
-
Hepatocellular carcinoma
- CDK:
-
Cyclin-dependent kinases
- RT-qPCR:
-
Revers transcription-quantitative PCR
- MST:
-
Median survival time
- HR:
-
Hazard ration
- TCGA:
-
The Cancer Genome Atlas
- GEO:
-
Gene expression omnibus
- GO:
-
Gene Ontology
- CHCC:
-
Chinese hepatocellular carcinoma
- T2DM:
-
Type 2 diabetes
- NAFLD:
-
Non-alcoholic fatty liver
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PPI:
-
Protein–protein interaction
- ROC:
-
Receiver operating characteristic
- OS:
-
Overall survival
- AUC:
-
Area under the curve
- GSEA:
-
Gene set enrichment analysis
- CI:
-
Confidence interval
- HBV:
-
Hepatitis B virus
- β:
-
Regression coefficient
- AFP:
-
α-Fetoprotein
References
Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021.
Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–14.
Younossi Z, Tacke F, Arrese M, et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology (Baltimore, MD). 2019;69(6):2672–82.
Facciorusso A. The influence of diabetes in the pathogenesis and the clinical course of hepatocellular carcinoma: recent findings and new perspectives. Curr Diabetes Rev. 2013;9(5):382–6.
Bellanti F, Villani R, Tamborra R, et al. Synergistic interaction of fatty acids and oxysterols impairs mitochondrial function and limits liver adaptation during nafld progression. Redox Biol. 2018;15:86–96.
Sherman M. Surveillance for hepatocellular carcinoma. Best Pract Res Clin Gastroenterol. 2014;28(5):783–93.
Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014;15(6):122.
Alexander SPH, Fabbro D, Kelly E, et al. The concise guide to pharmacology 2019/20: enzymes. Br J Pharmacol. 2019;176(Suppl 1):S297–396.
Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer. 2001;1(3):222–31.
Ravindran Menon D, Luo Y, Arcaroli JJ, et al. CDK1 interacts with Sox2 and promotes tumor initiation in human melanoma. Cancer Res. 2018;78(23):6561–74.
Piao J, Zhu L, Sun J, et al. High expression of CDK1 and BUB1 predicts poor prognosis of pancreatic ductal adenocarcinoma. Gene. 2019;701:15–22.
Yin X, Yu J, Zhou Y, et al. Identification of CDK2 as a novel target in treatment of prostate cancer. Future Oncol (London, England). 2018;14(8):709–18.
Peng C, Zeng W, Su J, et al. Cyclin-dependent kinase 2 (CDK2) is a key mediator for EGF-induced cell transformation mediated through the ELK4/c-Fos signaling pathway. Oncogene. 2016;35(9):1170–9.
Zhang Z, Huang A, Zhang A, Zhou C. HuR promotes breast cancer cell proliferation and survival via binding to CDK3 mRNA. Biomed Pharmacother. 2017;91:788–95.
Xiao T, Zhu JJ, Huang S, et al. Phosphorylation of NFAT3 by CDK3 induces cell transformation and promotes tumor growth in skin cancer. Oncogene. 2017;36(20):2835–45.
Wang L, Hu HY, Lin YL, et al. CDK3 expression and its clinical significance in human nasopharyngeal carcinoma. Mol Med Rep. 2014;9(6):2582–6.
Zhou Y, Shen JK, Yu Z, Hornicek FJ, Kan Q, Duan Z. Expression and therapeutic implications of cyclin-dependent kinase 4 (CDK4) in osteosarcoma. Biochim Biophys Acta Mol Basis Dis. 2018;1864(5 Pt A):1573–82.
Dai M, Zhang C, Ali A, et al. CDK4 regulates cancer stemness and is a novel therapeutic target for triple-negative breast cancer. Sci Rep. 2016;6:35383.
Kollmann K, Heller G, Schneckenleithner C, et al. A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell. 2016;30(2):359–60.
Scheicher R, Hoelbl-Kovacic A, Bellutti F, et al. CDK6 as a key regulator of hematopoietic and leukemic stem cell activation. Blood. 2015;125(1):90–101.
Goldman M, Craft B, Hastie M, et al. The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. bioRxiv 2019:326470.
Roessler S, Long EL, Budhu A, et al. Integrative genomic identification of genes on 8p associated with hepatocellular carcinoma progression and patient survival. Gastroenterology. 2012;142(4):957-966.e912.
Roessler S, Jia HL, Budhu A, et al. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 2010;70(24):10202–12.
Gao Q, Zhu H, Dong L, et al. Integrated proteogenomic characterization of HBV-related hepatocellular carcinoma. Cell. 2019;179(2):561-577.e522.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Warde-Farley D, Donaldson SL, Comes O, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38 (Web Server issue):W214–W220.
Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008;9(Suppl 1):S4.
Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607-d613.
Li T, Fu J, Zeng Z, et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48(W1):W509–14.
Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98-w102.
Menyhárt O, Nagy Á, Győrffy B. Determining consistent prognostic biomarkers of overall survival and vascular invasion in hepatocellular carcinoma. R Soc Open Sci. 2018;5(12):181006.
Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–50.
Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–73.
Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics (Oxford, England). 2011;27 (12):1739–40.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28(11):1947–51.
Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M, Tanabe M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49(D1):D545-d551.
Yang WX, Pan YY, You CG. CDK1, CCNB1, CDC20, BUB1, MAD2L1, MCM3, BUB1B, MCM2, and RFC4 may be potential therapeutic targets for hepatocellular carcinoma using integrated bioinformatic analysis. Biomed Res Int. 2019;2019:1245072.
Li R, Jiang X, Zhang Y, et al. Cyclin B2 overexpression in human hepatocellular carcinoma is associated with poor prognosis. Arch Med Res. 2019;50(1):10–7.
Li J, Gao JZ, Du JL, Huang ZX, Wei LX. Increased CDC20 expression is associated with development and progression of hepatocellular carcinoma. Int J Oncol. 2014;45(4):1547–55.
Wu SY, Lan SH, Liu HS. Degradative autophagy selectively regulates CCND1 (cyclin D1) and MIR224, two oncogenic factors involved in hepatocellular carcinoma tumorigenesis. Autophagy. 2019;15(4):729–30.
Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9(3):153–66.
Wang LL, Sun KX, Wu DD, et al. DLEU1 contributes to ovarian carcinoma tumourigenesis and development by interacting with miR-490-3p and altering CDK1 expression. J Cell Mol Med. 2017;21(11):3055–65.
Shi YX, Zhu T, Zou T, et al. Prognostic and predictive values of CDK1 and MAD2L1 in lung adenocarcinoma. Oncotarget. 2016;7(51):85235–43.
Zeestraten EC, Maak M, Shibayama M, et al. Specific activity of cyclin-dependent kinase I is a new potential predictor of tumour recurrence in stage II colon cancer. Br J Cancer. 2012;106(1):133–40.
Tian Z, Cao S, Li C, et al. LncRNA PVT1 regulates growth, migration, and invasion of bladder cancer by miR-31/CDK1. J Cell Physiol. 2019;234(4):4799–811.
Jiang Q, Mai C, Yang H, et al. Nuclear expression of CDK4 correlates with disease progression and poor prognosis in human nasopharyngeal carcinoma. Histopathology. 2014;64(5):722–30.
Banerjee J, Pradhan R, Gupta A, et al. CDK4 in lung, and head and neck cancers in old age: evaluation as a biomarker. Clin Transl Oncol. 2017;19(5):571–8.
Jin J, Valanejad L, Nguyen TP, et al. Activation of CDK4 triggers development of non-alcoholic fatty liver disease. Cell Rep. 2016;16(3):744–56.
Zhao J, Han SX, Ma JL, et al. The role of CDK1 in apoptin-induced apoptosis in hepatocellular carcinoma cells. Oncol Rep. 2013;30(1):253–9.
Chiu CY, Kuo KK, Kuo TL, Lee KT, Cheng KH. The activation of MEK/ERK signaling pathway by bone morphogenetic protein 4 to increase hepatocellular carcinoma cell proliferation and migration. MCR. 2012;10(3):415–27.
Facciorusso A, Licinio R, Carr BI, Di Leo A, Barone M. MEK 1/2 inhibitors in the treatment of hepatocellular carcinoma. Expert Rev Gastroenterol Hepatol. 2015;9(7):993–1003.
Jin J, Xu H, Li W, Xu X, Liu H, Wei F. LINC00346 acts as a competing endogenous RNA regulating development of hepatocellular carcinoma via modulating CDK1/CCNB1 axis. Front Bioeng Biotechnol. 2020;8:54.
Wu CX, Wang XQ, Chok SH, et al. Blocking CDK1/PDK1/β-catenin signaling by CDK1 inhibitor RO3306 increased the efficacy of sorafenib treatment by targeting cancer stem cells in a preclinical model of hepatocellular carcinoma. Theranostics. 2018;8(14):3737–50.
Bollard J, Miguela V, Ruiz de Galarreta M, et al. Palbociclib (PD-0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut. 2017;66 (7):1286–96.
Masaki T, Shiratori Y, Rengifo W, et al. Cyclins and cyclin-dependent kinases: comparative study of hepatocellular carcinoma versus cirrhosis. Hepatology (Baltimore, MD). 2003;37(3):534–43.
Lu JW, Lin YM, Chang JG, et al. Clinical implications of deregulated CDK4 and Cyclin D1 expression in patients with human hepatocellular carcinoma. Med Oncol (Northwood, London, England). 2013;30(1):379.
Wang H, Liao P, Zeng SX, Lu H. Co-targeting p53–R249S and CDK4 synergistically suppresses survival of hepatocellular carcinoma cells. Cancer Biol Ther. 2020;21(3):269–77.
Gan W, Zhao H, Li T, Liu K, Huang J. CDK1 interacts with iASPP to regulate colorectal cancer cell proliferation through p53 pathway. Oncotarget. 2017;8(42):71618–29.
Mathew S, Ali A, Abdel-Hafiz H, et al. Biomarkers for virus-induced hepatocellular carcinoma (HCC). Infect Genet Evol. 2014;26:327–39.
Luo P, Wu S, Yu Y, et al. Current status and perspective biomarkers in AFP negative HCC: towards screening for and diagnosing hepatocellular carcinoma at an earlier stage. POR. 2019.
Zhou F, Shang W, Yu X, Tian J. Glypican-3: a promising biomarker for hepatocellular carcinoma diagnosis and treatment. Med Res Rev. 2018;38(2):741–67.
Luo P, Yin P, Hua R, et al. A Large-scale, multicenter serum metabolite biomarker identification study for the early detection of hepatocellular carcinoma. Hepatology (Baltimore, MD). 2018;67(2):662–75.
Ren Z, Li A, Jiang J, et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut. 2019;68(6):1014–23.
Li L, Chen J, Chen X, et al. Serum miRNAs as predictive and preventive biomarker for pre-clinical hepatocellular carcinoma. Cancer Lett. 2016;373(2):234–40.
Acknowledgements
We would like to sincerely thank the contributors of The Cancer Genome Atlas, Gene Expression Omnibus, University of California, Santa Cruz Xena and The National Omics Data Encyclopedia for sharing the data with the public. In addition, we would like to acknowledge the helpful comments on this article received from our reviewers.
Funding
This work was supported in part by the National Natural Science Foundation of China (No.: 81560535, 81802874), Natural Science Foundation of Guangxi Province of China (Grant No. 2018GXNSFBA138013, 2018GXNSFAA050119, 2020GXNSFAA159127), Key laboratory of High-Incidence-Tumor Prevention & Treatment (Guangxi Medical University), Ministry of Education (GKE2018-01, GKE2019-11, GKE-ZZ202009), The Basic Ability Improvement Project for Middle-aged and Young Teachers in Colleges and Universities in Guangxi (2018KY0110), Guangxi Key Laboratory for the Prevention and Control of Viral Hepatitis (No.GXCDCKL201902), and 2018 Innovation Project of Guangxi Graduate Education (JGY2018037). As well as, the present study is also partly supported by Research Institute of Innovative Think-tank in Guangxi Medical University (The gene-environment interaction in hepatocarcinogenesis in Guangxi HCCs and its translational applications in the HCC prevention) and the central government in 2019 subsidized Guangxi's major and difficult disease clinical collaboration construction project of Chinese and Western medicine. We would also acknowledge the supported by the Key laboratory of High-Incidence-Tumor Prevention & Treatment (Guangxi Medical University), Ministry of Education.
Author information
Authors and Affiliations
Contributions
ZW and TP constructed the study design; ZW, CLL, HSS and XZ completed the RT-qPCR experiment. XL made acquisition of data; SM, YW made acquisition of data and made data analysis. ZW wrote the manuscript, and TP guided and supervised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was performed in accordance with the Declaration of Helsinki and have been approved by the Ethical Review Committee of the First Affiliated Hospital of Guangxi Medical University (2021-KY-E-032). Informed consent was provided by all HCC patients.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Figure S1: KEGG pathway map of CDK1-4, 6 in cell cycle pathway.
Additional file 2
. Figure S2: KEGG pathway map of CDK1-4, 6 in p53 signaling pathway.
Additional file 3
. Figure S3: Overall survival curves of CDK1 and CDK4 in CHCC cohort.
Additional file 4
. Figure S4: CDK1 gene expression in cancers from the TIMER2.0 website.
Additional file 5
. Figure S5: CDK4 gene expression in cancers from the TIMER2.0 website.
Additional file 6
. Figure S6: The association of CDK1 and CDK4 gene expression with TP53 gene mutation in cancers from TIMER 2.0 website.
Additional file 7
. Figure S7: Box plot, pathological stage plot and gene expression profile for CDK1 and CDK4 from GEPIA website.
Additional file 8
. Figure S8: GSEA results of CDK1 and CDK4 in HCC patients of TCGA cohort.
Additional file 9
. Figure S9: GSEA results of CDK1 and CDK4 in HCC patients of GSE14520 cohort.
Additional file 10
. Table S1: Clinical data of 159 HCC patients in the CHCC cohort.
Additional file 11
.Table S2. Joint effects analysis of CDK1 and CDK4 expression in CHCC cohort.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wei, Zl., Zhou, X., Lan, Cl. et al. Clinical implications and molecular mechanisms of Cyclin-dependent kinases 4 for patients with hepatocellular carcinoma. BMC Gastroenterol 22, 77 (2022). https://doi.org/10.1186/s12876-022-02152-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12876-022-02152-w