
Abstract
Background
It is unclear whether genetic variants identified from single nucleotide polymorphisms (SNPs) strongly associated with coronary heart disease (CHD) in genome-wide association studies (GWAS), or a genetic risk score (GRS) derived from them, can help stratify risk of recurrent events in patients with CHD.
Methods
Study subjects were enrolled at the close-out of the LIPID randomised controlled trial of pravastatin vs placebo. Entry to the trial had required a history of acute coronary syndrome 3–36 months previously, and patients were in the trial for a mean of 36 months. Patients who consented to a blood sample were genotyped with a custom designed array chip with SNPs chosen from known CHD-associated loci identified in previous GWAS. We evaluated outcomes in these patients over the following 10 years.
Results
Over the 10-year follow-up of the cohort of 4932 patients, 1558 deaths, 898 cardiovascular deaths, 727 CHD deaths and 375 cancer deaths occurred. There were no significant associations between individual SNPs and outcomes before or after adjustment for confounding variables and for multiple testing. A previously validated 27 SNP GRS derived from SNPs with the strongest associations with CHD also did not show any independent association with recurrent major cardiovascular events.
Conclusions
Genetic variants based on individual single nucleotide polymorphisms strongly associated with coronary heart disease in genome wide association studies or an abbreviated genetic risk score derived from them did not help risk profiling in this well-characterised cohort with 10-year follow-up. Other approaches will be needed to incorporate genetic profiling into clinically relevant stratification of long-term risk of recurrent events in CHD patients.
Similar content being viewed by others
Introduction
Genome-wide association studies (GWAS) have identified large numbers of loci reliably associated with prevalent CHD [1]. Genetic risk scores (GRS) derived from SNPs which had the strongest associations with CHD improved the prediction of first events [2], but attempts to use a GRS for genetic profiling to predict recurrent events in established CHD have yielded conflicting findings. This may be because they have been statistically underpowered with short-term periods of follow-up [3]. While polygenic risk scores (PRS) including thousands of SNPs have been shown to assist with risk stratification, their value in clinical application remains uncertain [4]. Therefore, in the present study, we tested whether individual SNPs or an abbreviated, previously validated GRS derived from SNPs known to be strongly associated with CHD could be applied clinically to predict long-term cardiovascular outcomes.
Methods
The LIPID genetic cohort
The LIPID (Long-term Intervention with Pravastatin in Ischaemic Disease) trial (Trial Registration ACTRN12616000535471) comparing pravastatin 40 mg per day with placebo was conducted between 1992 and 1998 in 9014 patients. Entry to the trial required a history of acute coronary syndrome (ACS) 3–36 months previously, and patients were in the trial for a mean of 36 months. The results have been reported previously [5]. The cohort was followed for a total of 10 years from the close-out of the randomised controlled trial (RCT) [6, 7].
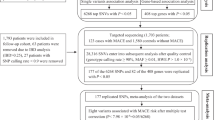
The LIPID Genetic cohort is described in Table 1. It included those patients alive at the end of the RCT in 1998, who had also given consent for collection of a blood sample and had high-quality DNA extracted. Whole blood was not available to enable DNA extraction from samples obtained at the time of patient randomisation. The LIPID Genetic cohort totalled 4932 patients. All fatal events were analysed in the 10 years of follow-up between 1997 and 2006, and all coronary events (fatal and non-fatal) in the first two years of cohort follow-up.
DNA extraction
DNA was extracted from whole blood samples from consenting patients at their close-out visit and stored at − 80 °C. The reasons for exclusion from the Genetic cohort included death during the trial (n = 1132), lack of consent for DNA extraction at close-out or DNA samples not of suitable quality for analysis (n = 2950).
Exploration and selection of SNPs
A literature review was undertaken using English language reports in PubMed to select SNPs for further exploration and was based on (1) SNPs with a significance of p < 5 × 10−8 in published GWAS reports of cardiovascular disease and (2) SNPs from known atherothrombotic pathways and other pathways related to rhythm and conduction disturbances, left ventricular dysfunction/cardiac failure and statin responsiveness. A custom designed Illumina GoldGate array of 384 SNPs with minor allele frequency (MAF) > 1% in European populations was used in this study (See Additional file 1: Table S1).
Exploration of a previously derived GRS
We explored the predictive value of a GRS derived by Mega et al. [8] This required an additional five SNPS to be included after amplification using Taq Man probe assays. In our testing of this GRS, we created a score for each patient by summing the number of risk alleles for each SNP weighted by the log of the ORs used by Mega et al. [8]. We created an unadjusted model and also used the same baseline variables as quoted in Mega et al. for multivariable adjustment.
Genotype quality control
Variants were excluded if they had a call rate < 95%, deviated substantially from Hardy–Weinberg equilibrium (p < 10−6), [9] or had a MAF of less than 1%. After quality control procedures, a total of 338 variants were available for analysis in all 4932 individuals.
Statistical analysis
Associations between SNPs and outcomes were assessed individually using adjusted proportional hazards regression models. The choice of potential covariates was based on our previous analyses which stratified risk for fatal as well as non-fatal outcomes [10]. SNPs that remained significant using a cut-off of p < 0.01 were reported. The Bonferroni method of adjusting for multiple comparisons suggested a cut-off of 0.0001. As no SNPs met the predetermined cut-off of p < 0.0001, the less conservative cut-off of 0.01 was used for retention of SNPs in the models.
The SNPs that were independent predictors for each cause of death in the LIPID data were used to create a risk score for each patient, applying the log of the hazard ratio from a model adjusted for clinical risk factors. The resulting scores were divided into quintiles and then the three middle groups were combined into one group. For the validation of the Mega et al. model, this procedure was repeated for CHD death using the SNPs and odds ratios previously reported in their manuscript [8].
Ethics
The LIPID trial was approved by the ethics committee at each participating site. All patients gave written informed consent for cohort follow-up, either in the clinic or remotely. The LIPID Genetic cohort study was approved by the Human Research Ethics Committee of Sir Charles Gairdner Hospital, Perth. The Long term follow-up of patients in the LIPID cohort study was approved by University of Sydney Human Research Ethics Committee Reference No: 01-2002/2454. The Genetic Cohort study was approved by The Human Research Ethics Committee of Sir Charles Gairdner Hospital, Perth HREC No 2011-060.
Patient involvement
Patients were not involved in the study design. Patient involvement in the study occurred at the time of informed consent, supervised by the Human Research Ethics Committee of each site.
Results
Characteristics of the study population
The baseline characteristics of the patients in the LIPID Genetic cohort (n = 4932) at the time of entry into the LIPID trial are summarised in Additional file 1: Table S2 and were similar to those of the full LIPID trial cohort [5,6,7].
Outcomes during follow-up
Table 2 shows cause-specific deaths in the 10-year follow-up of the LIPID Genetic cohort. There was a total of 1558 deaths, of which 898 were cardiovascular, including 727 related to CHD, and 375 due to cancer.
Association of individual SNPs with 10-year fatal outcomes in the LIPID Genetic cohort
The associations (unadjusted for baseline variables or multiple testing) of fatal outcomes over 10 years from the end of the double-blind phase of the RCT with the individual SNPs are presented in Table 2. After adjustment for baseline variables, and after further correction for multiple SNP testing, there were no statistically significant associations of individual SNPs with subsequent deaths. When testing for internal validation, the risk score for each patient was based on the hazard ratios discovered from our own data. In this internal validation, we found highly significant stratification of risk, including for deaths from cancer, which had an adjusted hazard ratio of 3.8 (95% confidence interval 2.59–5.57, p = 7.82 × 10−12) between the lowest and highest quintile (Table 3). The pattern for each of the 10-year fatal outcomes examined is displayed graphically in Fig. 1.

Plots of all-cause, coronary, cardiovascular, and cancer deaths over 10 years based on hazard ratios of risk for patients with high (top quintile), moderate (middle 3 quintiles) and low (lowest quintile) risk. Risk stratification derived from associations of SNPs with statistically significant hazard ratios with outcomes on unadjusted analyses
Prediction of 10-year risk of fatal CHD from a previously-derived GRS
The GRS described by Mega et al. [8] both unadjusted and after adjustment for the baseline risk factors listed in that publication, showed no statistically significant stratification for CHD death over 10 years. The results for CHD death are shown in Fig. 2. Before and after adjustment for baseline variables, the categories of risk based on each individual’s GRS did not distinguish between high (top quintile) moderate (middle 3 quintiles) and low (bottom quintile) risk of CHD death over 10 years (Table 4). The variables adjusted for are described in the Table.

Plot of coronary heart disease death over 10 years using the genetic risk score derived by Mega et al. [8]. High risk = top quintile, moderate risk = middle three quintiles, low risk = bottom quintile. Unadjusted for baseline variables
Two-year non-fatal outcomes
We also examined the value of the same SNPs for predicting non-fatal as well as fatal outcomes in the two years of open label follow-up, using a composite of CHD events (CHD death, non-fatal myocardial infraction, unstable angina, coronary artery bypass grafting and percutaneous coronary revascularisation) as the principal outcome measure, as described by Mega et al. [8] (Table 5).
Reclassification
When the 27 SNPs were added to a model with the baseline risk factors used by Mega et al. [8] there was moderate improvement in the net reclassification index (NRI) with a value of 0.097 and a very minor increase in the C-statistic from the ROC (receiver operating curve) curve from 0.69 to 0.70. When we added a history of prior CHD or prior MI (none, one or multiple) to the baseline model, with and without the SNPs, there were NRIs of 0.064 and 0.067, respectively (Table 5), and there was no significant change in the C-statistic.
Discussion
Our results do not support the hypothesis that individual SNPs strongly associated with prevalent or incident CHD on GWAS, or a previously validated 27-SNP GRS based on these SNPs [8] can predict long-term outcomes of patients with CHD who have had an ACS in the past.
The role of individual SNPs
In the early GWAS reports of associations of SNPs with CHD, p values < 10−10 were found for multiple SNPs, particularly the rs1333049 variant in the 9p21 gene [11]. We have significantly extended these previous observations by selecting a large number of other SNPs that have shown statistically strong associations with CHD or CHD pathways in previous GWAS reports. Our conclusion from this first part of our study is that selecting individual SNPs from strong associations with CHD in GWAS did not improve prediction of long-term cardiovascular risk in established CHD.
The role of a previously validated GRS based on 27 SNPs
We next tested a previously validated GRS for its potential for clinical application to improve risk prediction in known CHD patients [8]. Since the risk score for each patient was derived from the hazard ratios within the data set, it was expected that ranking of the risk scores would correlate strongly with cardiovascular risk, and this was indeed the case, even after adjustment for clinical variables. The strong correlation with cancer deaths was unexpected. However, a more stringent test of the role of a genetic influence on outcome is to test if an externally derived GRS is predictive.
GRS ranging from 19 to 300 SNPs [12] and more recently, PRS (Polygenic Risk Scores) of 50,000 [13] to 6 million SNPs [14] have been evaluated for their value in identifying risk of incident CHD. The larger panels have been shown to be superior to smaller scale scores in predicting events in people at high risk of incident CHD, but recent reports show only a modest improvement in prediction over clinical predictors [15].
GRSs developed for prediction of recurrent events in known CHD patients have been tested in smaller cohorts than the present study, less well characterised to enable full adjustment for confounding, or with shorter follow-up, and external validation has been infrequent [16]. Modest associations with recurrent events have been shown, but none have demonstrated clear-cut improvement in risk prediction in patients with established vascular disease [1, 16,17,18,19,20,21,22,23]. These are summarised in Table 6.
Because of the inherent appeal of a clinically applicable GRS with a limited number of SNPs we chose to evaluate the 27 SNP GRS which had been derived by Mega et al. [8] and which has been externally validated for predicting incident CHD [24]. This GRS was derived from large number of patients with established CHD including nearly 5000 who were in the CARE [25] and PROVE-IT [26] clinical trials of statin therapy. When the Mega et al. [8] score was applied to the LIPID cohort, it did not show any genetic contribution to prediction of recurrent CHD events or of fatal outcomes even before adjustment for clinical determinants of risk. We conclude from this second part of our study that there is a low likelihood of identifying CHD patients at high risk of recurrent events based on GRSs composed of an abbreviated SNP panel.
Limitations
There are several limitations of this study. The analyses are subject to recruitment bias which would be relevant if we were studying early survival after ACS but is less relevant for a longer-term study of a defined CHD cohort whose ACS was years distant.
We chose only one previously described GRS for validation. The GRS derived by Mega et al. [8] was the most relevant score for testing against outcomes in our cohort as it included patients with a similar clinical profile although the endpoints in the Mega GRS were only for the duration of the clinical trials. The Mega et al. database included primary prediction studies but also included 17,000 person years of follow-up in secondary prediction studies.
The reasons why this study of genetic polymorphisms of individual genes did not reveal an effect on the risk of recurrent events in patients with CAD remains unclear. Firstly, the sample size in this study may have been too small to detect a genetic effect on outcomes, but with a total of over 1500 deaths, this seems unlikely. Secondly, many of the clinical variables used for adjustment in the statistical models, are themselves subject to genetic influence, but it is striking that the lack of prediction by genetic variants was observed even before adjustment for clinical predictors. Finally, CHD, particularly when an ACS has occurred in the past, may simply be too complex a condition for genetics to influence survival.
It is important to recognise that our data do not exclude a genetic influence on survival in CHD patients. However, it is clear from these analyses that clinically applicable genetic profiling with single SNPs or a SNP-derived GRS with a limited number of highly selected SNPs did not add precision to the prediction of recurrent major CHD outcomes. It is conceivable that a polygenic risk score (PRS) with many thousands of SNPs will demonstrate a genetic influence on outcomes, but it remains to be established whether a PRS will have a clinically applicable role in enhancing the precision of recurrent event prediction beyond clinical markers of risk [27]. Further studies to clarify the genetic contribution to risk in established CHD will require the pooling of data from large numbers of individual cohorts [28], recognition of the limitations of GWAS [29] and possibly an omnigenic approach with exploration of regulatory genes undetected on GWAS [30].
Conclusion
In this large cohort of patients with CHD who had an ACS in the past, individual SNPs strongly associated with prevalent or incident CHD on GWAS, and a previously validated 27-SNP GRS based on these SNPs did not predict long-term outcomes.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request by emailing peter.thompson@health.wa.gov.au.
Abbreviations
- SNP:
-
Single nucleotide polymorphism
- GWAS:
-
Genome wide association study
- CHD:
-
Coronary heart disease
- ACS:
-
Acute coronary syndrome
- MI:
-
Myocardial infarction.
- GRS:
-
Genetic risk score
- PRS:
-
Polygenic risk score
- LIPID trial:
-
Long-Term Intervention with Pravastatin in Ischaemic Disease trial
- NRI:
-
Net reclassification index
- NHMRC:
-
National Health and Medical Research Council of Australia
- ROC:
-
Receiver operating curve
References
Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357(5):443–53.
Dainis AM, Ashley EA. Cardiovascular precision medicine in the genomics era. JACC Basic Transl Sci. 2018;3(2):313–26.
Labos C, Thanassoulis G. Genetic risk prediction for primary and secondary prevention of atherosclerotic cardiovascular disease: an update. Curr Cardiol Rep. 2018;20(5):36.
Dron JS, Hegele RA. The evolution of genetic-based risk scores for lipids and cardiovascular disease. Curr Opin Lipidol. 2019;30(2):71–81.
Long-Term Intervention with Pravastatin in Ischaemic Disease Study G. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med. 1998;339(19):1349–57.
Group LS. Long-term effectiveness and safety of pravastatin in 9014 patients with coronary heart disease and average cholesterol concentrations: the LIPID trial follow-up. Lancet. 2002;359(9315):1379–87.
Hague WE, Simes J, Kirby A, Keech AC, White HD, Hunt D, et al. Long-term effectiveness and safety of pravastatin in patients with coronary heart disease: Sixteen years of follow-up of the LIPID study. Circulation. 2016;133(19):1851–60.
Mega JL, Stitziel NO, Smith JG, Chasman DI, Caulfield M, Devlin JJ, et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet. 2015;385(9984):2264–71.
Stark AE. A clarification of the Hardy–Weinberg law. Genetics. 2006;174(3):1695–7.
Marschner IC, Colquhoun D, Simes RJ, Glasziou P, Harris P, Singh BB, et al. Long-term risk stratification for survivors of acute coronary syndromes. Results from the Long-term Intervention with Pravastatin in Ischemic Disease (LIPID) Study. LIPID Study Investigators. J Am Coll Cardiol. 2001;38(1):56–63.
Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106(23):9362–7.
Beaney KE, Ward CE, Bappa DA, McGale N, Davies AK, Hirani SP, et al. A 19-SNP coronary heart disease gene score profile in subjects with type 2 diabetes: the coronary heart disease risk in type 2 diabetes (CoRDia study) study baseline characteristics. Cardiovasc Diabetol. 2016;15(1):141.
Abraham G, Havulinna AS, Bhalala OG, Byars SG, De Livera AM, Yetukuri L, et al. Genomic prediction of coronary heart disease. Eur Heart J. 2016;37(43):3267–78.
Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50(9):1219–24.
Elliott J, Bodinier B, Bond TA, Chadeau-Hyam M, Evangelou E, Moons KGM, et al. Predictive accuracy of a polygenic risk score-enhanced prediction model vs a clinical risk score for coronary artery disease. JAMA. 2020;323(7):636–45.
Labos C, Martinez SC, Leo Wang RH, Lenzini PA, Pilote L, Bogaty P, et al. Utility of a genetic risk score to predict recurrent cardiovascular events 1 year after an acute coronary syndrome: a pooled analysis of the RISCA, PRAXY, and TRIUMPH cohorts. Atherosclerosis. 2015;242(1):261–7.
Christiansen MK, Nyegaard M, Larsen SB, Grove EL, Wurtz M, Neergaard-Petersen S, et al. A genetic risk score predicts cardiovascular events in patients with stable coronary artery disease. Int J Cardiol. 2017;241:411–6.
Jiang J, Zheng Q, Han Y, Qiao S, Chen J, Yuan Z, et al. Genetic predisposition to coronary artery disease is predictive of recurrent events: a Chinese prospective cohort study. Hum Mol Genet. 2020;29(6):1044–53.
Patel RS, Sun YV, Hartiala J, Veledar E, Su S, Sher S, et al. Association of a genetic risk score with prevalent and incident myocardial infarction in subjects undergoing coronary angiography. Circ Cardiovasc Genet. 2012;5(4):441–9.
Pereira A, Mendonca MI, Sousa AC, Borges S, Freitas S, Henriques E, et al. Genetic risk score and cardiovascular mortality in a Southern European population with coronary artery disease. Int J Clin Pract. 2017;71(6):e12956.
Vaara S, Tikkanen E, Parkkonen O, Lokki ML, Ripatti S, Perola M, et al. Genetic risk scores predict recurrence of acute coronary syndrome. Circ Cardiovasc Genet. 2016;9(2):172–8.
Weijmans M, de Bakker PI, van der Graaf Y, Asselbergs FW, Algra A, Jan de Borst G, et al. Incremental value of a genetic risk score for the prediction of new vascular events in patients with clinically manifest vascular disease. Atherosclerosis. 2015;239(2):451–8.
Wirtwein M, Melander O, Sjogren M, Hoffmann M, Narkiewicz K, Gruchala M, et al. Relationship between selected DNA polymorphisms and coronary artery disease complications. Int J Cardiol. 2017;228:814–20.
Tada H, Melander O, Louie JZ, Catanese JJ, Rowland CM, Devlin JJ, et al. Risk prediction by genetic risk scores for coronary heart disease is independent of self-reported family history. Eur Heart J. 2016;37(6):561–7.
Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med. 1996;335(14):1001–9.
Cannon CP, Braunwald E, McCabe CH, Rader DJ, Rouleau JL, Belder R, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350(15):1495–504.
Mosley JD, Gupta DK, Tan J, Yao J, Wells QS, Shaffer CM, et al. Predictive accuracy of a polygenic risk score compared with a clinical risk score for incident coronary heart disease. JAMA. 2020;323(7):627–35.
Patel RS, Tragante V, Schmidt AF, McCubrey RO, Holmes MV, Howe LJ, et al. Subsequent event risk in individuals with established coronary heart disease. Circ Genom Precis Med. 2019;12(4):e002470.
Tam V, Patel N, Turcotte M, Bosse Y, Pare G, Meyre D. Benefits and limitations of genome-wide association studies. Nat Rev Genet. 2019;20(8):467–84.
Boyle EA, Li YI, Pritchard JK. An expanded view of complex traits: From polygenic to omnigenic. Cell. 2017;169(7):1177–86.
Acknowledgements
We thank the LIPID study investigators and coordinators, patients and relatives at each of the participating centres, for their long commitment to the study.
Funding
The study was designed, conducted, analysed, and reported by the investigators and coordinated by the National Health and Medical Research Council (NHMRC) Clinical Trials Centre under the auspices of the National Heart Foundation of Australia, independently of funders of the study. The study drug (including placebo) and financial support for the LIPID randomised controlled trial were provided by Bristol-Myers Squibb (for the first 8 years). Subsequently, funding related to follow-up of the cohort was from NHMRC research grants 1037786 and 490968. The genetic analyses reported here were funded by NHMRC grant 1010279.
Author information
Authors and Affiliations
Contributions
PLT conceived, planned and supervised the conduct, analysis and write-up of the study. JH and JB conducted the GWAS literature review and selection of SNPs and design of the chip and conducted the DNA extraction and genetic analyses. LJP, GFW, MJW and DRS advised on interpretation of genetic aspects of the results. AK and AM conducted the data extraction, planned and conducted the statistical analyses with PLT and RJS. HDW, RS, RJS and AMT advised on the non-genetic aspects of the results and interpretation. All authors were involved in discussion of the results and reviewed and commented on the final manuscript. MJW was deceased at the time of final submission but reviewed near-final versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The LIPID trial was approved by the ethics committee at each participating site. All patients gave written informed consent for cohort follow-up, either in the clinic or remotely. The LIPID Genetic cohort study was approved by the Human Research Ethics Committee of Sir Charles Gairdner Hospital, Perth. The Long term follow-up of patients in the LIPID cohort study was approved by University of Sydney Human Research Ethics Committee Reference No: 01-2002/2454. The Genetic Cohort study was approved by The Human Research Ethics Committee of Sir Charles Gairdner Hospital, Perth HREC No 2011-060.
Consent for publication
All authors and participating patients consented to publication.
Competing interests
Some authors have received research grants, travel grants or honoraria from companies that might have an interest in the submitted work: Dr Thompson from Amgen, AstraZeneca, Bristol-Myers Squibb, Merck, Pfizer; Dr White from Sanofi and Regeneron Pharmaceuticals, Omthera Pharmaceuticals, American Regent, Eisai Inc, DalCor Pharma UK Inc, CSL Behring, Sanofi Australia Pty Ltd and Esperion Therapeutics Inc. He was on the Advisory Board for Genentech Inc. Dr Tonkin from Amgen, AstraZeneca, Bayer, Bristol-Myers Squibb, Merck, Pfizer and Sanofi-Aventis; the other authors report no conflicts.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
M. J. West: Deceased
Supplementary Information
Additional file 1:
SNPs included in the custom designed Illumina Gold Gate array of 384 SNPs with minor allele frequency (MAF) > 1 plus additional SNPs included in the panel derived by Mega et al [8].
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Thompson, P.L., Hui, J., Beilby, J. et al. Common genetic variants do not predict recurrent events in coronary heart disease patients. BMC Cardiovasc Disord 22, 96 (2022). https://doi.org/10.1186/s12872-022-02520-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12872-022-02520-0