
Abstract
Background
Fabry disease is caused by mutations in the α-galactosidase A (GLA) gene, which is located in X-chromosome coding for the lysosomal enzyme of GLA. Among many gene mutations, E66Q mutation is under discussion for its pathogenicity because there is no clinical report showing pathological evidence of Fabry disease with E66Q mutation.
Case presentation
A 65-year-old Japanese female was referred to our hospital for chest discomfort on effort. Transthoracic echocardiography showed severe left ventricular (LV) hypertrophy with LV outflow obstruction. Maximum LV outflow pressure gradient was 87 mmHg, and Valsalva maneuver increased the pressure gradient up to 98 mmHg. According to medical interview, one of her younger sister and a nephew died suddenly at age 42 and 36, respectively. Another younger sister also presented LV hypertrophy with outflow obstruction. Maximum LV outflow pressure gradient was 100 mmHg, and the E66Q mutation was detected similar to the case. Endomyocardial biopsy specimens presented vacuolation of cardiomyocytes, in which zebra bodies were detected by electron microscopic examination. Although the enzymatic activity of GLA was within normal range, the c. 196G>C nucleotide change, which lead to the E66Q mutation of GLA gene, was detected. We initially diagnosed her as cardiac Fabry disease based on the findings of zebra body. However, immunostaining showed few deposition of globotriaosylceramide in left ventricular myocardium, and gene mutations in the disease genes for hypertrophic cardiomyopathy (HCM), MYBPC3 and MYH6, were detected. Although the pathogenicity of the E66Q mutation cannot be ruled out, hypertrophic obstructive cardiomyopathy (HOCM) was more reasonable to explain the pathophysiology in the case.
Conclusions
This is the confusable case of HOCM with Fabry disease with the GLA E66Q mutation. We have to take into consideration the possibility that some patients with the E66Q mutation may have similar histological findings of Fabry disease, and should be examed the possibility for harboring gene mutations associated with HCM.
Similar content being viewed by others


Background
Fabry disease is caused by mutations in the α-galactosidase A (GLA) gene, which is located in X-chromosome coding for the lysosomal enzyme of GLA. To date, more than 500 gene mutations have been identified [1], and clinical manifestations are varied based on the location of the mutation and gender. Among many gene mutations, E66Q mutation is relatively common, especially in Asian people [2]. The E66Q mutation is thought to be a functional single-nucleotide polymorphism and not a disease-causing mutation, because residual enzymatic activity is relatively preserved and the mutation site is distant from the enzymatic active site [3]. Histological examination is helpful to diagnose Fabry disease, and zebra body is the strong evidence to suggest an accumulation of globotriaosylceramide (Gb3). In the present paper, we demonstrated confusable case of hypertrophic obstructive cardiomyopathy (HOCM) with cardiac Fabry disease associated with the E66Q mutation.
Case presentation
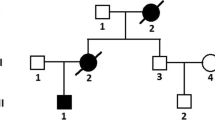
A 65-year-old female was referred to our hospital with chest discomfort on effort. On auscultation, a grade III/VI systolic murmur at the 4th left intercostal space was detected. Her blood pressure was 122/66 mmHg. A chest X-ray showed cardiomegaly (cardiothoracic ratio; 0.55) without pulmonary congestion, and an electrocardiogram showed regular sinus rhythm, poor R wave progression in leads V1 to V4, an inverted T wave, and left ventricular (LV) high voltage (SV1 + RV5 = 6.52 mV). She presented no angiokeratoma on her skin, and had no history of acroparesthesia. A medical interview revealed that her father, one of her younger sisters, and a nephew died suddenly at age 58, 42, and 36, respectively (Fig. 1). Another younger sister also presented with LV hypertrophy with outflow obstruction. Transthoracic echocardiography showed diffuse LV hypertrophy with more than 20 mm wall thickness and hyper contraction with LV outflow obstruction. The maximum LV outflow pressure gradient was 87 mmHg, and Valsalva maneuver increased the pressure gradient up to 98 mmHg (Fig. 2a-c). Moderate mitral valve regurgitation was detected due to systolic anterior motion of the anterior mitral leaflet. Laboratory data displayed preserved renal function and no proteinuria. Cardiac catheterization revealed a systolic pressure gradient between mid LV and outflow tract by 82 mmHg, and biopsy specimens presented vacuolation of cardiomyocytes, which were stained by periodic acid-Schiff (PAS) stain (Fig. 3a and b). Zebra bodies were detected by electron microscopic examination in the cells with vacuolation (Fig. 3c and d). Zebra body is a specific finding of Fabry disease, but is also found in other lysosome diseases, such as Niemann-Pick disease, Landing’s disease, Sandhoff’s disease, and mucopolysaccharidosis. The patient had no clinical findings consistent with those diseases. Cationic amphiphilic drugs, including gentamycin, hydroxychloroquine, and amiodarone, are capable of inducing phospholipidosis, leading to deposition of zebra bodies in various cells [4–6], but she had not taken any of these drugs. Although the enzymatic activity of leukocyte GLA was within normal range (62 nmol/mg/h), we initially diagnosed the case with cardiac Fabry disease with the E66Q mutation based on the histological findings. But because the distribution and the density of zebra body were much less than typical cases of Fabry disease, we added the immunostaining against Gb3 in the specimen of LV myocardium. As compared to a case of typical Fabry disease (Fig. 4a), there was few deposition of Gb3, even in vacuolated cells (Fig. 4b). We also analyzed plasma levels of Gb3, but it was within normal range (2.8 μg/ml). In contrast, when we analyzed the case for mutations in the disease genes for HCM [7, 8], it was revealed that the patient carried heterozygous mutations of MYBPC3 (Gly1009Val) and MYH6 (Ser624del). Although both mutations were novel, they were not found in the public sequence databases including dbSNP [9], 1000 genomes [10], and Human Genetic Variation database [11]. In addition, the MYBPC3 mutation was predicted to be a disease-causing mutation by three different in silico studies; predicted to be disease-causing by Mutation Taster [12], probably damaging (score 1.000) by PolyPhen-2 [13], and damaging (score 0) by SIFT [14]. As for the MYH6 mutation, one amino acid at 624th position in the head domain was deleted, which might be deleterious for the function of α-myosin heavy chain. However, because MYH6 is mainly expressed in the atrial muscles and not in the ventricular muscles in human adult hearts, pathological role of the MYH6 mutation in cardiac hypertrophy in this case might be less significant than the MYBPC3 mutation. From these observations, HOCM was more reasonable to explain the pathophysiology in the case, although the disease modifying effect of the E66Q mutation cannot be ruled out.

Pedigree of the case. The numbers indicate current age or age at death

Echocardiographic findings. a, b Echocardiographic images of para-sternal long axis view of LV showing diffuse cardiac hypertrophy at diastolic (a) and systolic (b) phases. Systolic anterior motion of anterior mitral leaflet was observed. c Measurement of the pressure gradient between mid-LV and LV outflow tract

Histological findings of left ventricular endomyocardial biopsy. a Vacuolated cardiomyocytes in the left ventricle (hematoxylin and eosin staining). Bar, 50 μm. b PAS stain-positive cardiomyocytes with vacuolation. Bar, 50 μm. c Zebra body in vacuolated cardiomyocytes. d Magnified image of C

Immunostaining of myocardium against Gb3. a Immunostaining of myocardium in typical Fabry disease’s case. Brown staining indicates Gb3 accumulation. Bar, 50 μm. b Immunostaining of myocardium in the present case. Bar, 50 μm
Several reports have already shown that E66Q mutation is accompanied with LV hypertrophy and/or renal dysfunction [15, 16], but histological evidence of Fabry disease has not yet been demonstrated. One report showed typical findings of classic-type Fabry disease with E66Q mutation, but the patient also had another R112C mutation [17]. In the present case, both PAS-positive vacuolated cardiomyocytes and small number of zebra bodies were observed in LV myocardium, but no accumulation of Gb3, indicating that those histological changes were not associated with Fabry disease. A recent study showed that the frequency of the E66Q allele was not different between hemodialysis patients and the general Japanese population [18]. Therefore, thorough genetic analysis is needed to perform differential diagnosis of Fabry disease. Although E66Q is considered as functional polymorphism, not pathogenic mutation, Nakamura et al. showed that the frequency of the E66Q mutation was significantly higher in patients with cerebral small vessel occlusion [19]. Thus, we need to accumulate the clinical data of patients with E66Q mutation in order to elucidate the pathological roles of E66Q mutation.
Conclusions
This is the confusable case of HOCM with Fabry disease. We have to take into consideration the possibility that some patients with the E66Q mutation may have similar histological findings of Fabry disease, and should be examined the possibility for harboring HCM-related gene mutations.
Consent
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal. The study was approved by the Ethics Committee of the Fukushima Medical University School of Medicine.
Abbreviations
- Gb3:
-
globotriaosylceramide
- GLA:
-
α-galactosidase A
- HCM:
-
hypertrophic cardiomyopathy
- HOCM:
-
hypertrophic obstructive cardiomyopathy
- LV:
-
left ventricle
- PAS:
-
periodic acid-Schiff
References
Nagueh SF. Anderson-Fabry disease and other lysosomal storage disorders. Circulation. 2014;130:1081–90.
Lee BH, Heo SH, Kim GH, Park JY, Kim WS, Kang DH, et al. Mutations of the GLA gene in Korean patients with Fabry disease and frequency of the E66Q allele as a functional variant in Korean newborns. J Hum Genet. 2010;55:512–7.
Togawa T, Tsukimura T, Kodama T, Tanaka T, Kawashima I, Saito S, et al. Fabry disease: Biochemical, pathological and structural studies of the α-galactosidase A with E66Q amino acid substitution. Mol Genet Metab. 2012;105:615–20.
Pintavorn P, Cook WJ. Progressive renal insufficiency associated with amiodarone-induced phospholipidosis. Kidney Int. 2008;74:1354–7.
Reasor MJ, Kacew S. Drug-Induced phospholipidosis: are there functional consequences? Exp Biol Med. 2001;226:825–30.
Albay D, Adler SG, Philipose J, Calescibetta CC, Romansky SG, Cohen AH. Chloroquine-induced lipidosis mimicking Fabry disease. Mod Pathol. 2005;18:733–8.
Kimura A. Molecular genetics and pathogenesis of cardiomyopathy. J Hum Genet. 2016;61:41–50.
Otsuka H, Arimura T, Abe T, Kawai H, Aizawa Y, Kubo T, et al. Prevalence and distribution of sarcomeric gene mutations in Japanese patients with familial hypertrophic cardiomyopathy. Circ J. 2012;76:453–61.
dbSNP. http://www.ncbi.nlm.nih.gov/snp. Accessed 24 Mar 2016.
1000 genomes. http://www.1000genomes.org/. Accessed 24 Mar 2016.
Human Genetic Variation database. http://www.genome.med.kyoto-u.ac.jp/SnpDB/. Accessed 24 Mar 2016.
Mutation Taster. http://www.mutationtaster.org/. Accessed 24 Mar 2016.
PolyPhen-2. http://genetics.bwh.harvard.edu/pph2/. Accessed 24 Mar 2016.
SIFT. http://sift.jcvi.org/. Accessed 24 Mar 2016.
Fujii H, Kono K, Goto S, Onishi T, Kawai H, Hirata K, et al. Prevalence and cardiovascular features of Japanese hemodialysis patients with Fabry disease. Am J Nephrol. 2009;30:527–35.
Kikumoto Y, Sugiyama H, Morinaga H, Inoue T, Takiue K, Kitagawa M, et al. The frequency of Fabry disease with the E66Q variant in the α-galactosidase A gene in Japanese dialysis patients: a case report and a literature review. Clin Nephrol. 2012;78:224–9.
Ishii S, Sakuraba H, Suzuki Y. Point mutations in the upstream region of the alpha-galactosidase A gene exon 6 in an atypical variant of Fabry disease. Hum Genet. 1992;89:29–32.
Watanabe H, Goto S, Miyashita A, Maruyama H, Wakasugi M, Yokoseki A, et al. Role of the p.E66Q variant of GLA in the progression of chronic kidney disease. Clin Exp Nephrol. 2015;19:225–30.
Nakamura K, Sekijima Y, Nakamura K, Hattori K, Nagamatsu K, Shimizu Y, et al. p.E66Q mutation in the GLA gene is associated with a high risk of cerebral small-vessel occlusion in elderly Japanese males. Eur J Neurol. 2014;21:49–56.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MO performed instrumental, clinical analyses and patient care; NS performed instrumental and clinical analyses; AKo performed the echocardiography; SS performed the cardiac catheterization; AY coordinated the study and obtained the informed consent and ethics approval to conduct the study; TY practiced patient care; KN participated in acquisition and interpretation of data; HS participated in acquisition and interpretation of data; SS participated the review of the manuscript; YK performed the histological analysis; HN carried out the genetic analysis; TH carried out the genetic analysis; AKi carried out the genetic analysis; YT participated in the review of the manuscript. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Oikawa, M., Sakamoto, N., Kobayashi, A. et al. Familial hypertrophic obstructive cardiomyopathy with the GLA E66Q mutation and zebra body. BMC Cardiovasc Disord 16, 83 (2016). https://doi.org/10.1186/s12872-016-0262-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12872-016-0262-y