
Abstract
Background
Aceruloplasminaemia is a very rare autosomal recessive disorder caused by a mutation in the ceruloplasmin gene, which is clinically manifested by damage to the nervous system and retinal degeneration. This classical clinical picture can be preceded by diabetes mellitus and microcytic anaemia, which are considered to be early manifestations of aceruloplasminaemia.
Case presentation
In our report, we describe the case of a patient with aceruloplasminaemia detected in an early stage (without clinical symptoms of damage to the nervous system) during the search for the cause of hepatopathy with very low values of serum ceruloplasmin.
Molecular genetic examination of the CP gene for ceruloplasmin identified a new variant c.1664G > A (p.Gly555Glu) in the homozygous state, which has not been published in the literature or population frequency databases to date. Throughout the 21-month duration of chelatase treatment, the patient, who is 43 years old, continues to be without neurological and psychiatric symptomatology. We observed a decrease in the serum concentration of ferritin without a reduction in iron deposits in the brain on magnetic resonance imaging.
Conclusion
Currently, there is no unequivocal recommendation of an effective treatment for aceruloplasminaemia. Early diagnosis is important in the neurologically asymptomatic stage.
Similar content being viewed by others


Background
Aceruloplasminaemia is a rare autosomal recessive disorder caused by a mutation in the ceruloplasmin gene linked to the accumulation of iron in an organism, mainly in the liver, brain, retina and pancreas. It is clinically manifested by retinal degeneration, diabetes mellitus, microcytic anaemia and neurological symptomatology [1]. The diagnosis of aceruloplasminaemia is often not confirmed before the origin of irreversible neurological symptomatology.
In our report, we describe the case of a patient with aceruloplasminaemia, which was identified in an early, neurologically asymptomatic stage while searching for the cause of chronic hepatopathy with low values of serum ceruloplasmin with originally supposed Wilson’s disease.
Case presentation
A 41-year-old patient with several years of anamnesis of microcytic hypochromic anaemia after recurring miscarriages was initially examined in a district hepatology outpatient clinic because of slightly increased values of ALT activity. The physical examination was, apart from obesity (body mass index 30), in a normal range. Ultrasonographic examination revealed hyperechogenicity of the liver; therefore, we assumed the presence of liver steatosis. The basic laboratory examination confirmed microcytic hypochromic anaemia of a light degree. Virus and autoimmune hepatitis were excluded. The ceruloplasmin (CP) level was measured by the immune nephelometry assay in a BN II and BN ProSpec system (Siemens): human serum was mixed with anti-human CP immunoglobulin fraction, and the resulting immune complexes were measured by immune nephelometry (immune complexes can scatter the light; the source of incident light is the infrared high performance LED, and the scattered light intensity at a fixed angle of 13–24 degrees is measured by a photodiode detector) [2]. Regarding the low level of immunoreactive CP (measured by immune nephelometry) concentration (0.02 g/l), Wilson’s disease was suspected. The patient was sent to our outpatient clinic for the purpose of diagnostic final solution and treatment.
In the laboratory, hyposideraemia with paradoxically increased values of serum ferritin was found in addition to the abovementioned slightly increased ALT activity and microcytic hypochromic anaemia (complete laboratory and histological findings in the present patient at first examination are available in Table 1). The absence of Kayser-Fleischer ring, normal values of excretion of copper in urine (value: 346 nmol/24 h), genetic examination (including complete sequence analysis of the ATP7B chromosomal gene) and normal concentration of copper in dry matter of the liver (33 μg copper/g of dry liver tissue) excluded the supposed diagnosis of Wilson’s disease.
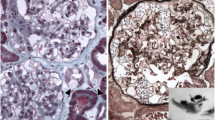
Histological examination of the liver revealed minimum lobular inflammation without fibrosis. The presence of liver steatosis was not confirmed (Fig. 1). Strong positivity of parenchymatous iron (Fig. 2) was a dominant finding. The accumulation of iron was confirmed by its quantitative determination (14,602 μg of iron/g of dry liver tissue, where the standard is up to 1600 μg/g of dry tissue).

Histology of the liver. Haematoxylin-eosin staining. Almost no lobular inflammation, without steatosis and without fibrosis. 400x

Liver histology. Perl’s stain. Strong positivity for parenchymatous iron. 600x
Mutations were not found by extended genetic examination of hereditary haemochromatosis (STRIP method, 12 most frequent mutations in the HFE gene and 4 most frequent mutations in the TRF2 and FPN1 genes) and genetic examination of aceruloplasminaemia (two most frequent mutations of 125insTACAC and W858X).
The CP gene was sequenced by next-generation sequencing (MiSeq), which revealed a new mutation in the ceruloplasmin gene c.1664 > A (p.Gly555Glu).
Although all in silico prediction tools (SIFT: Deleterious; Polyphen 2: Probably damaging; LTR: Deleterious; Mutation Taster: Disease causing; Mutation Assessor: High risk; FATHMM: Deleterious; SVM: Deleterious; LR: Deleterious) show deleterious effects, this change affects a conserved domain (the third cupredoxin domain of ceruloplasmin). MRI of the brain revealed excessive deposition of iron in the area of the basal ganglia, thalamus, mesencephalic nuclei and nucleus dentatus bilaterally (Fig. 3). The first neurological examination (and repeatedly during the follow-up) did not show any typical manifestation of the disease (ataxia, involuntary movements, cognitive dysfunction, Parkinsonism). Ophthalmological examination (including fundoscopy and visual field tests) didn’tn show retinal degeneration. Her visual acuity was not disturbed during the follow-up.

MRI of the brain. Susceptibility weighted imaging. Hypo-intensive changes in the basal ganglia and thalamus caused by deposits of iron
Our patient had normal fasting blood glucose values after repeated examinations, and an oral glucose tolerance test did not confirm the diagnosis of diabetes mellitus. The HOMA index was not calculated.
Chelatase treatment with deferasirox at a dose of 350 mg once daily during the first 9 months was started, and subsequently, from the 10th month of treatment, the dose was changed to 700 mg once daily. We recorded a progressive reduction in the serum concentration of ferritin in the laboratory. The patient continues to be without neurological and psychiatric symptomatology during the 21 months of chelatase treatment. Table 2 shows the dynamics of laboratory parameters during treatment. Control MRI (after 21 months of treatment) did not show changes in the iron concentration in the brain. During the indicated observation, no neuropsychiatric symptomatology was found.
Discussion and conclusions
Ceruloplasmin is a polypeptide composed of 1046 amino acid residues, which is encoded by a gene on the 3rd chromosome [3, 4].
In humans, ceruloplasmin occurs in two isoforms, which arise by alternative RNA “splicing” [5].
The soluble (secretory) form is expressed only in hepatocytes [6].
It constitutes the main transporter of copper in the plasma, has antioxidant properties and participates in the homeostasis of nitric oxide (NO) [6, 7].
The membrane-bound form of ceruloplasmin, associated with glycosylphosphatidylinositol (GPI-ceruloplasmin), is expressed in astrocytes, Sertoli cells, retinal cells, lungs, liver, kidneys, macrophages and immune system cells [8, 9].
GPI-ceruloplasmin participates in maintaining iron homeostasis in the brain. Under physiological conditions, iron is continuously transferred between astrocytes and neurons by means of transferrin, which acts as a pendulum. GPI-ceruloplasmin oxidizes Fe2+ on astrocyte protrusions, enabling binding and transport of Fe3+ to transferrin to meet the needs of neurons. When GPI-ceruloplasmin is absent, astrocytes are not capable of transporting iron through transferrin to neurons, which is why they catch it from alternative sources (citrate, ascorbate). The surplus of iron in astrocytes leads to oxidizing damage and a reduction in protective function in relation to neurons [1, 7].
Serum CP may be decreased in conditions with marked renal or enteric protein loss, malabsorption syndromes or severe end-stage liver disease and fulminant liver failure as a result of decreased hepatic protein synthesis [10].
An association between the low concentration and activity of CP and non-alcoholic fatty liver disease (NAFLD) with a high NAFLD score (NAS ≥ 5) in children was reported by Nobili et al. CP was associated with the odds of NASH, ballooning, inflammation and steatosis [11].
An inherent reduction in CP is typical of Wilson’s disease (due to mutations in the ATP7B gene, which encodes a copper-transporting P-type ATPase, with subsequent accumulation of copper in affected tissues) and of Menkes disease (due to mutations in the ATB7A gene causing a disorder of intestinal copper uptake) [10]. TMEM199-CDG is a rare genetic liver disease with abnormal glycosylation, chronically elevated serum transaminases, steatosis and low serum ceruloplasmin [12, 13].
In the present case, we repeatedly detected very low serum ceruloplasmin values in a patient with obesity. However, the patient had no arterial hypertension, impaired glucose tolerance or elevation of triacylglycerol (typical of NAFLD). Ultrasonographic examination of the liver revealed increased liver echogenicity. However, histological examination did not confirm liver steatosis, hepatocyte ballooning or fibrosis, and copper accumulation in the liver (typical of Wilson’s disease) was also excluded. Genetic examination revealed a mutation in the CP gene.
Aceruloplasminaemia is a rare, autosomal recessive hereditary disorder caused by mutations of the ceruloplasmin gene. The prevalence is estimated to be 1/2,000,000 in the Japanese population [14]. The prevalence in other parts of the world is not known.
Clinical manifestations of aceruloplasminaemia include retinal degeneration, diabetes mellitus, microcytic anaemia and neuropsychiatric symptomatology (cerebellar ataxia, dysarthria, nystagmus, dystonia, tremor, chorea, rigidity, akinesia, behaviour malfunctions, cognitive deficit, dementia) [14, 15].
Neuropsychiatric symptomatology mostly arises in the fifth life decade, and it can often be preceded by diabetes mellitus and microcytic anaemia, which are considered to be early manifestations of aceruloplasminaemia [16, 17].
The basic laboratory finding is a very low measurable concentration of ceruloplasmin in serum. Other laboratory findings include a low concentration of serum iron, low saturation of transferrin (10%), elevated concentration of serum ferritin and microcytic anaemia of light degree [18]. Visual examination is performed to diagnose peripheral degeneration of the retina [16]. MRI of the brain is used to find the accumulation of iron in the basal ganglia, thalamus, mesencephalon and cerebellum [19].
In the case of our patient, aceruloplasminaemia was not found before searching for the cause of chronic hepatopathy with low concentrations of serum ceruloplasmin. The initial supposed diagnosis of Wilson’s disease was ruled out by complex diagnostics. The finding of parenchymatous accumulation of iron in the liver led to the genetic examination focused on diseases causing accumulation of iron.
Extended genetic analysis revealed the variant c.1664G > A (p.Gly555Glu) of ceruloplasmin in the homozygous state. This mutation has not been previously described in the literature or population frequency databases and has unknown clinical significance according to the American College of Medical Genetics and Genomics standards [20, 21].
However, all in silico prediction tools showed this variant to be pathogenic or disease causing. Moreover, the 555 AA position in the CP protein belongs to the third cupredoxin domain of CP, a very important functional domain [22]. However, neither functional assays nor co-segregation of the variant in families have been performed to reclassify it as a likely pathogenic variant. We do not currently have a genetic examination of the patient’s relatives. Thus, we cannot determine the enzymatic or biological properties of altered CP and their relationship with the disease phenotype. We plan to add this to our investigation after obtaining informed consent.
Recently, there has not been a definite recommendation for aceruloplasminaemia treatment. Medical possibilities include chelatase substances (deferoxamine, deferasirox, deferiprone), vitamin E, vitamin C, zinc preparations, fresh frozen plasma and minocycline. Experience with these substances has been, however, limited to the results of individual case histories of patients [23].
Considering the most often used chelatase treatment, a reduction in serum ferritin and a reduction in the content of iron in the liver were observed. MRI mostly did not show changes in iron deposits of iron in the brain during chelatase treatment [24]. The changes in neurological symptomatology covered a wide range from improvement to stabilisation to deterioration [24, 25].
When the patient is without significant neurological symptomatology, ideally when they are asymptomatic, an early start to chelatase treatment is considered to be a basic factor for its success [17, 26]. In accordance with the literature data, we recorded a reduction in the concentration of serum ferritin. MRI did not show a reduction in the iron content in the brain.
Neurological symptomatology arises in patients with aceruloplasminaemia, usually in the fifth decade. Therefore, it has not yet been possible to assess the effect of treatment on its postponement.
Availability of data and materials
Patient documentation is available from the corresponding author on request.
Abbreviations
- ATPase 7B:
-
Adenosine triphosphatase 7B
- ATPase 7A:
-
Adenosine triphosphatase 7A
- ALT:
-
Alanine aminotransferase
- CDG:
-
Congenital disorder of glycosylation
- CP:
-
Ceruloplasmin
- GPI:
-
Glycosylphosphatidylinositol
- MRI:
-
Magnetic resonance imaging
- NO:
-
Nitrogen oxide
- NAFLD:
-
Nonalcoholic fatty liver disease
- NAS:
-
Nonalcoholic fatty liver disease score
- RNA:
-
Ribonucleic acid
- STRIP:
-
STRucture In Populations
- TRF2:
-
Transferrin receptor 2
- FPN1:
-
Ferroportin-1
References
Miyajima H. Aceruloplasminemia. Neuropathology. 2015;35:83–90.
Stanley J, et al. Essentials of immunology and serology, vol. 2002. Florence: Thomson Delmar Learning; 2002. p. 143–4. ISBN 0-7668-1064-X.
Takahashi N, Ortel TL, Putnam FW. Single-chain structure of human ceruloplasmin: the complete amino acid sequence of the whole molecule. Proc Natl Acad Sci U S A. 1984;81:390–4.
Yang F, Naylor SL, Lum JB, Cutshaw S, McCombs JL, Naberhaus KH, et al. Characterization, mappiong, and expression of the human ceruloplasmin gene. Proc Natl Acad Sci U S A. 1986;83:3257–61.
Yang F, Friedrichs WE, Cupples RL, Bonifacio MJ, Sanford JA, Horton WA, et al. J Biol Chem. 1990;265:10780–5.
Hellman NE, Gitlin JD. Ceruloplasmin metabolism and function. Annu Rev Nutr. 2002;22:439–58.
Shiva S, Wang X, Ringwood LA, Xu X, Yuditskaya S, Annavajihala V, et al. Ceruloplasmis is a NO oxidase and nitrite synthase that determines endocrine NO homeostasis. Nat Chem Biol. 2006;2:486–93.
Kono S, Yoshida K, Tomosugi N, Terada T, Hamaya Y, Kanaoka S, et al. Biological effects of mutant ceruloplasmin on hepicidin-mediated internalization of ferroportin. Biochim Biophys Acta. 1802;2010:968–75.
Musci G, Polticelli F, Bonaccorrsi di Patti MC. Ceruloplasmin-ferroportin system of iron traffic in vertebrates. World J Biol Chem. 2014;5:204–15.
Ferenci P, et al. EASL clinical practice guidelines: Wilson s disease. J Hepatol. 2012;56:671–85.
Nobili V, Siotto M, Bedogni G, et al. Levels of serum ceruloplasmin associate with pediatric nonalcoholic fatty LIver disease. J Pediatr Gastroenterol Nutr. 2013;56:370–5.
Vajro P, et al. Three unreported cases of TMEM199-CDG, a rare genetic liver disease with abnormal glycosylation. Orphanet J Rare Dis. 2018;13:4. https://doi.org/10.1186/s13023-017-0757-3.
Jansen JC, Timal S, van Scherpenzeel M, et al. TMEM199 deficiency is a disorder of golgi homeostasis characterized by elevated aminotransferases, alkaline phosphatase, and cholesterol and abnormal glycosylation. Am J Hum Genet. 2016;98:322–32.
Miyajima H, Kohno S, Takahashi Y, Yonekawa O, Kanno T. Estimation of the gene frequency of aceruloplasminemia in Japan. Neurology. 1999;3:617–9.
Vroegindeweijl LH, Langendonk JG, Lengeveld M, Hoogendoorn M, Kievit JA, Do Raimondo D, et al. New insights in the neurological phenotype of aceruloplasminemia in Caucasian patients. Parkinsonism Relat Disord. 2017;36:33–40.
Vroegindeweij LH, van der Beek EH, Boon AJ, Hoogendoorn M, Kievit JA, Wilson JH, et al. Aceruloplasminemia presents as type 1 diabetes in non-obese adults: a detailed case series. Diabet Med. 2015. https://doi.org/10.1111/dme.12712.
Pelucchi S, Mariani R, Ravasi G, Pelloni L, Marano M, Tremolizzo L, et al. Phenotypic heterogenity in seven Italian cases aceruloplasminemia. Parkinsonism Relat Disord. 2018;51:36–42.
Marchi G, Busti F, Zidanes AL, Castagna A, Girelli D. Aceruloplasminemia: a severe neurodegenerative disorder deserving an early diagnosis. Front Neurosci. 2019. https://doi.org/10.3389/fnins.2019.00325.
Parks NE, Vandorpe RA, Moeller JJ. Teaching neuroimages: neurodegeneration with brain iron accumulation in aceruloplasminemia. Neurology. 2013. https://doi.org/10.1212/01.wnl.0000435557.21319.ad.
Stenson PD, Mort M, Ball EV, Shaw K, Philips A, Cooper DN. The human gene mutation database: a building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Gene Ther. 2014;131:1–9.
Landrum MJ, Lee JM, Bensom M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:862–8.
Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2004;32:115–9.
Piperno A, Alessio M. Aceruloplasminemia: waiting for an efficient therapy. Front Neurosci. 2018. https://doi.org/10.3389/fnins.2018.00903.
Finkenstedt A, Wolf E, Hofner E, Gasser BL, Bosch S, Bakry R, et al. Hepatic but not brain iron is rapidly chelated by deferasirox in aceruloplasminemia due to a novel gene mutation. J Hepatol. 2010;53:1101–7.
Skidmore FM, Drago V, Foster P, Schmalfuss IM, Heilman KM, Streiff RR. Aceruloplasminemia with progressive atrophy without brain iron overload: treatment with oral chelation. J Neurol Neurosurg Psychiatry. 2008;79:467–70.
Doyle A, Rusli F, Bhathal P. Aceruloplasminaemia: a rare but important cause of iron overload. BMJ Case Rep. 2015. https://doi.org/10.1136/bcr-2014-207541.
Acknowledgements
Not applicable.
Funding
No funding.
Author information
Authors and Affiliations
Contributions
MO collected clinical data and drafted the manuscript. SD collected clinical data. MD participated in the haematological treatment. JLS performed the genetic analysis and contributed to revision of the manuscript. RZM and PJ performed and coordinated the genetic analysis. MF analysed the brain MRI. BR performed histological analysis of the liver. MZ supervised the case and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The case report was approved by the ethics committee of the University Hospital in Bratislava. The patient was given standard care. The patient gave the consent to the publication of this case report.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ondrejkovičová, M., Dražilová, S., Drakulová, M. et al. New mutation of the ceruloplasmin gene in the case of a neurologically asymptomatic patient with microcytic anaemia, obesity and supposed Wilson’s disease. BMC Gastroenterol 20, 95 (2020). https://doi.org/10.1186/s12876-020-01237-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12876-020-01237-8