
Abstract
Background
Studies of microbiota composition of infants with small intestinal ostomy due to various etiologies are limited. Here, we characterized the intestinal microbiota of neonates with ileostomy resulting from distinct primary diseases.
Methods
Fifteen patients with necrotizing enterocolitis, eight patients with meconium peritonitis, and seven patients with Hirschsprung’s disease were included in the study. The small intestinal microbiota composition in infants with ileostomy caused by these diseases was studied.
Results
Microbial diversity in neonatal ileostomy fluid was generally low, and was dominated by members of the Proteobacteria and Firmicutes phyla. At the genus level, the most abundant were Klebsiella, Escherichia-Shigella, Streptococcus, Clostridium sensu stricto 1, Enterococcus, and Lactobacillus. Streptococcus and Veillonella are related to carbohydrate metabolism and immunity, and breastfeeding can increase the proportion of these potentially beneficial bacteria. The proportion of Bifidobacterium in the breastfeeding group was higher than that in the non-breastfeeding group, and incidence of colitis and sepsis was reduced in the breastfeeding group. The proportion of Bifidobacterium increased and incidence of colitis and sepsis decreased in the breastfeeding group compared with the non- breastfeeding group, but there was no significant difference. The increase in body weight in the breastfeeding group was observed to be higher than in the non-breastfeeding group.
Conclusions
Excessive Klebsiella and Escherichia-Shigella and low abundance of Streptococcus, Veillonella and Faecalibacterium suggests that the small intestinal microbiota is in an unhealthy state after ileostomy. However, Streptococcus, Faecalibacterium, and Veillonella species were frequently present, suggesting that expansion of these bacteria might assist the development of the immune system after surgery.
Similar content being viewed by others
Background
Enterostomy formation is one of the important surgical treatments for infants with acute abdominal disease. It is commonly performed in response to a variety of intestinal conditions, including necrotizing enterocolitis (NEC), meconium ileus, focal intestinal perforation, intestinal atresia and volvulus. Extensive resections of the small intestine may lead to development of functional short bowel syndrome (SBS). SBS is a malabsorption disorder characterized by fluid and electrolyte imbalances that are consequent to extensive bowel resection. These infants are placed on long-term parenteral nutrition (PN) and enteral nutrition (EN) to compensate for their nutritional deficiencies, which may have a considerable impact on microbiota structure in the small intestine.
The small intestine is the main site for digestion and absorption of nutrients, including almost all proteins, lipids, monosaccharides, and starch [1]. Although the abundance of microbial species in the small intestinal is relatively low, because of rapid peristalsis and secretion of bactericidal substances [2], the small intestinal microbiota has functions that include regulation of immunity, metabolism, and the endocrine system. The impact of these effects on host health can be substantial [3]. Hayashi reported that the majority of microbes in the jejunum and ileum were aerobic bacteria and facultative anaerobes, which included Streptococcus, Lactobacillus, Enterococcus, and γ-Pseudomonas [4]. Wang et al. reported that Streptococcus accounted for more than 60% of the jejunal microbiota, and Clostridium clusters IVa and IV were the dominant species in the distal ileum [5]. It has been shown that Streptococcus, Veillonella, and Lactobacillus are the main microorganisms in the small intestine and that they are involved in intestinal immune regulation [6]. Therefore, alterations of small intestinal microbiota composition might affect disease symptoms. Due to the location of the small intestine, its microbiota is difficult to sample. As a result, only a few studies have focused on small intestinal microbiota.
Multiple primary diseases can lead to ileostomy, including Hirschsprung’s disease (HD), meconium peritonitis (MP), and NEC. During infancy, the composition of the intestinal microbiota is relatively simple and can be influenced by a variety of factors. In this study, we characterized the intestinal microbiota of infants with an ileostomy resulting from defined primary diseases. The association between clinical symptoms, intestinal microbiota composition and therapeutic effects were also comprehensively analyzed. This study provides valuable data for informing postoperative care and future clinical practice.
Methods
Study subjects and sample collection
We recruited 30 infants with small intestinal ostomy caused by various primary diseases. All patients were recruited from the Children’s Hospital of Fudan University, as shown in Tables 1 and S1. All patients were categorized into the following three groups according to their primary diseases: HD (8 cases), MP (7 cases), and NEC (15 cases). After the infants had reached full enteral feeding (daily enteral feeding > 120 ml/kg), samples of ileostomy fluid were collected and stored at − 80 °C until microbiota analysis. The infants received oral formula or breast milk, and were not fed any solid food. This study was approved by the Human Investigation Committee of the Children’s Hospital of Fudan University. Written informed consent was obtained from all parents.
16S rRNA gene sequencing
Bacterial genomic DNA was extracted from the ileostomy fluid samples using the E.Z.N.A. Stool DNA Isolation Kit (Omega Bio-Tek, Inc., GA, USA). The 16S rDNA V3-V4 region was amplified by PCR using barcoded Illumina adapter-containing primers 341F (5′-CCTACGGGNGGCWGCAG-3′) and 805R (5′-GACTACHVGGGTATCTAATCC-3′) [7]. The final 16S rRNA gene amplicon library was sequenced on the MiSeq platform (Illumina) using a 2 × 300 bp paired-end protocol. Illumina MiSeq sequencing was performed by Shanghai Mobio Biomedical Technology Co., Ltd. (China). Raw sequencing data have been submitted to the NCBI Sequence Read Archive under accession number PRJNA553095.
Clean data was extracted from raw data using USEARCH 8.0 with the following criteria: (i) sequences of each sample were extracted using each index with zero mismatch, (ii) sequences with overlaps of less than 50 bp and with error rate of the overlap greater than 0.1 were discarded, (iii) sequences less than 400 bp after merging were also discarded. Quality-filtered sequences were step-wise clustered into operational taxonomic units (OTUs) at a similarity of 97% using UPARSE (version 7.1 http://drive5.com/uparse/) [8]. The phylogenetic affiliation of each 16S rRNA gene sequence was analyzed by RDP Classifier (http://rdp.cme.msu.edu/) against the Silva (SSU123)16S rRNA database using a confidence threshold of 70% [9].
Statistical analyses
Estimates of alpha diversity were calculated with standard methods using QIIME 1.9.0 [10]. The structure and characteristics of the microbial communities after each treatment step were analyzed using R. Hierarchical clusters at the genus level were generated with the Bray-Curtis average distance method using R. Student’s t-test was performed to analyze normally distributed data. Chi-square test or Fisher’s exact test was used for analysis of categorical variables. The built in function p.adjust in R was used (method = “BH”) to control the false discovery rate. Genera with relative abundances higher than 1% in at least one of the samples were included. A canonical correspondence analysis (CCA), which describes the relationships between the corresponding environmental parameters and relatively abundant genera in microbial communities, was performed using Canoco 4.5.
Results
Low microbial diversity and chaotic microbial succession
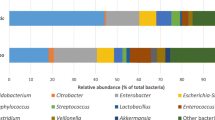
To understand the relationship between the distinct primary diseases and shifts in intestinal microbiota composition, the patients were classified into three groups according to their primary disease; namely, the HD, MP, and NEC groups. There were 8, 7, and 15 patients in the HD, MP, and NEC groups, respectively (Table 1). Shannon diversity index was relatively low in infants with ileostomy as shown in Tables 1, S1 and Figure S1. Clinical profiles of patients are summarized in Table S1. Proteobacteria and Firmicutes were the most prevalent phyla, followed by Actinobacteria and Bacteroidetes (Figure S1). These four phyla accounted for more than 99.9% of the microbial communities in all samples. In these patient samples, Enterobacteriaceae (56.4%) was the dominant taxonomic family, followed by Streptococcaceae (11.8%), Clostridiaceae 1 (9.6%), Enterococcaceae (8.7%), Lactobacillaceae (5.5%), and Bifidobacteriaceae (1.6%). These six most common taxonomic families accounted for more than 93.6% of the microbial communities in all samples (Fig. 1a).

Microbial communities in infants with ileostomy. Panel a shows the proportion of family within the microbiota in patients with HD (H01-H08), MP (M01-M07), and NEC (N01-N15). Panel b shows the proportion of genus within the microbiota in patients with HD (H01-H08), MP (M01-M07), and NEC (N01-N15). HD: Hirschsprung’s disease; MP: meconium peritonitis; NEC: necrotizing enterocolitis
Structural differences in intestinal microbiota from individuals
To further explore microbial composition of the three groups, core genera were examined in detail (Fig. 1b and S2). One hundred and nineteen OTUs were obtained from all the samples. The most abundant bacteria were Klebsiella (26.0% on average), followed by Escherichia–Shigella (24.6%), Streptococcus (11.8%), Clostridium sensu stricto 1 (9.6%), Enterococcus (8.7%), and Lactobacillus (5.5%). Cluster analysis demonstrated that the relative abundance of Klebsiella was higher in some patients (Fig. 2), and Klebsiella and Enterobacter always appeared simultaneously (indistinguishable based on the 16S V4 region). Cluster analysis of intestinal microbiota composition in patients was also performed at the level of genera (Fig. 2). Microbial community structures were not well grouped according to the different etiologies. Although different patients have diverse small intestinal microbiota profiles, Escherichia-Shigella and Klebsiella are commonly detected and are relatively high (Figs. 2 and S2). In addition, Clostridium sensu stricto 1 was the main species in samples M06 and N14, and Raoultella was the main species in sample N11 (Fig. 2). The microbiota composition analysis of ileostomy fluid thus reveals that individual subjects have distinct microbiota structures. However, despite these distinct characteristics, the microbiotas contained common species. For example, Streptococcus was detected in almost every sample, albeit with variable relative abundance (Figs. 2 and S2).

Heatmap of the relative abundance of the signature genera in infants with ileostomy. Each parallel lane corresponds to one sample. Relative abundance percentage of each genus in the corresponding whole community is indicated by the color scale. Blue: 0, white: 0.01 (1%), and yellow: 0.5 (50%)
Bifidobacterium and breastfeeding
Similarly to Faecalibacterium, Bifidobacterium was also detectable in some samples at low abundance. Eight of the 30 patients were breast-fed (Table S1). Streptococcus is associated with carbohydrate metabolism, and breastfeeding can increase the proportion of this beneficial bacterium in the small intestine. Breast milk usually contains Bifidobacterium, Lactobacillus, and Streptococcus, and another probiotic species Staphylococcus is often found on areola skin; therefore, these bacteria can be transferred directly from mother to baby during breastfeeding [11, 12]. We found that the incidence of colitis and sepsis was lower in the breast-fed group than in the non-breast-fed group, while the weight gain was higher in the breast-fed group than in the non-breast-fed group, but there was no significant difference (Table 2). CCA analysis revealed that Bifidobacteria, Streptococci, and Veillonella were closely associated with breastfeeding (Fig. 3).

Canonical correspondence analysis (Canoco 4.5 CCA) of the effects of different factors. a Ordination diagram of canonical correspondence analysis exhibits bacterial species and environmental variables and primary diseases (arrows). b Influence of environmental variables and primary diseases on samples, which plotted on the same axes as the first panel. Axis 1 explains 42.9% of the variation, whereas axis 2 explains an additional 19.3%. HD: Hirschsprung’s disease; MP: meconium peritonitis; NEC: necrotizing enterocolitis; DAW: duration of antibiotic withdrawal, BF: breastfeeding; PN: parenteral nutrition; RIL: residual intestinal length
Other influences on intestinal microbiota composition
To investigate potential effects of other external factors on intestinal microbiota structure, we performed CCA analysis. As shown in Fig. 3, more than 60% of the variability in microbiota structure was explained by the CCA axes. According to our data, different etiologies have a significant influence on the microbial community structure. Taking account of external factors, we grouped infants with ileostomy into three different primary diseases (Fig. 3). In addition to the etiologies, the intestinal microbiotas were influenced by several factors, which included: duration of antibiotic withdrawal, breastfeeding, PN duration, and residual intestine length.
Discussion
The intestinal microbiota represents an important factor affecting human health, exhibiting evolutionarily conserved roles in host metabolism, immunity, development, and behavior [13, 14]. It has been reported that the fecal microbiota of babies born by caesarean section is rich in Enterobacter, Streptococcus australis, and Veillonella, which are mainly derived from the skin, oral cavity, and the surrounding environment during birth. The intestinal microbiota of neonates born by vaginal delivery is rich in Escherichia-Shigella, Bacteroides, and Bifidobacterium, with Escherichia-Shigella being the most abundant genus [15]. It has been reported that enrichment of genes involved in vitamin K2 synthesis in newborns is associated with a high abundance of Escherichia-Shigella, an organism with the ability to synthesize vitamin K2 [15, 16].
Streptococcus and Veillonella often coexist in the intestinal ecosystem [17, 18], and might cooperate in the metabolic process, so their combined immunoregulatory properties have also been investigated [6]. Since the source of these bacteria in newborns is maternal, it can be implied that they play a role in clinical recovery and immune development of ileostomy patients after surgery. Data suggest that microorganisms in the small intestine are mainly involved in simple carbohydrate metabolism, a task predominantly performed by specific species including Streptococcus and Veillonella. Our results are consistent with findings of previous studies; that is, enterobacteria always include Streptococcus and Veillonella spp. in the ecosystem, and these are the most active members of the ileal microbiota [3]. Functions of Streptococcus and Veillonella are often complementary, however, in our study we found a lower abundance of Veillonella in patients. Streptococci can metabolize a variety of carbohydrates, while Veillonella can utilize lactic acid as a carbon and energy source, and are proposed to metabolize lactic acid produced by Streptococcus [19, 20]. Metatranscriptomic analysis of ileostomy fluid also revealed that the presence of Streptococcus in the small intestine could be related to the transportation and metabolism of carbohydrate substrates [17]. Escherichia or other microbes can substitute for this role when the abundance of Streptococcus is insufficient [17]. Further studies identifying underlying mechanisms are needed to explain the contribution of Streptococcus and Veillonella to immunity and homeostasis in pediatric patients after ileostomy, and this could help guide future clinical practice. Faecalibacterium, a prominent member of the Firmicutes phylum, has been reported to possess immunoregulatory and anti-inflammatory properties [21]. For example, Faecalibacterium prausnitzii produces anti-inflammatory proteins, and Crohn’s disease (CD), a disorder associated with disrupted microbial ecology, is characterized by decreased abundance of F. prausnitzii. Moreover, the decrease in F. prausnitzii is associated with an increased risk of symptom recurrence after ileal CD surgery [21, 22]. A limitation of our study is that the sample size is too small to link specific bacterial populations to the 3 primary diseases. Grouping microbial community structure according to different etiologies was not effective. In contrast, different patients have diverse small intestinal microbiota profiles, and dominated by Escherichia-Shigella and Klebsiella. In addition, many of the clinical parameters we described could have influenced the dataset, including duration of antibiotic removal, breastfeeding, PN duration, and residual intestine length.
Nutritional factors have a significant influence on the composition of the intestinal microbiota. Breast milk is an optimal source of nutrition for infants, providing proteins, carbohydrates, lipids, fats, and some micronutrients essential for infant growth. Breast milk also contains several biologically active components that include immunoglobulins and oligosaccharides, and resulting microbiota plays a critical role in infant intestinal homeostasis and immune development [23]. Our study found a trend of increased growth rate of body mass and decreased prevalence of colitis and sepsis in breastfeeding patients compared to non-breastfeeding patients. Thus, breastfeeding is likely to play a role in maintaining the intestinal function and improving postoperative recovery of such patients.
Excessive Klebsiella and Escherichia-Shigella, and low abundance of Streptococcus, Veillonella, and Faecalibacterium, indicated that the small intestinal microbiota of our donors was in an unhealthy state. It has been reported that the relative abundance of Enterobacter or Klebsiella is significantly higher in breast milk of mothers of infants infected with rotavirus as well as in the intestine of the infected neonates, whereas the abundance of Streptococcus and Staphylococcus was significantly lower [24]. Our data suggest a correlation between the presence of Klebsiella/Enterobacter and neonatal gastrointestinal disease, as well as a potential protective effect of Staphylococcus or Streptococcus. A frequent occurrence of Klebsiella species has also been reported in pathogenic genera identified in SBS II patients [25]. Therefore, it would be beneficial to study further the effects of Klebsiella/Enterobacter in ileostomy and its prognosis, in order to provide novel hypotheses that guide future clinical practice.
Conclusions
This is the first report to define characteristics of the intestinal microbiota of neonates with ileostomy resulting from distinct etiologies. The results reveal that patients with small intestinal ostomy subsequent to different etiologies have dissimilar microbial community characteristics. In addition, we observed relatively individualized small intestinal microbiota profiles. The diversity of species in the small intestinal fluid of neonates with ostomy was low, reflected by low Shannon indexes. Bacteria were mainly distributed within the phyla Proteobacteria and Firmicutes, and less frequent within Actinobacteria and Bacteroidetes. More research is warranted to understand the relationship between intestinal dysbiosis and primary disease, and the associated influences of microbiota on the prognosis of infants with ileostomy.
Availability of data and materials
The datasets used and analysed in the current study are available from the corresponding author upon reasonable request.
Abbreviations
- NEC:
-
Necrotizing enterocolitis
- SBS:
-
Short bowel syndrome
- PN:
-
Parenteral nutrition
- EN:
-
Enteral nutrition
- HD:
-
Hirschsprung’s disease
- MP:
-
Meconium peritonitis
- CCA:
-
Canonical correspondence analysis
- CD:
-
Crohn’s disease
References
Krajmalnik-Brown R, Ilhan ZE, Kang DW, DiBaise JK. Effects of gut microbes on nutrient absorption and energy regulation. Nutr Clin Pract. 2012;27:201–14.
Leser TD, Mølbak L. Better living through microbial action: the benefits of the mammalian gastrointestinal microbiota on the host. Environ Microbiol. 2009;11:2194–206.
El Aidy S, van den Bogert B, Kleerebezem M. The small intestine microbiota, nutritional modulation and relevance for health. Curr Opin Biotechnol. 2015;32:14–20.
Hayashi H, Takahashi R, Nishi T, Sakamoto M, Benno Y. Molecular analysis of jejunal, ileal, caecal and recto-sigmoidal human colonic microbiota using 16S rRNA gene libraries and terminal restriction fragment length polymorphism. J Med Microbiol. 2005;54:1093–101.
Wang M, Ahrné S, Jeppsson B, Molin G. Comparison of bacterial diversity along the human intestinal tract by direct cloning and sequencing of 16S rRNA genes. FEMS Microbiol Ecol. 2005;54:219–31.
van den Bogert B, Meijerink M, Zoetendal EG, Wells JM, Kleerebezem M. Immunomodulatory properties of Streptococcus and Veillonella isolates from the human small intestine microbiota. PLoS One. 2014;9:e114277.
Hugerth LW, Wefer HA, Lundin S, Jakobsson HE, Lindberg M, Rodin S, et al. DegePrime, a program for degenerate primer design for broad-taxonomicrange PCR in microbial ecology studies. Appl Environ Microbiol. 2014;80:5116–23.
Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–8.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6.
Hunt KM, Foster JA, Forney LJ, Schütte UM, Beck DL, Abdo Z, et al. Characterization of the diversity and temporal stability of bacterial communities in human milk. PLoS One. 2011;6:e21313.
Pannaraj PS, Li F, Cerini C, Bender JM, Yang S, Rollie A, et al. Association between breast milk bacterial communities and establishment and development of the infant gut microbiome. JAMA Pediatr. 2017;171:647–54.
Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148:1258–70.
Erkosar B, Storelli G, Defaye A, Leulier F. Host-intestinal microbiota mutualism: “learning on the fly”. Cell Host Microbe. 2013;13:8–14.
Bäckhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015;17:690–703.
Wang M, Song F, Wu R, Allen KN, Mariano PS, Dunaway-Mariano D. Co-evolution of HAD phosphatase and hotdog-fold thioesterase domain function in the menaquinone-pathway fusion proteins BF1314 and PG1653. FEBS Lett. 2013;587:2851–9.
Zoetendal EG, Raes J, van den Bogert B, Arumugam M, Booijink CC, Troost FJ, et al. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J. 2012;6:1415–26.
Egland PG, Palmer RJ Jr, Kolenbrander PE. Interspecies communication in Streptococcus gordonii-Veillonella atypica biofilms: signaling in flow conditions requires juxtaposition. Proc Natl Acad Sci U S A. 2004;101:16917–22.
van den Bogert B, Erkus O, Boekhorst J, de Goffau M, Smid EJ, Zoetendal EG, et al. Diversity of human small intestinal Streptococcus and Veillonella populations. FEMS Microbiol Ecol. 2013;85:376–88.
Van den Bogert B, Boekhorst J, Smid EJ, Zoetendal EG, Kleerebezem M. Draft genome sequence of Veillonella parvula HSIVP1, isolated from the human small intestine. Genome Announc. 2013;1:e00977–13.
Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008;105:16731–6.
Quévrain E, Maubert MA, Michon C, Chain F, Marquant R, Tailhades J, et al. Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn's disease. Gut. 2016;65:415–25.
Bode L. Human milk oligosaccharides: every baby needs a sugar mama. Glycobiology. 2012;22:1147–62.
Ramani S, Stewart CJ, Laucirica DR, Ajami NJ, Robertson B, Autran CA, et al. Human milk oligosaccharides, milk microbiome and infant gut microbiome modulate neonatal rotavirus infection. Nat Commun. 2018;9:5010.
Huang Y, Guo F, Li Y, Wang J, Li J. Fecal microbiota signatures of adult patients with different types of short bowel syndrome. J Gastroenterol Hepatol. 2017;32:1949–57.
Acknowledgements
We thank to Ms. Lili Ma for helping of stool collecting.
Funding
This work was supported by a grant from Shanghai Science and Technology commission (18ZR1405200) and a grant from National Natural Science Foundation of China (81873849). The funders had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
TQ, RZ and CS contributed to conceptualization. TQ and RZ contributed to methodology. TQ, HZ and LZ contributed to investigation. TQ contributed to writing the original draft. HZ, LZ and CC contributed to writing the review and to editing. RZ and CS contributed to funding acquisition and supervision. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was reviewed and approved by the Human Investigation Committee of Children’s Hospital of Fudan University, China. All the written consents were obtained from the parents.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qian, T., Zhu, H., Zhu, L. et al. Small intestinal microbiota composition and the prognosis of infants with ileostomy resulting from distinct primary diseases. BMC Gastroenterol 20, 224 (2020). https://doi.org/10.1186/s12876-020-01366-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12876-020-01366-0