
Abstract
Background
Percutaneous coronary intervention (PCI) with primary stenting, which stands for stent implantation regardless of obtaining satisfactory results with balloon angioplasty, has superseded conventional plain old balloon angioplasty with provisional stenting. With drug-coated balloon (DCB), primary DCB angioplasty with provisional stenting has shown non-inferiority to primary stenting for de novo coronary small vessel disease. However, the long-term efficacy and safety of such a strategy to the primary stenting on clinical endpoints in de novo lesions without vessel diameter restrictions remain uncertain.
Study design
The REC-CAGEFREE I is an investigator-initiated, multicenter, randomized, open-label trial aimed to enroll 2270 patients with acute or chronic coronary syndrome from 43 interventional cardiology centers in China to evaluate the non-inferiority of primary paclitaxel-coated balloons angioplasty to primary stenting for the treatment of de novo, non-complex lesions without vessel diameter restrictions. Patients who fulfill all the inclusion and exclusion criteria and have achieved a successful lesion pre-dilatation will be randomly assigned to the two arms in a 1:1 ratio. Protocol-guided DCB angioplasty and bailout stenting after unsatisfactory angioplasty are mandatory in the primary DCB angioplasty group. The second-generation sirolimus-eluting stent will be used as a bailout stent in the primary DCB angioplasty group and the treatment device in the primary stenting group. The primary endpoint is the incidence of Device-oriented Composite Endpoint (DoCE) within 24 months after randomization, including cardiac death, target vessel myocardial infarction, and clinically and physiologically indicated target lesion revascularization.
Discussion
The ongoing REC-CAGEFREE I trial is the first randomized trial with a clinical endpoint to assess the efficacy and safety of primary DCB angioplasty for the treatment of de novo, non-complex lesions without vessel diameter restrictions. If non-inferiority is shown, PCI with primary DCB angioplasty could be an alternative treatment option to primary stenting.
Trial registration
Registered on clinicaltrial.gov (NCT04561739).
Similar content being viewed by others


Introduction
Since the bare-metal stent era, primary plain old balloon angioplasty (POBA) with provisional stenting has been superseded by routine coronary stenting for the treatment of de novo coronary lesions because POBA had a higher risk of repeat revascularization [1]. This notion was maintained in the drug-eluting stents (DES) era, even for patients who require urgent non-cardiac surgery or high bleeding risks, as short-duration dual antiplatelet therapy may be reasonable with both strategies [2,3,4]. However, stent implantation continues to face notable challenges as there is a permanent metallic scaffold left behind in the vessel. Stents may distort and constrain the coronary vessel, limit vessel pulsatility, and adaptive remodeling [5], and promote chronic inflammation, which in turn increases the risk of late stent thrombosis and restenosis by approximately 2% per year [2].
Drug-coated balloons (DCB) represent a contemporary therapeutic effort in the treatment of coronary artery disease (CAD) [6]. Upon reaching the target lesion, the expansion of DCB can rapidly deliver antiproliferative drugs into the arterial wall through a lipophilic matrix during angioplasty without the necessity of implanting a scaffold [7]. This feature of DCB has the potential to minimize the negative effects associated with stent-related maladaptive biologic response [8]. Currently, the management of in-stent restenosis (ISR) by DCB is considered a Class IA recommendation [2]. The safety and effectiveness of the DCB strategy have also been demonstrated in de novo small vessels [9], acute coronary syndromes [10, 11], and high-bleeding risk patients [12]. Moreover, the application of DCB is gradually expanding to include all de novo coronary arteries without diameter restrictions. However, the use of DCB in such cases is still controversial [13, 14] due to the lack of randomized study with powered clinical endpoints.
To fill the knowledge gap, we designed the REC-CAGEFREE I trial to revive the longstanding debate between angioplasty and stenting in contemporary settings. The trial will investigate the potential non-inferiority of the primary paclitaxel-coated balloon angioplasty with provisional stenting compared to the primary second‐generation sirolimus-eluting stenting for the treatment of de novo coronary lesions without vessel diameter restrictions. The evaluation will be conducted through a randomized controlled trial, focusing on a composite clinical endpoint comprising cardiac death, target vessel myocardial infarction, and clinically indicated target lesion revascularization at the 24-month follow-up.
Study design
Objectives and hypothesis
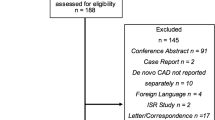
The REC-CAGEFREE I trial (ClinicalTrials.gov, NCT04561739) is an investigator-initiated, multicenter, prospective, randomized, open-label trial aimed to enroll 2270 patients from ≥ 40 interventional cardiology centers in China. The primary objective of the trial is to test the non-inferiority of the primary balloon angioplasty strategy with paclitaxel-coated balloons (Experimental arm) to the primary stenting strategy with second‐generation sirolimus-eluting stents (Reference group) for the treatment of de novo lesions without vessel diameter restrictions in the setting of non-complex percutaneous coronary intervention (PCI). The incidence of Device-oriented Composite Endpoint (DoCE) at 24 months will be assessed as the primary endpoint (Fig. 1).

Study flow chart. CAG, Coronary Angiography; PCI, percutaneous coronary intervention; CAD, coronary artery disease; TIMI, Thrombolysis In Myocardial Infarction; POBA, plain old balloon angioplasty; NCB, non-compliant balloon; DoCE, Device-oriented Composite Endpoint; TV-MI, target vessel myocardial infarction; CPI-TLR, clinically and physiologically indicated target lesion revascularization
Study organization and funding
This trial is investigator-initiated and obtained grant support from Xijing Hospital (Xi’an, China; Grant No. XJZT24LY36) and unrestricted grant support from Shenqi Medical (Shanghai, China) and Microport Medical Group (Shanghai, China). Apart from this sponsorship, Shenqi and Microport will not be involved in the design, execution, or decision to publish the study. The steering committee has a pivotal role with overall responsibility for the concept, design, and execution of the study progress in accordance with scientific, medical, ethical, and practical elements. The committee will convene a series of meetings to ensure the effective management and execution of the study, including data acquisition, quality control, security, analysis, and reporting. The study will follow the ethical principles outlined in the Declaration of Helsinki and has received approval for its protocol from the institutional review board at each participating center.
Study population
The complete inclusion and exclusion criteria are shown in Table 1. Patients indicated for PCI either due to acute (including STEMI, NSTEMI, and unstable angina) or chronic coronary syndrome are eligible [15, 16]. To be considered suitable for enrollment, the target lesion must be de novo, non-complex, and successfully pre-dilatated. Therefore, patients will be consented before the angiography but are formally included and randomized if it is confirmed that all angiographic criteria are met and the target lesion has been successfully pre-dilatated (Fig. 1). To ensure that eligible patients fully comprehend the purpose and procedures of the investigation without encountering any language barriers, the study may opt to enroll patients of Chinese nationality and ethnicity exclusively. To ensure adherence to the study protocol, a training course was organized at each center, led by TL and CG, to ensure that the investigators comprehended and followed the protocol effectively. The eligibility review committees of each participating site, comprising of TL, CG, and the investigators of each site, will conduct a thorough online assessment of cases being screened at the site (after the completion of pre-dilatation) to ensure that all enrolled participants have fulfilled the angiographic and lesion preparation criteria.
Randomization
Eligible patients who have signed the informed consent will be randomized at a 1:1 ratio using web-based software to be assigned to either the primary DCB angioplasty or primary stenting group. Web-response dynamic-block randomization, utilizing varying blocks of 2, 4, or 6, will allocate random assignment stratified by center.
Patients who fulfill the angiographic criteria (de novo and non-complex lesions) but have unsuccessful pre-dilatation will not be randomized. These patients will be included in a separate parallel cohort study (Fig. 1). The cohort study will be implemented only in sites that agree to join. PCI with DES is recommended for these patients.
Study procedures
Investigators may exercise discretion in utilizing antithrombotic medications, glycoprotein IIb/IIIa inhibitors, intravascular imaging, or fractional flow reserve. Complete revascularization in one PCI session is recommended. If a staged procedure becomes necessary, it will be documented during the index procedure, and the patient shall use the same allocated strategy and revascularized within 45 days post-index procedure. Any revascularization that is unplanned or beyond the indicated period will be considered a potential event and adjudicated by the independent clinical-event adjudication committee (CEC).
Selecting de novo, non-complex lesion and lesion pre-dilatation
As aforementioned, to be considered suitable for enrollment, the target lesion must be de novo, non-complex, and successfully pre-dilatated. Non-complex lesion is defined as fulfilling all of the following criteria [17]: (1) Planned numbers of lesions/vessel to be treated < 3, planned DES/DCB implanted < 3, or planned total DES/DCB length < 60 mm; (2) Bifurcation does not require two DES/DCB; (3) Non-left main lesion; (4) Non-venous or arterial graft lesion; (5) Non-chronic total occlusion lesion; (6) Do not require the use of an atherectomy device.
Optimized and successful pre-dilatation includes the requirement of with or without a plain old balloon angioplasty (POBA), a pre-dilation prior to DCB angioplasty shall be performed with a non-compliant balloon, cutting balloon, or scoring balloon at 0.8-1.0 balloon/vessel size ratio. After lesion preparation, a 10-minute observational period should be conducted, followed by an angiogram to ensure satisfactory lesion preparation, which consists of the following criteria: 1) ≤ 30% residual stenosis (visual); 2) Thrombolysis In Myocardial Infarction (TIMI) flow grade 3; 3) the absence of a dissection type D, E, and F according to NHLBI classification; and 4) without serious complications. If the pre-dilatation is deemed unsuccessful, patients will be disqualified from entering the randomization.
Randomization to the primary DCB angioplasty strategy
The performance of DCB angioplasty adheres to the recommendations of the German Consensus Group on DCB interventions [18] and the Third Report of the International DCB Consensus Group with adjustments [19]. The DCB angioplasty should only be used after successful pre-dilatation. Subsequently, the DCB, on each side longer than the DCB by at least 2–3 mm (visual) to avoid geographical mismatch, is inflated at nominal pressure for 30–45 s. Similarly, a 10-minute observational period should be conducted, followed by an angiogram to ensure satisfactory DCB angioplasty. After DCB angioplasty, if there is a deterioration of blood flow (TIMI grade flow ≤ 2) after DCB angioplasty, it is recommended to give intracoronary medication (e.g., nitroprusside) and wait approximately 5 min before making the final assessment. In cases when subjects experience dissection type D, E, and F (NHLBI classification) or visual residual stenosis > 30%, a second-generation sirolimus-eluting stent, which is the same stenting used in the Reference arm, will be implanted mandatory as the bailout stent. These participants will be considered as a part of the primary DCB angioplasty strategy and included in the primary analyses.
The device used for primary DCB angioplasty is the Swide DCB (Shenqi Medical, Shanghai, China), which is a balloon coated with a paclitaxel-iopromide formulation (3 µg Paclitaxel per 1 mm2 of the balloon surface) using a proprietary dipping process that deposited the formulation preferentially in the folds of the balloon [20]. The spray coating of the mixture of paclitaxel and iopromide of the DCB is via ultrasound, with the crystal size < 2 μm. Previously, the Swide DCB has demonstrated non-inferiority to the SeQuent Please DCB, which is also a paclitaxel-iopromide coated DCB (3 µg Paclitaxel per 1 mm2), for the primary endpoint of 9-month in-segmentlate loss in patients with in-stent restenosis [21].
Randomization to the primary stenting strategy
The performance of primary stenting adheres to the routine local clinical practice and established guidelines [2, 15, 16]. In the primary stenting group, the Firebird 2 DES will be used. The Firebird2 DES (MicroPort, Shanghai, China) is a sirolimus-eluting coronary stent with an L605 Co-Cr alloy platform and durable polymer. The strut is 86 μm in thickness, and 80% of the drug is released within 30 days. The effectiveness and safety of the Firebird2 DES have been confirmed in a real-world population and randomized cohorts [22,23,24]. If delivery failure occurs, an alternative stent may be utilized [25].
Concomitant medication and treatment
All study patients are administered antithrombotic drugs according to international guidelines [26]. All subjects must receive DAPT, being aspirin and either clopidogrel or ticagrelor for at least one month after index PCI, followed by aspirin, clopidogrel, or ticagrelor monotherapy indefinitely. Detailed recommendations for pre-procedural and post-procedural antiplatelet regimens are shown in Supplementary Table 1. While the physician has discretion over other medical treatments, it is strongly advised to implement guideline-directed medical therapy to address the patient’s specific condition, such as controlling hypertension or diabetes mellitus, prescribing high-intensity statins, discontinuing cigarette smoking, and providing optimal pharmacologic treatment for heart failure. All antiplatelet medications (including start and stop times of interrupted DAPT) and other cardiac medications will be recorded in the eCRF at each visit.
Follow-up
Scheduled follow-up visits occur at 1 (± 14 days), 3, 6, 12, 18, and 24 (± 30 days) months post-randomization. After 24 months, the follow-up will be conducted annually and kept for up to 10 years. All follow-up visits are preferably scheduled on-site. If the patients are unable or unwilling to visit the outpatient clinic, the scheduled visit can be replaced by a telephone call except for the 30-day, 1- and 2-year visits. At each visit, self-reported adherence to the prescribed medications is collected with the assessment of any cardiac or cerebrovascular ischemic or bleeding occurrences or any serious adverse event. Each participant’s WeChat account will be documented for record-keeping purposes. To facilitate the acquisition of patient-reported outcomes and adherence to the prescribed medications, we developed a mobile application that functions through the WeChat platform. All participants will be contacted monthly through this application and receive a questionnaire to evaluate their health status and adherence.
Study endpoints
The study endpoints are listed in Table 2. The primary endpoint is the Device-oriented Composite Endpoint (DoCE) within 24 months after randomization. DoCE is defined as a composite clinical endpoint of cardiovascular death, target vessel myocardial infarction (TV-MI), and clinically indicated target lesion revascularization (CPI-TLR) [27]. The definition of Academic Research Consortium (ARC)-2 will be followed [27]. Cardiovascular death is defined as any death due to a cardiac cause, unwitnessed death, death of unknown cause, and all study procedure-related deaths [27]. MI will be defined using the SCAI consensus for peri-procedure MI within 48 h of the index procedure [28], and the Fourth Universal Definition of MI > 48 h after the index procedure [29]. TV-MI is defined as MI that cannot clearly be attributable to a non-target vessel. TLR is defined as a repeat percutaneous intervention of the target lesion or bypass surgery of the target vessel performed for restenosis or other complications of the target lesion. Clinically and physiologically indicated TLR will be adjudicated based on the assessment of a positive functional ischemia test by either Wire-based or angiographic-derived Fractional Flow Reserve or Quantitative Coronary Analysis, with explicit criteria provided in the Supplementary Methods. The definition of device and procedure success are also provided in the Supplementary Methods. For secondary endpoints, stroke is defined as any non-convulsive focal or global neurological deficit of abrupt onset lasting for more than 24 h or leading to death, which is caused by ischemia or hemorrhage within the brain. The Neuro-ARC definition and classification will be used [30]. Bleeding will be defined by the Bleeding Academic Research Consortium (BARC) criteria [31], and other definitions [32,33,34,35,36] will used for exploratory purposes. The adherence to the medication will be assessed according to the Non-adherence Academic Research Consortium (NARC) [37] definitions.
Suspected adverse events will be reported promptly on an electronic case report form, with source documents centrally collected. After collecting adverse events centrally, any record that could lead to the unblinding of treatment assignment will be obliterated before submission to the clinical event committee (CEC). All adverse events will be categorized according to predefined criteria by an independent clinical-event adjudication committee whose members are unaware of the assignment group. However, if the CEC members reviewed the angiogram, due to the absence of a metallic scaffold in the primary DCB group (unless the patient had bailout stenting), the blinding of the assignment group might not be feasible.
Off-line quantitative coronary angiographic measurements (QCA) and quantitative flow ratio (QFR) measurement
The off-line quantitative coronary angiography (QCA) [38] and quantitative flow ratio (QFR) [39] assessment by an independent Corelab will be performed at baseline (pre- and post-PCI). Routine follow-up angiography in the absence of symptoms was not recommended. For the purpose of adjudicating clinically indicated or physiologically indicated revascularization, QCA, and QFR measurements will be conducted if the angiogram of revascularization is assessable.
Sample size calculation
This study compares treatment groups at the individual patient level. Our hypothesis is that for the treatment of de novo, non-complex lesions, the primary DCB angioplasty group would be non-inferior to the primary stenting group in terms of Device-oriented composite endpoint within 24 months after PCI.
Due to the limited availability of dedicated data on the occurrence rate of DoCE at two years in patients of non-complex lesions, the event rate of the primary stenting group in this trial was estimated by referring to the findings of the GLOBAL LEADERS subgroup analysis of complex/non-complex PCI [17], in which the patients with non-complex PCI had 2-year cumulative event rate of DoCE of 6.7%, and the findings of the contemporary all-comers DES trials, including TARGET AC [40], TALENT [41], DESSOLVE III [42], BIONYX trials [43], in which patients had 2-year cumulative event rate of DoCE ranging from 6.9 to 8.7%. It was assumed that patients treated with different second-generation DES would have a similar cumulative event rate of DoCE. Therefore, it is anticipated that 6.7% of patients in the primary stenting group will reach the primary endpoint of DoCE at two years. The non-inferiority margin of 2.68%, which was 40% of the cumulative event rate of DoCE, was chosen based on clinically acceptable relevance according to the margins in previous major trials of comparing DCB to DES [44], or one DES comparing to another DES [40,41,42,43], and the feasibility of patient enrolment. With a total of 2156 patients (1078 per group), the study is estimated to have 80% power to show non-inferiority with a 5% one-sided α error rate [3, 40,41,42,43]. Accounting for an attrition rate of approximately 5%, the final sample size was determined to be at least 2270 patients (1135 per group).
Statistical considerations
The demographic and clinical variables at baseline will be summarized for each treatment group, considering the intention-to-treat (ITT), per-protocol, and as-treated populations. Categorical data will be described as numbers (percentages). Continuous variables will be expressed as mean ± standard deviation or median (interquartile range) for normal or skewed distributions.
The primary endpoint of the trial is DoCE at 24 months after randomization. The primary analysis will be performed based on a crude measurement of treatment difference between groups in the primary endpoint, without adjusting for any covariates, using the intention-to-treat (ITT) population. To estimate the cumulative event rate of DoCE at 24 months in each group, the Kaplan-Meier (KM) method will be employed. The one-sided 95% confidence interval (CI) for the difference in the cumulative event rate at 24 months between the primary DCB angioplasty group and the primary stenting group will be calculated using Greenwood’s formula for the variance of the KM estimates. If the upper limit of the one-sided 95%CI is below 2.68%, it will be concluded that the primary DCB angioplasty group is non-inferior to the primary stenting group. If the non-inferiority testing for the primary endpoint is met, the superiority testing of the primary endpoint will be further tested.
In addition, a covariate-adjusted analysis of the primary endpoint using the inverse probability of treatment weighting approach, considering the covariates at baseline and center effect, will be performed in the ITT population as a sensitivity analysis. The crude and adjusted analyses will be repeated in the per-protocol and as-treated population to support the primary results. For secondary endpoints, the difference in cumulative event rate and their two-sided 95%CIs will be reported, and Cox proportional hazard ratios (HR) will also be provided.
The prespecified subgroup analyses will also be conducted for clinically relevant factors such as age, sex, body mass index, diabetes mellitus or smoking, and other risk indicators, with details described in Supplementary Table 2. Stratum-specific HRs and corresponding 95% CI will be calculated for each subgroup using a Cox proportional hazards model. Interaction testing will be performed using the subgroup X treatment as an additional term in the Cox model. A prespecified landmark analysis of the primary endpoint will be performed from 0 to 12 and 12 to 24 months. Unless otherwise specified, a two-sided test will be utilized for testing at a 5% significance level. All analyses will be described in detail in the statistical analysis plan, which will be developed and finalized before the database lockup.
Safety monitoring
The Data and Safety Monitoring Board (DSMB), in conjunction with the steering committee responsible for ensuring participant safety, will act in an advisory capacity to monitor participant safety, evaluate the study progress, and review procedures for maintaining data confidentiality. A biannual DSMB meeting will be held, either in-person or via teleconference, to discuss study progress, ensure proper execution of study procedures, maintain data quality and security, and review any safety concerns related to participants. Although no interim analysis was initially planned, the DSMB holds the power to terminate the study process and scrutinize relevant events during the trial in the event of any safety issues.
Discussion
In 1996, the US Food and Drug Administration approved the first 2 bare-metal stents (BMS) for the treatment of de novo lesions to prevent recoil and for treatment of acute artery closure after plain old balloon angioplasty (POBA) [45]. Meanwhile, pivotal randomized trials compared BMS with POBA, and the results indicated that the use of BMS led to a reduction of adverse events by 30% within the first six months after PCI. This reduction was primarily attributed to a 50% decrease in the risk of repeat revascularization [45, 46]. Furthermore, these trials established an important concept: stents could effectively decrease restenosis by providing significant initial angiographic gain and by preventing early recoil and late negative remodeling of the treated vessel [47, 48].
However, there was concern that primary stenting without first trying to obtain satisfactory results with POBA alone would increase the occurrence of in-stent restenosis. Consequently, in 2000, studies were conducted to compare primary stenting using BMS versus POBA with the provisional use of BMS [49,50,51]. These trials revealed several common findings, one of which was the difficulty in attaining a satisfactory result solely with POBA. Using angiographic criteria alone, the OCBAS trial [49] showed that 13.5% of the POBA group crossed over to stent implantation. When both angiographic and physiologic criteria were employed to determine an optimal outcome, up to 50% of patients failed to achieve a satisfactory result. Furthermore, the clinical outcome with primary stenting is as good or better than that achieved with a strategy of provisional stenting [45].
After the mechanical era in interventional cardiology, as represented by POBA and BMS, the local dispensing era emerged with the delivery of anti-restenotic drugs directly into the coronary artery [52]. The introduction of DES revolutionized the field and established itself as the preferred treatment for patients undergoing PCI [2]. However, the risk of late stent thrombosis and restenosis after DES is still approximately 2% per year and leads to a target-lesion failure rate of approximately 14% after 5 years [2, 53].
DCBs were initially introduced in the European market in 2007, with the aim of being an alternative strategy for ISR instead of DES [2, 54]. The rationale behind the utilization of DCB is founded on the concept that highly lipophilic drugs can achieve effective drug delivery even with short contact durations between the balloon surface and the vessel wall, thus theoretically avoiding the side effects associated with the maladaptive biological response induced by permanent prosthesis implantation [2]. Based on these theoretical foundations, DCB has demonstrated a correlation with faster vascular healing, a favorable effect on preventing late negative remodeling, and even provides late lumen enlargement [55, 56].
Two decades after the early trials comparing primary stenting with BMS vs. POBA with provisional stenting, in the era of DES and DCB, together with the introduction of newer balloons such as cutting and scoring balloons for achieving satisfactory angiographic results, DCB angioplasty with provisional stenting has been explored for the treatment of de novo coronary lesions in some specific settings, demonstrating notable advantages, particularly in SVD [57, 58]. The BASKET-SMALL 2 trial [6, 9], which enrolled 758 participants randomly allocated to DES or DCB, is the largest study to date investigating de novo SVD. The study demonstrated that DCB angioplasty with provisional DES implantation was non-inferior to primary DES in terms of major adverse cardiovascular events over a period of 3 years. Similarly, the PICCOLETO II trial, which also focused on patients with SVD, found no significant difference in clinical outcomes at 12 months between the DCB and DES arms. However, the DCB arm showed significantly lower late lumen loss (LLL) than the DES arm [59]. Furthermore, DCBs have also exhibited potential advantages in other de novo settings, including patients with a higher risk of bleeding or those encountering high thrombus burden and inflammatory states [11, 12].
However, there remains a scarcity of randomized data that compares the clinical outcomes associated with the use of DCB angioplasty and DES in the context of de novo disease with all vessel diameters. The available findings show significant variability, with most of the existing data coming from small-scale angiographic investigations or clinical follow-ups that solely focus on the DCB arm [19]. The findings of the REVELATION trial [11] demonstrated that DCB was comparable to DES in terms of the primary endpoint, fractional flow reserve (FFR), during a 9-month angiographic follow-up of 120 patients who underwent primary PCI for ST-elevation myocardial infarction. The trial also revealed similar angiographic late lumen loss (LLL) and clinical outcomes between the two treatment modalities. Nishiyama et al. [60] have also reported no significant difference in MLD or late lumen loss between the DES and DCB in large vessels. Conversely, there are also studies [61] showing a higher LLL in patients with acute coronary syndrome when treated with DCB compared to DES.
Considering the limitations of stents and the basis of promising evidence indicating the effectiveness of DCB angioplasty in treating ISR and SVD, there is potential for primary DCB with provisional stenting to serve as a viable substitute for primary DES implantation in treating de novo lesions across all vessel diameters. To validate this hypothesis, we designed the REC-CAGEFREE I study, which aimed to enroll a large cohort to investigate the non-inferiority of primary DCB angioplasty to primary stenting in patients with de novo lesions without any limitations on vessel diameter.
Current status of the REC-CAGEFREE I trial
The REC-CAGEFREE I trial commenced with the enrollment of the first patient in February 2021 and concluded with the enrollment of the last patient in May 2022 (Supplementary Fig. 1). A total of 2,272 patients were ultimately enrolled from 43 participating sites. Follow-up for the primary endpoint will be finalized in June 2024, and all the participants will be monitored for up to 10 years after randomization. The findings from the primary analyses are anticipated to be published in the third quarter of 2024.
Conclusion
The REC-CAGEFREE I trial is the first randomized trial with a large cohort and clinical endpoint to assess the non-inferiority of primary DCB angioplasty to primary stenting to treat de novo, non-complex lesions in all vessel diameters. If non-inferiority is shown, PCI with primary DCB angioplasty could provide as an alternative treatment option to primary stenting.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Brophy JM, Belisle P, Joseph L. Evidence for use of coronary stents. A hierarchical bayesian meta-analysis. Ann Intern Med. 2003;138(10):777–86.
Neumann FJ, Sousa-Uva M, Ahlsson A, Alfonso F, Banning AP, Benedetto U, Byrne RA, Collet JP, Falk V, Head SJ, et al. : 2018 ESC/EACTS guidelines on myocardial revascularization. Eur Heart J. 2019;40(2):87–165.
Windecker S, Latib A, Kedhi E, Kirtane AJ, Kandzari DE, Mehran R, Price MJ, Abizaid A, Simon DI, Worthley SG, et al. Polymer-based or polymer-free stents in patients at high bleeding risk. N Engl J Med. 2020;382(13):1208–18.
Urban P, Meredith IT, Abizaid A, Pocock SJ, Carrie D, Naber C, Lipiecki J, Richardt G, Iniguez A, Brunel P, et al. Polymer-free drug-coated coronary stents in patients at high bleeding risk. N Engl J Med. 2015;373(21):2038–47.
Kawai T, Watanabe T, Yamada T, Morita T, Furukawa Y, Tamaki S, Kawasaki M, Kikuchi A, Seo M, Nakamura J, et al. Coronary vasomotion after treatment with drug-coated balloons or drug-eluting stents: a prospective, open-label, single-centre randomised trial. EuroIntervention: J EuroPCR Collab Working Group Interventional Cardiol Eur Soc Cardiol. 2022;18(2):e140–8.
Jeger RV, Farah A, Ohlow MA, Mangner N, Mobius-Winkler S, Leibundgut G, Weilenmann D, Wohrle J, Richter S, Schreiber M, et al. Drug-coated balloons for small coronary artery disease (BASKET-SMALL 2): an open-label randomised non-inferiority trial. Lancet. 2018;392(10150):849–56.
Gray WA, Granada JF. Drug-coated balloons for the prevention of vascular restenosis. Circulation. 2010;121(24):2672–80.
Her AY, Shin ES, Kim S, Kim B, Kim TH, Sohn CB, Choi BJ, Park Y, Cho JR, Jeong YH. Drug-coated balloon-based versus drug-eluting stent-only revascularization in patients with diabetes and multivessel coronary artery disease. Cardiovasc Diabetol. 2023;22(1):120.
Jeger RV, Farah A, Ohlow MA, Mangner N, Mobius-Winkler S, Weilenmann D, Wohrle J, Stachel G, Markovic S, Leibundgut G, et al. Long-term efficacy and safety of drug-coated balloons versus drug-eluting stents for small coronary artery disease (BASKET-SMALL 2): 3-year follow-up of a randomised, non-inferiority trial. Lancet. 2020;396(10261):1504–10.
Merinopoulos I, Gunawardena T, Corballis N, Bhalraam U, Reinhold J, Wickramarachchi U, Maart C, Gilbert T, Richardson P, Sulfi S, et al. Assessment of Paclitaxel Drug-Coated Balloon only Angioplasty in STEMI. JACC Cardiovasc Interventions. 2023;16(7):771–9.
Vos NS, Fagel ND, Amoroso G, Herrman JR, Patterson MS, Piers LH, van der Schaaf RJ, Slagboom T, Vink MA. Paclitaxel-Coated Balloon Angioplasty Versus Drug-Eluting Stent in Acute myocardial infarction: the REVELATION Randomized Trial. JACC Cardiovasc Interventions. 2019;12(17):1691–9.
Rissanen TT, Uskela S, Eranen J, Mantyla P, Olli A, Romppanen H, Siljander A, Pietila M, Minkkinen MJ, Tervo J, et al. Drug-coated balloon for treatment of de-novo coronary artery lesions in patients with high bleeding risk (DEBUT): a single-blind, randomised, non-inferiority trial. Lancet. 2019;394(10194):230–9.
Yerasi C, Case BC, Forrestal BJ, Torguson R, Weintraub WS, Garcia-Garcia HM, Waksman R. Drug-coated balloon for De Novo Coronary Artery Disease: JACC State-of-the-art review. J Am Coll Cardiol. 2020;75(9):1061–73.
Colombo A, Leone PP, Ploumen EH, von Birgelen C. Drug-coated balloons as a first choice for patients with de novo lesions: pros and cons. EuroIntervention: J EuroPCR Collab Working Group Interventional Cardiol Eur Soc Cardiol. 2024;20(2):e120–2.
Byrne RA, Rossello X, Coughlan JJ, Barbato E, Berry C, Chieffo A, Claeys MJ, Dan GA, Dweck MR, Galbraith M, et al. 2023 ESC guidelines for the management of acute coronary syndromes. Eur Heart J. 2023;44(38):3720–826.
Virani SS, Newby LK, Arnold SV, Bittner V, Brewer LC, Demeter SH, Dixon DL, Fearon WF, Hess B, Johnson HM et al. 2023 AHA/ACC/ACCP/ASPC/NLA/PCNA Guideline for the Management of Patients With Chronic Coronary Disease: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation 2023, 148(9):e9-e119.
Serruys PW, Takahashi K, Chichareon P, Kogame N, Tomaniak M, Modolo R, Chang CC, Komiyama H, Soliman O, Wykrzykowska JJ, et al. Impact of long-term ticagrelor monotherapy following 1-month dual antiplatelet therapy in patients who underwent complex percutaneous coronary intervention: insights from the global leaders trial. Eur Heart J. 2019;40(31):2595–604.
Kleber FX, Rittger H, Bonaventura K, Zeymer U, Wöhrle J, Jeger R, Levenson B, Möbius-Winkler S, Bruch L, Fischer D, et al. Drug-coated balloons for treatment of coronary artery disease: updated recommendations from a consensus group. Clin Res Cardiology: Official J German Cardiac Soc. 2013;102(11):785–97.
Jeger RV, Eccleshall S, Wan Ahmad WA, Ge J, Poerner TC, Shin ES, Alfonso F, Latib A, Ong PJ, Rissanen TT, et al. Drug-coated balloons for coronary artery disease: third report of the International DCB Consensus Group. JACC Cardiovasc Interventions. 2020;13(12):1391–402.
Zhu Z, Han H, Zhu J, Zhang J, Du R, Ni J, Ying C, An X, Zhang R. Safety and efficacy of a novel iopromide-based paclitaxel-eluting balloon following bare metal stent implantation in rabbit aorta abdominalis. Biomed Mater Eng. 2015;26(1–2):79–88.
Zhu J, Liu L, Zhu Z, Yang Z, Hu J, Ding F, Zhou Y, Su X, Ge J, Liu X, et al. A randomized comparison of a novel iopromide-based paclitaxel-coated balloon shenqi versus SeQuent please for the treatment of in-stent restenosis. Coron Artery Dis. 2021;32(6):526–33.
Xu B, Zhang Q, Yang YJ, Qiao SB, Zhang RY, Zhang JS, Hu J, Qin XW, Hong T, Li JP, et al. Sirolimus-eluting cobalt-chromium stents: two-year clinical results from first-in-man study on the Firebird 2 stent. Chin Med J (Engl). 2008;121(6):492–7.
Han Y, Xu B, Jing Q, Lu S, Yang L, Xu K, Li Y, Li J, Guan C, Kirtane AJ, et al. A randomized comparison of novel biodegradable polymer- and durable polymer-coated cobalt-chromium sirolimus-eluting stents. JACC Cardiovasc Interventions. 2014;7(12):1352–60.
Ge JB, Zhang F, Qian JY, Ge L, Liu XB, Zhou J. Six-month clinical outcomes of Firebird 2TM sirolimus-eluting stent implantation in real-world patients with coronary artery diseases. Chin Med J (Engl). 2011;124(6):831–5.
Chang CC, Kogame N, Onuma Y, Byrne RA, Capodanno D, Windecker S, Morel MA, Cutlip DE, Krucoff MW, Stone GW, et al. Defining device success for percutaneous coronary intervention trials: a position statement from the European Association of Percutaneous Cardiovascular Interventions of the European Society of Cardiology. EuroIntervention: J EuroPCR Collab Working Group Interventional Cardiol Eur Soc Cardiol. 2020;15(13):1190–8.
Valgimigli M, Bueno H, Byrne RA, Collet JP, Costa F, Jeppsson A, Juni P, Kastrati A, Kolh P, Mauri L, et al. 2017 ESC focused update on dual antiplatelet therapy in coronary artery disease developed in collaboration with EACTS: the Task Force for dual antiplatelet therapy in coronary artery disease of the European Society of Cardiology (ESC) and of the European Association for Cardio-thoracic surgery (EACTS). Eur Heart J. 2018;39(3):213–60.
Garcia-Garcia HM, McFadden EP, Farb A, Mehran R, Stone GW, Spertus J, Onuma Y, Morel MA, van Es GA, Zuckerman B, et al. Standardized end point definitions for coronary intervention trials: the Academic Research Consortium-2 Consensus Document. Eur Heart J. 2018;39(23):2192–207.
Moussa ID, Klein LW, Shah B, Mehran R, Mack MJ, Brilakis ES, Reilly JP, Zoghbi G, Holper E, Stone GW. Consideration of a new definition of clinically relevant myocardial infarction after coronary revascularization: an expert consensus document from the Society for Cardiovascular Angiography and interventions (SCAI). J Am Coll Cardiol. 2013;62(17):1563–70.
Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, White HD. Executive Group on behalf of the Joint European Society of Cardiology /American College of Cardiology /American Heart Association /World Heart Federation Task Force for the Universal Definition of Myocardial I: fourth universal definition of myocardial infarction (2018). J Am Coll Cardiol. 2018;72(18):2231–64.
Lansky AJ, Messé SR, Brickman AM, Dwyer M, van der Bart H, Lazar RM, Pietras CG, Abrams KJ, McFadden E, Petersen NH, et al. Proposed standardized neurological endpoints for Cardiovascular clinical trials: an Academic Research Consortium Initiative. Eur Heart J. 2018;39(19):1687–97.
Mehran R, Rao SV, Bhatt DL, Gibson CM, Caixeta A, Eikelboom J, Kaul S, Wiviott SD, Menon V, Nikolsky E, et al. Standardized bleeding definitions for cardiovascular clinical trials: a consensus report from the Bleeding Academic Research Consortium. Circulation. 2011;123(23):2736–47.
Bovill EG, Terrin ML, Stump DC, Berke AD, Frederick M, Collen D, Feit F, Gore JM, Hillis LD, Lambrew CT, et al. Hemorrhagic events during therapy with recombinant tissue-type plasminogen activator, heparin, and aspirin for acute myocardial infarction. Results of the Thrombolysis in myocardial infarction (TIMI), phase II trial. Ann Intern Med. 1991;115(4):256–65.
investigators G. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med. 1993;329(10):673–82.
Schulman S, Kearon C, Subcommittee on Control of Anticoagulation of the S, Standardization Committee of the International Society on T., Haemostasis: Definition of major bleeding in clinical investigations of antihemostatic medicinal products in non-surgical patients. Journal of thrombosis and haemostasis: JTH 2005, 3(4):692–694.
Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S, Neumann FJ, Ardissino D, De Servi S, Murphy SA, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357(20):2001–15.
Urban P, Mehran R, Colleran R, Angiolillo DJ, Byrne RA, Capodanno D, Cuisset T, Cutlip D, Eerdmans P, Eikelboom J, et al. Defining high bleeding risk in patients undergoing percutaneous coronary intervention. Circulation. 2019;140(3):240–61.
Valgimigli M, Garcia-Garcia HM, Vrijens B, Vranckx P, McFadden EP, Costa F, Pieper K, Vock DM, Zhang M, Van Es GA, et al. Standardized classification and framework for reporting, interpreting, and analysing medication non-adherence in cardiovascular clinical trials: a consensus report from the non-adherence Academic Research Consortium (NARC). Eur Heart J. 2019;40(25):2070–85.
He Y, Wang R, Liu J, Li F, Li J, Li C, Zhou J, Zhao Z, Yang W, Mou F, et al. A Randomized comparison of the Healing Response between the Firehawk Stent and the Xience Stent in patients with ST-Segment Elevation myocardial infarction at 6 months of Follow-Up (TARGET STEMI OCT China Trial): an optical coherence Tomography Study. Front Cardiovasc Med. 2022;9:895167.
Fan Y, Fezzi S, Sun P, Ding N, Li X, Hu X, Wang S, Wijns W, Lu Z, Tu S. In vivo validation of a Novel Computational Approach to assess Microcirculatory Resistance based on a single angiographic view. J Pers Med 2022, 12(11).
Xu B, Saito Y, Baumbach A, Kelbaek H, van Royen N, Zheng M, Morel MA, Knaapen P, Slagboom T, Johnson TW, et al. 2-Year clinical outcomes of an Abluminal groove-filled biodegradable-polymer sirolimus-eluting stent compared with a durable-polymer everolimus-eluting stent. JACC Cardiovasc Interventions. 2019;12(17):1679–87.
Gao C, Kogame N, Sharif F, Smits PC, Tonino P, Hofma S, Moreno R, Choudhury A, Petrov I, Cequier A, et al. Prospective Multicenter Randomized all-comers trial to assess the safety and effectiveness of the Ultra-thin Strut Sirolimus-Eluting Coronary Stent Supraflex: two-year outcomes of the TALENT trial. Circ Cardiovasc Interv. 2021;14(3):e010312.
Katagiri Y, Onuma Y, Lurz P, Buszman P, Piek JJ, Wykrzykowska JJ, Asano T, Kogame N, Takahashi K, Chang CC, et al. Clinical outcomes of bioabsorbable polymer sirolimus-eluting stents versus durable polymer everolimus-eluting stents: two-year follow-up of the DESSOLVE III trial. EuroIntervention: J EuroPCR Collab Working Group Interventional Cardiol Eur Soc Cardiol. 2020;15(15):e1366–74.
Buiten RA, Ploumen EH, Zocca P, Doggen CJM, Jessurun GAJ, Schotborgh CE, Roguin A, Danse PW, Benit E, Aminian A, et al. Thin composite-wire-strut zotarolimus-eluting stents versus ultrathin-strut sirolimus-eluting stents in BIONYX at 2 years. JACC Cardiovasc Interventions. 2020;13(9):1100–9.
Jeger RV, Farah A, Ohlow MA, Mangner N, Möbius-Winkler S, Leibundgut G, Weilenmann D, Wöhrle J, Richter S, Schreiber M, et al. Drug-coated balloons for small coronary artery disease (BASKET-SMALL 2): an open-label randomised non-inferiority trial. Lancet. 2018;392(10150):849–56.
Al Suwaidi J, Berger PB, Holmes DR Jr. Coronary artery stents. JAMA. 2000;284(14):1828–36.
Serruys PW, de Jaegere P, Kiemeneij F, Macaya C, Rutsch W, Heyndrickx G, Emanuelsson H, Marco J, Legrand V, Materne P, et al. A comparison of balloon-expandable-stent implantation with balloon angioplasty in patients with coronary artery disease. Benestent Study Group. N Engl J Med. 1994;331(8):489–95.
Kuntz RE, Safian RD, Carrozza JP, Fishman RF, Mansour M, Baim DS. The importance of acute luminal diameter in determining restenosis after coronary atherectomy or stenting. Circulation. 1992;86(6):1827–35.
Kuntz RE, Gibson CM, Nobuyoshi M, Baim DS. Generalized model of restenosis after conventional balloon angioplasty, stenting and directional atherectomy. J Am Coll Cardiol. 1993;21(1):15–25.
Rodriguez A, Ayala F, Bernardi V, Santaera O, Marchand E, Pardinas C, Mauvecin C, Vogel D, Harrell LC, Palacios IF. Optimal coronary balloon angioplasty with provisional stenting versus primary stent (OCBAS): immediate and long-term follow-up results. J Am Coll Cardiol. 1998;32(5):1351–7.
Weaver WD, Reisman MA, Griffin JJ, Buller CE, Leimgruber PP, Henry T, D’Haem C, Clark VL, Martin JS, Cohen DJ, et al. Optimum percutaneous transluminal coronary angioplasty compared with routine stent strategy trial (OPUS-1): a randomised trial. Lancet. 2000;355(9222):2199–203.
Serruys PW, de Bruyne B, Carlier S, Sousa JE, Piek J, Muramatsu T, Vrints C, Probst P, Seabra-Gomes R, Simpson I, et al. Randomized comparison of primary stenting and provisional balloon angioplasty guided by flow velocity measurement. Doppler endpoints Balloon Angioplasty Trial Europe (DEBATE) II study Group. Circulation. 2000;102(24):2930–7.
Cortese B, Bertoletti A. Paclitaxel coated balloons for coronary artery interventions: a comprehensive review of preclinical and clinical data. Int J Cardiol. 2012;161(1):4–12.
Madhavan MV, Kirtane AJ, Redfors B, Genereux P, Ben-Yehuda O, Palmerini T, Benedetto U, Biondi-Zoccai G, Smits PC, von Birgelen C, et al. Stent-related adverse events > 1 year after percutaneous coronary intervention. J Am Coll Cardiol. 2020;75(6):590–604.
Baan J Jr., Claessen BE, Dijk KB, Vendrik J, van der Schaaf RJ, Meuwissen M, van Royen N, Gosselink ATM, van Wely MH, Dirkali A, et al. A Randomized comparison of Paclitaxel-Eluting Balloon Versus Everolimus-Eluting Stent for the treatment of any In-Stent restenosis: the DARE Trial. JACC Cardiovasc Interventions. 2018;11(3):275–83.
Kleber FX, Schulz A, Waliszewski M, Hauschild T, Bohm M, Dietz U, Cremers B, Scheller B, Clever YP. Local paclitaxel induces late lumen enlargement in coronary arteries after balloon angioplasty. Clin Res Cardiol. 2015;104(3):217–25.
Cortese B, Silva Orrego P, Agostoni P, Buccheri D, Piraino D, Andolina G, Seregni RG. Effect of drug-coated balloons in native coronary artery Disease Left with a dissection. JACC Cardiovasc Interv. 2015;8(15):2003–9.
Tang Y, Qiao S, Su X, Chen Y, Jin Z, Chen H, Xu B, Kong X, Pang W, Liu Y, et al. Drug-coated balloon Versus Drug-Eluting Stent for Small-Vessel Disease: the RESTORE SVD China Randomized Trial. JACC Cardiovasc Interv. 2018;11(23):2381–92.
Latib A, Colombo A, Castriota F, Micari A, Cremonesi A, De Felice F, Marchese A, Tespili M, Presbitero P, Sgueglia GA, et al. A randomized multicenter study comparing a paclitaxel drug-eluting balloon with a paclitaxel-eluting stent in small coronary vessels: the BELLO (Balloon Elution and late loss optimization) study. J Am Coll Cardiol. 2012;60(24):2473–80.
Cortese B, Di Palma G, Guimaraes MG, Piraino D, Orrego PS, Buccheri D, Rivero F, Perotto A, Zambelli G, Alfonso F. Drug-coated balloon Versus Drug-Eluting Stent for Small Coronary Vessel Disease: PICCOLETO II randomized clinical trial. JACC Cardiovasc Interventions. 2020;13(24):2840–9.
Nishiyama N, Komatsu T, Kuroyanagi T, Fujikake A, Komatsu S, Nakamura H, Yamada K, Nakahara S, Kobayashi S, Taguchi I. Clinical value of drug-coated balloon angioplasty for de novo lesions in patients with coronary artery disease. Int J Cardiol. 2016;222:113–8.
Zhong PY, Ma Y, Shang YS, Niu Y, Bai N, Wang ZL. Efficacy of drug-coated balloon approaches for de novo Coronary Artery diseases: a bayesian network Meta-analysis. Front Cardiovasc Med. 2022;9:899701.
Acknowledgements
Not applicable.
Author information
Authors and Affiliations
Contributions
CG and XH drafted the manuscript. JL, ZJ, JX, DW provided statistical comments and contributed to the study design. YL, BZ, XQ, YX, TZ, PW, RZ provided consultations on study design. YO and PS provided key consultations on study design and concept. CG and LT designed the trial. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study will be conducted according to the principles outlined in the Declaration of Helsinki, the Ethical Guidelines for Medical and Health Research Involving Human Subjects, and all other applicable laws and regulations in China. All patients must provide written informed consent. The institutional review board at Xijing Hospital (KY20202070-C-1) and the other 43 participating centers in China have approved this study protocol.
Consent for publication
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Financial support
This trial is investigator-initiated and obtained grant support from Xijing Hospital (Xi’an, China; Grant No. XJZT24LY36) and unrestricted grant support from Shenqi Medical (Shanghai, China) and Microport Medical Group (Shanghai, China). Apart from this sponsorship, Shenqi and Microport were not involved in the design, execution, or decision to publish the study.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gao, C., He, X., Liu, Y. et al. Drug-coated balloon angioplasty with provisional stenting versus primary stenting for the treatment of de novo coronary artery lesions: REC-CAGEFREE I trial rationale and design. BMC Cardiovasc Disord 24, 319 (2024). https://doi.org/10.1186/s12872-024-03974-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12872-024-03974-0