
Abstract
Background
To determine the safety and efficacy of different doses of tolvaptan for treating Chinese cirrhotic patients with or without hyponatraemia who still had ascites after routine therapy with diuretics.
Methods
In the present placebo-controlled, randomized, double-blinded, multicentre clinical trial, patients with cirrhotic ascites who failed to adequately respond to a combination of an aldosterone antagonist plus an orally administered loop diuretic were randomly placed at a 4:2:1 ratio into 3 groups [the 15 mg/day tolvaptan group (N = 301), 7.5 mg/day tolvaptan group (N = 153) and placebo group (N = 76)] for 7 days of treatment. The effects and safety were evaluated on days 4 and 7. A change in body weight from baseline on day 7 of treatment was the primary endpoint.
Results
The administration of 7.5 or 15 mg/day tolvaptan significantly decreased body weight from baseline on day 7 of treatment compared to that with placebo treatment (P = 0.026; P = 0.001). For the secondary endpoints, changes in abdominal circumference from baseline and improvements in ascites were markedly different in the treatment groups and the placebo group on day 7 (P7.5 = 0.05, P15.0 = 0.002 and P7.5 = 0.037, P15.0 = 0.003), but there was no difference between the 7.5 mg/day and 15 mg/day dosage groups. The 24-h cumulative urine volume was higher in the 7.5 mg/day and 15 mg/day tolvaptan groups than the placebo group (P = 0.002, P < 0.001) and was greater in the 15 mg/day tolvaptan group than the 7.5 mg/day tolvaptan group (P = 0.004). Sodium serum concentrations were higher in patients with hyponatraemia after tolvaptan treatment, with no significant difference between the two dosage groups. The incidence of serious adverse drug reactions was not different between the groups (P = 0.543).
Conclusions
Tolvaptan treatment at 7.5 mg per day might be a good therapeutic choice for Chinese cirrhotic patients with ascites who did not achieve satisfactory clinical responses to previous treatment regimens with combination therapy with an aldosterone antagonist and an orally administered loop diuretic.
Trial registration
NCT01349348. Retrospectively registered May 2011.
Similar content being viewed by others


Background
A very common complication of liver cirrhosis is ascites, which usually leads to a poor prognosis for patients [1]. The underlying pathophysiological mechanisms involved in cirrhotic ascites are complex and remain to be fully elucidated. It is known, however, that the activation of the renin–angiotensin–aldosterone system, sympathetic nervous system, and arginine-vasopressin interactions are intimately involved in the formation of ascites. Therefore, drugs that suppress these neurohormones should be used to treat cirrhotic ascites patients. Aldosterone antagonists, such as spironolactone, administered alone or together with a loop diuretic, such as furosemide, are recommended as the first-line treatment [2].
Unfortunately, a proportion of patients do not adequately respond to this combination therapy [3], and this conventional combination is associated with side effects such as renal failure, electrolyte disturbance, gynaecomastia and muscle cramps [4]. Tolvaptan, a vasopressin V2 receptor antagonist, has emerged as a new treatment choice for patients with ascites. Under the brand names of SAMSCA, JINARC, and JYNARQUE, tolvaptan has been licensed in many countries as therapy for euvolemic and hypervolemic hyponatraemia [5,6,7]. In September 2013, Japan approved 7.5 mg/day tolvaptan for the treatment of patients with ascites who failed to adequately respond to conventional diuretics [8]. The Japan Liver Cirrhosis guidelines recommend the treatment of ascites with a dosage range from 3.75 to 7.5 mg/day [9].
Tolvaptan was approved in China in September 2011 to treat hypervolemic and euvolemic hyponatraemia caused by liver cirrhosis, heart failure or syndrome of inappropriate antidiuretic hormone in a dose range of 15–60 mg/day [10]. From March 2009 to February 2010, China carried out a phase II ascites trial that showed that tolvaptan was effective and safe for treating Chinese ascites patients, with no differences between a 15 mg/day or 30 mg/day therapy regimen [11]. At the time of the initiation of this study (October 2010), tolvaptan had not been approved in Japan for the treatment of ascites. However, a dose finding trial in Japan revealed that compared to 15 or 30 mg/day, 7.5 mg/day tolvaptan produced maximal changes in body weight and abdominal circumference measurements with good patient tolerance [12]. The aims of our clinical trial were to confirm the safety and effectiveness of 7 days of tolvaptan therapy for cirrhotic patients with ascites in China who had insufficiently responded to standard diuretic therapy and to determine the optimal tolvaptan dose.
Methods
Trial setting
This phase 3 trial was performed at 39 centres in China between October 5, 2010, and January 20, 2012, and followed the principles outlined in the Declaration of Helsinki. The study was approved by the ethics committee of Shanghai Renji Hospital or the ethics committees of individual participating institutions. Informed signed consent forms were obtained from all subjects who agreed to be enrolled in the trial. The registration number of the clinical trial was NCT01349348.
Trial population
Inclusion criteria Hospitalized cirrhotic patients (18–75 years old) who were clinically or pathologically diagnosed and still presented with ascites after routine treatment, including a combination of oral loop diuretics and aldosterone antagonists with fixed doses for at least 4 days.
Exclusion criteria Patients suffering from hepatic encephalopathy (coma classification ≥ stage 2), cancerous ascites, or uncontrolled spontaneous bacterial peritonitis; patients likely to have gastrointestinal bleeding during the trial; or those receiving albumin or other blood preparations. In addition, patients with anuria (less than 100 mL of urine per day) and patients with dysuria caused by urinary tract stenosis, calculi and tumours were also excluded. Detailed information about the exclusion criteria is available in Additional file 1.
Trial design
The clinical trial consisted of a ≤ 10-day screening period, observations for 3 days before treatment, a 7-day treatment period, and a ≤ 14-day follow-up period. The doses and methods of administration of conventional diuretics remained unchanged for 4 days prior to the initiation of tolvaptan therapy until 1 day after the treatment period. Patients restricted their salt intake but not their water intake. Patients whose body weight before breakfast was stable (< ± 1.0 kg) were randomly allocated in a 1:2:4 ratio to receive placebo, 7.5 mg tolvaptan, or 15 mg tolvaptan once daily for 7 consecutive days. A preliminary randomized drug code was designated for each dose, and the drug trial manager allocated each patient a specific therapy code that corresponded to the trial drug code.
The sample size was based on the results of previous studies and met the Chinese health authority’s requirements. To date, this is the largest tolvaptan randomized controlled trial (RCT) of liver cirrhosis with a total of 535 randomization patients, with 76, 154 and 305 patients in the various groups.
Day 1 was defined as the period from the first administration until the second administration of tolvaptan. Days 2–7 were similarly defined. The primary evaluation time was day 7, but day 4 evaluation was added to look for potentially unresponsive cases or worsened cases in the placebo group. If this situation occurred, contingency methods such as albumin infusion or paracentesis were applied.
Efficacy assessment
Since there was a correlation between changes in body weight and ascites volume in patients with cirrhosis of the liver [13], a change in body weight is widely accepted as a useful marker for significant improvements in ascites and hepatic oedema. An alteration in patient body weight from baseline to the last dosage day (day 7) was the primary endpoint.
A change in abdominal circumference was the secondary endpoint. Abdominal circumference was measured with the patient in the supine position, legs straight and relaxed. If the patient had difficulties with a supine position, a prone position was also acceptable. The abdomen was totally relaxed. A tape with scale was placed under the patient’s back, perpendicular to the spine at the level of the umbilicus, touching but not compressing the skin and without twisting. When the patient had resumed regular normal breathing in a calm and relaxed manner, a measurement was taken at the end phase of exhalation. The date and measured abdominal circumference (accurate to 0.1 cm) were recorded in the original patient chart. If the abdominal circumference was reduced by ≥ 2 cm, the ascites condition was considered to have improved; other measurements were considered to be no change (increase or a reduction of < 2 cm) and deterioration (increase of > 2 cm). The percentage of improved cases among the total cases was defined as the improvement rate.
For patients who had lower limb oedema at baseline, lower limb oedema improvement rates were also evaluated. We assessed the degree of lower limb oedema as none, mild, moderate or severe. Changes were characterized as markedly improved (completely resolved or improved by ≥ 2 grades), improved (≥ 1 grade), unchanged or worsened (by ≥ 1 grade). The percentage of greatly improved or improved cases at baseline was defined as the improvement rate.
Serum electrolyte concentrations (Na+ and K+) were measured at baseline and 4–8 h after the first test drug dose as well as on days 1, 4 and 7. Cumulative 24-h urine volume and fluid intake were recorded daily.
Most parameters were measured in the morning after urination but before breakfast.
Safety assessments
Lab test results of renal and liver function were analysed at baseline and on days 4 and 7. Vital signs were recorded every day. A 12-lead electrocardiogram was recorded for each patient at baseline and on day 7. Adverse events were assessed during the whole study and followed up until they were resolved.
Statistical analysis
ANOVA (linear model) was used to look for any differences in efficacy between the 2 tolvaptan dosage groups and the placebo group, and the respective 95% confidence intervals (CIs) were calculated. Moreover, the correlation coefficient between changes in abdominal circumference and body weight on day 7 was calculated for both tolvaptan dosage groups. The regression equation and correlation coefficient were evaluated. Continuous variables were evaluated using ANOVA, while categorical variables were compared using either the Fischer exact or Kruskal–Wallis rank-sum tests. A difference was considered statistically significant at a P value < 0.05 (two-sided). Statistical analyses were conducted using SAS (ver. 9.2; Cary, NC, US).
For efficacy analysis, all patients who were randomized in the trial were included in the full analysis set (FAS). Any missing value data in the FAS were replaced using the last observation carried forward (LOCF) algorithm. After each patient had completed or discontinued their participation in the trial, the time point was the end of treatment (EOT) 7 (+ 3) days after the last treatment.
Results
Clinical characteristics and demographic parameters of enrolled patients
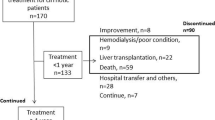
Of the 639 enrolled patients from 39 centres who insufficiently responded to primary at least 4 days of combination therapy with routine diuretic treatments, 535 were eligible to participate in the trial, with 76, 154 and 305 patients randomly allocated to the placebo, 7.5 mg or 15 mg tolvaptan groups, respectively. One patient in each of the tolvaptan dosage groups did not receive the study drug, and 3 in the 15 mg tolvaptan dosage group were lost to follow-up (Additional file 1: Fig. 1). The baseline clinical and demographic characteristics of the trial patients are presented in Table 1. There were significant differences in body weight and abdominal circumference (P = 0.008 and P < 0.001), and most patients had hepatitis B (64.5–66.4%), were in the Child–Pugh class B or C (97.7–98.7%) and nearly 100% had ascites without hepatic encephalopathy. Biochemical measurements of serum creatinine (Cr), blood urea nitrogen (BUN) and albumin, as well as all other characteristics, were well balanced among the 3 groups of patients (Table 1).
Changes in body weight
The changes in body weight from baseline to day 7 were − 1.2 ± 2.2 kg in the placebo group, − 2.0 ± 2.4 kg in the 7.5 mg tolvaptan group and − 2.2 ± 2.5 kg in the 15 mg tolvaptan group (Table 2). The difference in body weight changes on day 7 for the 7.5 mg tolvaptan and placebo groups was − 0.8 kg (95% CI − 1.4 to − 0.1; P = 0.026) and − 1.0 kg for the 15 mg tolvaptan and the placebo groups (95% CI − 1.6 to − 0.4; P = 0.001) (Table 2). It is worth noting that there were no significant differences in body weight changes between the 7.5 mg and 15 mg tolvaptan groups at this time point.
Daily changes in body weight showed significant differences for most time points between the placebo and the 7.5 mg and 15 mg tolvaptan groups (both P < 0.05) (Fig. 1, Additional file 1: Table 1).

Change in body weight from baseline at each time point in the placebo, 7.5 mg and 15 mg tolvaptan groups. Data are expressed as the mean ± standard deviation (SD). The comparison between the tolvaptan and placebo groups was performed using ANOVA. End of treatment (EOT)
Analysis of factors predicting responsiveness
Patients who had a body weight loss > 1.5 kg during treatment were defined as responders. Linear regression analysis showed that tolvaptan treatment (P = 0.036 for 7.5 mg, P = 0.004 for 15 mg), baseline body weight (P = 0.019), baseline albumin < 2.8 g/dL (vs > 3.5 g/dL) (P = 0.016) and baseline BUN levels (P = 0.020) were associated with responsiveness (Additional file 1: Table 2). Multivariable regression analysis also showed that tolvaptan treatment (P = 0.017 for 7.5 mg vs placebo, P = 0.003 for 15 mg vs placebo), baseline body weight (P = 0.047), albumin levels < 2.8 g/dL (vs > 3.5 g/dL) (P = 0.027) and baseline BUN levels (P = 0.027) were significantly correlated with responsiveness (Table 3).
Improvement of ascites and lower extremity oedema
The change in the abdominal circumference from baseline to day 7 was − 1.7 ± 3.5 cm, − 2.7 ± 3.4 cm and − 3.2 ± 3.8 cm for the placebo, tolvaptan 7.5 mg and tolvaptan 15 mg groups, respectively. Compared to the placebo group, the 7.5 mg and 15 mg tolvaptan groups had significantly decreased abdominal circumference from baseline to day 4 and day 7 (P = 0.012 and P = 0.05, and P < 0.001 and P = 0.002). However, no significant difference was observed between the two tolvaptan groups.
Compared to the placebo group, the improvement rate of ascites in the 15 mg tolvaptan group was significantly higher on both day 4 and day 7 (P = 0.023 and 0.003) but was significant on only day 7 for the 7.5 mg tolvaptan group. This may imply that initiating treatment with a lower dose requires a longer time for improvements to be observed. The improvement rate of lower limb oedema was higher in the tolvaptan groups but was not significantly different compared to that in the placebo group. This was mainly because most patients did not have significant lower limb oedema (Table 4).
Serum sodium and potassium concentrations
The serum Na+ concentration decreased from baseline in the placebo group, while in both tolvaptan dosage groups, the Na+ concentration significantly increased at each time point analysed (P < 0.001). The Na+ concentration increased more in the 15 mg tolvaptan group than in the 7.5 mg tolvaptan group, with significant differences on day 1 and day 4 (P < 0.001 and P = 0.002), but the difference gradually diminished by day 7 (P = 0.075) (Table 5).
This trend was also observed in hyponatraemia patients. Although 7.5 mg tolvaptan also increased sodium, overall sodium levels were still below 135 mmol/L at the end of treatment, while 15 mg tolvaptan normalized overall sodium levels starting on day 1. Comparing tolvaptan effects on hyponatraemia and normonatraemia patients, tolvaptan had stronger effects on hyponatraemia patients by increasing the absolute value of sodium more in these patients (Additional file 1: Table 3).
There were no significant changes in serum K+ concentrations from baseline for all groups throughout the treatment period (Table 5).
24-h urine volume and water intake
The 24-h urine volumes in the tolvaptan groups increased from baseline, most obviously at day 1. It is also worth noting that urine output during tolvaptan treatment was dose-dependent (P = 0.009 and P = 0.004 on day 4 and day 7, respectively). The tolvaptan patient groups also had a higher fluid intake but still a more negative water balance than the placebo group (P < 0.001 for both). There was no difference between the tolvaptan groups regarding water balance (Table 6, Fig. 2).

Change in the water balance from baseline at each time point in the placebo, 7.5 mg and 15 mg tolvaptan groups. Data are expressed as the mean ± SD
Safety assessments
Renal and liver function
Tolvaptan caused significant increases in serum Cr compared with placebo but these increases were far from worsening the renal failure criteria, which is defined as a ≥ 0.3 mg/dL increase in serum Cr. There was no significant difference between the 7.5 mg/day tolvaptan and placebo groups on day 7 (Table 7).
There was no significant increase in the liver function enzymes aspartate transaminase (AST) and alanine transaminase (ALT) or total bilirubin (TB) and BUN for either tolvaptan group compared with the placebo group.
The number of patients who met the definition of Hy’s Law (ALT or AST > 3 × upper limit of normal (ULN) and TB > 2 × ULN) was 1 (1.3%) in the placebo group, 2 (1.3%) in the tolvaptan 7.5 mg/day group and 1 (0.3%) in the tolvaptan 15 mg/day group, with no imbalance among the groups.
Adverse events
No significant difference was found in the overall rate of adverse events in the 3 trial groups (Table 8). Adverse events with an incidence of 5% or more were dry mouth and hypokalaemia in all 3 groups, abdominal bloating in only the placebo group (7.9%) and thirst in only the 15 mg tolvaptan group, but not in the placebo or 7.5 mg tolvaptan groups.
The rate of adverse drug reactions was higher in the tolvaptan groups; these reactions consisted mainly of thirst and dry mouth. Hypernatraemia occurred in 0%, 0.7% and 1.0% (P = 0.667) of patients in the placebo, 7.5 mg and 15 mg tolvaptan groups, respectively. Interestingly, upper gastrointestinal bleeding occurred to a lesser extent in the high tolvaptan dosage group (P = 0.029). However, since this study excluded any patients who had potential risks of bleeding during treatment and the total number of cases was low, any conclusions should be cautiously drawn.
No notable abnormalities were detected regarding vital signs and 12-lead electrocardiograms, and there were no serious adverse drug reactions in any of the patient groups.
In this study, a total of 15 patients died: 3/76 (3.9%), 4/153 (2.6%), and 8/304 (2.6%) died in the placebo, 7.5 mg tolvaptan and 15 mg tolvaptan groups, respectively. All deaths occurred after the treatment period and were determined to be unrelated to the trial drug (Additional file 1: Table 4).
Discussion
This clinical trial was a multicentre, randomized, placebo-controlled, double-blind study comprising the largest possible sample size for patients being treated with tolvaptan for hepatic cirrhosis. This confirmed the results of earlier studies showing that ascites in cirrhosis patients who did not respond to conventional diuretic therapy could be improved by tolvaptan therapy [11, 12, 14]. The improvement in ascites was mainly reflected in changes in body weight and abdominal circumference. In Japan, a phase III study used computer tomography (CT) to calculate ascites volume as the secondary endpoint, but this technology has not been generally adopted in China. Abdominal circumference has been widely used in China for decades, especially in grassroots hospitals, has a good correlation with body weight changes and ascites volume [13], and actually has more value for guidance in real-world clinical practice in China [15].
In the present study, doses of both 15 mg and 7.5 mg per day were effective, and significant differences were not observed among the dosage groups or in renal function indicators. The serum Cr concentration increased in both tolvaptan dose groups in the present trial, but it was more obvious in the 15 mg tolvaptan treatment group. Therefore, a relatively low dose (7.5 mg/day) of tolvaptan may be an optimal and safe treatment for cirrhotic patients with ascites. However, the Japanese ascites guidelines recommend starting with 3.75 mg/d and showed that tolvaptan at 3.75 mg/day exerts some effects, but 7.5 mg/day may be more beneficial [16]. Based on a continuous but consistent decrease in body weight and great improvement in the ascites volume at 7.5 mg/day, it is unlikely that 3.75 mg/day should be the clinical choice for Chinese ascites patients, but further clinical trials are needed to unequivocally confirm this view.
Cirrhosis, especially in advanced stages, is associated with a decrease in plasma albumin, and low albumin levels play a role in the formation of ascites. One of the functions of albumin is to enhance the diuretic effect of furosemide. However, studies have shown that combination therapy with loop diuretics and albumin increased urine output for the first 8 h, an effect that was no longer significant after 24 h [17]. This study is consistent with others since tolvaptan was shown to have good efficacy in patients with liver cirrhosis, with regard to both low and high serum albumin concentrations [18]. The present study showed that patients with lower albumin levels had even greater body weight reductions. This negative correlation trend was found in a pilot study. A possible explanation is that patients with lower albumin concentrations had more serous volume overload [19].
In our study, the baseline sodium concentration of 143 (27%) patients was below 135 mmol/L. For both the overall population and hyponatraemia subgroup, placebo further decreased the sodium level, while a low sodium level was an independent risk factor for poor prognosis in cirrhosis patients, and restoration was related to significantly improved 6-month survival rates [20]. Thus, tolvaptan is beneficial to both hyponatraemia and normal natraemia patients. It is also noteworthy that although both tolvaptan groups had increased sodium serum concentrations, only the 15 mg dosage group reached overall sodium normalization for hyponatraemia patients. It is possible that hyponatraemia patients need dose adjustments during treatment, as reported in the SALT study [21].
The occurrence of hepatic injury in 3 patients with autosomal dominant polycystic kidney disease who were treated with tolvaptan in a double-blind placebo-controlled trial [22, 23] led the FDA to worry about its safety in patients with liver disease [24]. However, an independent, hepatic adjudication committee reviewed the data from autosomal dominant polycystic kidney disease (ADPKD) and non-ADPKD tolvaptan trials and concluded that no imbalance in hepatic events was observed between the tolvaptan and placebo groups in lower-dose clinical trials of patients with cirrhosis, hyponatraemia or heart failure [25]. The present study also did not find any imbalance in liver function between the tolvaptan groups and the placebo group.
Responses to tolvaptan treatment not only reflect its short-term effectiveness but are also linked to significantly improved overall survival of patients with cirrhotic livers. This action was independent of the response definition or the presence of hepatocellular carcinoma [26, 27]. Different studies have identified different factors to predict tolvaptan responsiveness, such as urinary excretion of aquaporin 2, free water clearance, urinary sodium excretion, portal vein pressure, the BUN/Cr ratio, urine Na+/K+ ratio, etc. [28,29,30,31,32]; the most commonly identified factor was the baseline BUN level. The definitions of response were not the same in these studies, from increasing 500 mL urine to reducing 2 kg body weight. Some of the studies further explored the cut-off value of baseline BUN levels, and although the cut-off values were not the same, they were very close (from 22.4 to 29.0 mg/dL) [33,34,35,36,37,38,39]. This study set body weight loss equal to or greater than 1.5 kg in 7 days as the response criteria and verified that the baseline BUN level was a predictive factor.
The present trial had several limitations. First, the determination of clinically meaningful parameters (e.g., ascites volume and ascites-related symptoms) may be required to evaluate any future treatment of cirrhotic patients with tolvaptan. Second, several evaluations were conducted during only short-term treatment with tolvaptan.
Conclusions
Tolvaptan at doses of 7.5 mg/day or 15 mg/day significantly reduced the body weight and abdominal circumference of cirrhotic patients with ascites. Our findings indicate that 7.5 mg/day tolvaptan may be an optimal initial dose for Chinese cirrhotic patients with ascites who responded poorly to conventional diuretic therapy.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ADPKD:
-
Autosomal dominant polycystic kidney disease
- ALT:
-
Alanine transaminase
- AST:
-
Aspartate transaminase
- BUN:
-
Blood urea nitrogen
- Cr:
-
Creatinine
- CT:
-
Computer tomography
- RCT:
-
Randomized controlled trial
- TB:
-
Total bilirubin
- ULN:
-
Upper limit of normal
References
Gines P, Quintero E, Arroyo V, Teres J, Bruguera M, Rimola A, et al. Compensated cirrhosis: natural history and prognostic factors. Hepatology. 1987;7(1):122–8.
European Association for the Study of the Liver. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J Hepatol. 2018;69(2):406–60.
Hoorn EJ, Ellison DH. Diuretic Resistance. Am J Kidney Dis. 2017;69(1):136–42.
Santos J, Planas R, Pardo A, Durández R, Cabré E, Morillas RM, et al. Spironolactone alone or in combination with furosemide in the treatment of moderate ascites in nonazotemic cirrhosis. A randomized comparative study of efficacy and safety. J Hepatol. 2003;39(2):187–92.
Otsuka America Pharmaceutical, Inc. Tolvaptan (SamscaTM) Prescribing information 2012. https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/022275s007lbl.pdf.
Otsuka Pharmaceutical Europe Ltd. Tolvaptan (JinarcTM) Summary of product Characteristics 2019. https://www.ema.europa.eu/en/documents/product-information/jinarc-epar-product-information_en.pdf.
Otsuka America Pharmaceutical, Inc. Tolvaptan (JynarqueTM) Prescribing information 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/204441lbl.pdf.
Otsuka Pharmaceutical Co., Ltd. Tolvaptan (Samsca) Summary of investigation results. 2018. https://www.pmda.go.jp/files/000223416.pdf.
Fukui H, Saito H, Ueno Y, Uto H, Obara K, Sakaida I, et al. Evidence-based clinical practice guidelines for liver cirrhosis 2015. J Gastroenterol. 2016;51(7):629–50.
Liu Sde S-L, Yao T, Diehl AM, Zern MA. Vitamin E therapy of acute CC14-induced hepatic injury in mice is associated with inhibition of nuclear factor kappa B binding. Hepatology. 1995;22:7.
Wang YF, Tang JT, Han T, Ding HG, Ye WJ, Wang MR, et al. Tolvaptan in Chinese cirrhotic patients with ascites: A randomized, placebo-controlled phase 2 trial. J Dig Dis. 2018;19(3):144–54.
Okita K, Kawazoe S, Hasebe C, Kajimura K, Kaneko A, Okada M, et al. Dose-finding trial of tolvaptan in liver cirrhosis patients with hepatic edema: A randomized, double-blind, placebo-controlled trial. Hepatol Res. 2014;44(1):83–91.
Sakaida I, Okita K. Correlation between changes in bodyweight and changes in ascites volume in liver cirrhosis patients with hepatic edema in short-term diuretic therapy. Hepatol Res. 2014;44(7):735–9.
Sakaida I. Tolvaptan for the treatment of liver cirrhosis oedema. Expert Rev Gastroenterol Hepatol. 2014;8(5):461–70.
An J, Cai D, Chen G, Chen H, Chen X, Ding H, et al. Chinese guidelines on the management of ascites and its related complications in cirrhosis. Hepatol Int. 2019;13(1):1–21.
Sakaida I, Yanase M, Kobayashi Y, Yasutake T, Okada M, Okita K, et al. The pharmacokinetics and pharmacodynamics of tolvaptan in patients with liver cirrhosis with insufficient response to conventional diuretics: a multicentre, double-blind, parallel-group, phase III study. J Int Med Res. 2012;40(6):2381–93.
Kitsios GD, Mascari P, Ettunsi R, Gray AW. Co-administration of furosemide with albumin for overcoming diuretic resistance in patients with hypoalbuminemia: a meta-analysis. J Crit Care. 2014;29(2):253–9.
Sakaida I, Nakajima K, Okita K, Hori M, Izumi T, Sakurai M, et al. Can serum albumin level affect the pharmacological action of tolvaptan in patients with liver cirrhosis? A post hoc analysis of previous clinical trials in Japan. J Gastroenterol. 2015;50(10):1047–53.
Okabe T, Yakushiji T, Igawa W, Ono M, Kido T, Ebara S, et al. The efficacy of tolvaptan in congestive heart failure patients with and without hypoalbuminemia: a pilot study. Cardiovasc Ther. 2015;33(5):275–81.
Wang S, Zhang X, Han T, Xie W, Li Y, Ma H, et al. Tolvaptan treatment improves survival of cirrhotic patients with ascites and hyponatremia. BMC Gastroenterol. 2018;18(1):137.
Schrier RW, Gross P, Gheorghiade M, Berl T, Verbalis JG, Czerwiec FS, et al. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med. 2006;355(20):2099–112.
Muto S, Kawano H, Higashihara E, Narita I, Ubara Y, Matsuzaki T, et al. The effect of tolvaptan on autosomal dominant polycystic kidney disease patients: a subgroup analysis of the Japanese patient subset from TEMPO 3:4 trial. Clin Exp Nephrol. 2015;19(5):867–77.
Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367(25):2407–18.
FDA Drug Safety Communication: FDA limits duration and usage of Samsca (tolvaptan) due to possible liver injury leading to organ transplant or death 2013. https://www.fda.gov/Drugs/DrugSafety/ucm350062.htm.
Watkins PB, Lewis JH, Kaplowitz N, Alpers DH, Blais JD, Smotzer DM, et al. Clinical pattern of tolvaptan-associated liver injury in subjects with autosomal dominant polycystic kidney disease: analysis of clinical trials database. Drug Saf. 2015;38(11):1103–13.
Bellos I, Kontzoglou K, Psyrri A, Pergialiotis V. Tolvaptan response improves overall survival in patients with refractory ascites: a meta-analysis. Dig Dis. 2020;38(4):320–8.
Atsukawa M, Tsubota A, Takaguchi K, Toyoda H, Iwasa M, Ikegami T, et al. Analysis of factors associated with the prognosis of cirrhotic patients who were treated with tolvaptan for hepatic edema. J Gastroenterol Hepatol. 2020;35(7):1229–37.
Nakanishi H, Kurosaki M, Hosokawa T, Takahashi Y, Itakura J, Suzuki S, et al. Urinary excretion of the water channel aquaporin 2 correlated with the pharmacological effect of tolvaptan in cirrhotic patients with ascites. J Gastroenterol. 2016;51(6):620–7.
Miyaaki H, Nakamura Y, Ichikawa T, Taura N, Miuma S, Shibata H, et al. Predictive value of the efficacy of tolvaptan in liver cirrhosis patients using free water clearance. Biomed Rep. 2015;3(6):884–6.
Uojima H, Kinbara T, Hidaka H, Sung JH, Ichida M, Tokoro S, et al. Close correlation between urinary sodium excretion and response to tolvaptan in liver cirrhosis patients with ascites. Hepatol Res. 2017;47(3):E14-e21.
Nakagawa A, Atsukawa M, Tsubota A, Kondo C, Okubo T, Arai T, et al. Usefulness of portal vein pressure for predicting the effects of tolvaptan in cirrhotic patients. World J Gastroenterol. 2016;22(21):5104–13.
Kogiso T, Yamamoto K, Kobayashi M, Ikarashi Y, Kodama K, Taniai M, et al. Response to tolvaptan and its effect on prognosis in cirrhotic patients with ascites. Hepatol Res. 2017;47(9):835–44.
Iwamoto T, Maeda M, Hisanaga T, Saeki I, Fujisawa K, Matsumoto T, et al. Predictors of the effect of tolvaptan on the prognosis of cirrhosis. Intern Med. 2016;55(20):2911–6.
Yamada T, Ohki T, Hayata Y, Karasawa Y, Kawamura S, Ito D, et al. Potential effectiveness of tolvaptan to improve ascites unresponsive to standard diuretics and overall survival in patients with decompensated liver cirrhosis. Clin Drug Investig. 2016;36(10):829–35.
Chishina H, Hagiwara S, Nishida N, Ueshima K, Sakurai T, Ida H, et al. Clinical factors predicting the effect of tolvaptan for refractory ascites in patients with decompensated liver cirrhosis. Dig Dis. 2016;34(6):659–64.
Sakaida I, Terai S, Nakajima K, Shibasaki Y, Tachikawa S, Tsubouchi H. Predictive factors of the pharmacological action of tolvaptan in patients with liver cirrhosis: a post hoc analysis. J Gastroenterol. 2017;52(2):229–36.
Kawaratani H, Fukui H, Moriya K, Noguchi R, Namisaki T, Uejima M, et al. Predictive parameter of tolvaptan effectiveness in cirrhotic ascites. Hepatol Res. 2017;47(9):854–61.
Atsukawa M, Tsubota A, Kato K, Abe H, Shimada N, Asano T, et al. Analysis of factors predicting the response to tolvaptan in patients with liver cirrhosis and hepatic edema. J Gastroenterol Hepatol. 2018;33(6):1256–63.
Sakaida I, Terai S, Kurosaki M, Okada M, Hirano T, Fukuta Y. Real-world effectiveness and safety of tolvaptan in liver cirrhosis patients with hepatic edema: results from a post-marketing surveillance study (START study). J Gastroenterol. 2020;55(8):800–10.
Acknowledgements
The authors would like to thank other PIs and all of the subjects who participated in the present trial. The authors would also like to acknowledge the following investigators who participated in the present trial and were responsible for patient selection, recruitment and clinical evaluation during the study period: Maorong Wang (Nanjing Bayi Hospital), Weiping Ye (XiXi Hospital of Hangzhou), Haihang Zhu (Northern Jiangsu People's Hospital), Yonggang Li (302 Military Hospital of China), Zibin Tian (The Affiliated Hospital of Qingdao University), Bangmao Wang (Tianjin Medical University General Hospital), Yongtao Sun (TangDu Hospital), Zhongtao Gai (Jinan Infectious Disease Hospital), Heping Hu (Shanghai Eastern Hepatobiliary Hospital), Shumei Lin (The First Affiliated Hospital of Xian Jiaotong University), Ping Feng (West China Hospital of Medicine, SCU), Xiaozhong Guo (The General Hospital of Shenyang Military), Jia Wei (First Affiliated Hospital of Kunming Medical University), Wu Ni (Shanghai Changzheng Hospital), Jianting Cai (The Second Affiliated Hospital of Zhejiang University School of Medicine), Jizhong Guo (Wuxi People's Hospital), Yongwen He (Wuhan Union Hospital Affiliated to Tongji Medical College of Huazhong University), Haohu Chen (First Affiliated Hospital of Sun Yat-sen University), Yanyan Ji (JiangAn District Center Hospital of Shanghai), Yongping Chen (The First Affiliated Hospital of Wenzhou Medical University), Lungen Lu (Shanghai General Hospital), Yanlin Yu (The First Affiliated Hospital of Wannan Medical College), and Cheng Xu (Shenzhen Third People’s Hospital).
Funding
The Zhejiang Otsuka Pharmaceuticals Co., Ltd. and the Otsuka Beijing Research Institute funded this research. The funders had no role in the study design, data collection or analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
JTT and YFW performed the trial, analyzed the data and wrote the first draft of the manuscript. YMM conducted the present trial and contributed to the concept. YMM wrote the first draft of the manuscript, and MDZ, TH, QM, JC, HGD, JS, QZ, JN, FJ, CWC, JDJ, XJJ, NHL, YQG, ZHW, ZW, YXC and YMM were involved in critically revising the manuscript and all gave final approval for the version of the manuscript submitted.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This trial was reviewed and approved by the Ethics Committee of Shanghai Renji Hospital or the ethics committees of individual participating institutions. All eligible patients provided written informed consent prior to enrollment in the trial.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1
. Exclusion criteria of the study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tang, J., Wang, Y., Han, T. et al. Tolvaptan therapy of Chinese cirrhotic patients with ascites after insufficient diuretic routine medication responses: a phase III clinical trial. BMC Gastroenterol 20, 391 (2020). https://doi.org/10.1186/s12876-020-01536-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12876-020-01536-0