
Abstract
Sugars will be eventually effluxed transporters (SWEETs) have been confirmed to play diverse physiological roles in plant growth, development and stress response. However, the characteristics and functions of the SWEET genes in Hemerocallis citrina remain unclear and poorly elucidated. In this study, the whole genome of Hemerocallis citrina was utilized to conduct bioinformatics analysis and a total of 19 HcSWEET genes were successfully identified. Analysis of the physicochemical properties indicated dominant differences among these HcSWEETs. A phylogenetic analysis revealed that HcSWEET proteins can be divided into 4 clades ranging from Clade I to IV, where proteins within the same clade exhibited shared conserved motifs and gene structures. Five to six exons were contained in the majority of HcSWEET genes, which were unevenly distributed across 11 chromosomes. The gene duplication analysis showed the presence of 4 gene pairs. Comparative syntenic maps revealed that the HcSWEET gene family might present more closed homology in monocotyledons than dicotyledons. Cis-acting element analysis of HcSWEET genes indicated key responsiveness to various hormones, light, and stresses. Additionally, transcriptome sequencing analysis suggested that most HcSWEET genes had a relatively higher expression in roots, and HcSWEET4a was significantly up-regulated under salt stress. Overexpression further verified the possibility that HcSWEET4a was involved in response to salt stress, which provides novel insights and facilitates in-depth studies of the functional analysis of HcSWEETs in resistance to abiotic stress.
Similar content being viewed by others

Background
Growth and development processes are often severely affected when plants are exposed to various abiotic stresses such as drought and salinity due to their sessile characteristics [1, 2]. Sugars, as major sources of carbon skeletons for cell wall and energy metabolism, are crucial signals that regulate the expression of related genes under abiotic stress [3, 4]. Cell-to-cell transport of sugars is mediated by transport proteins. Thus far, three transporters have been identified, including MSTs (monosaccharide transporters), SUTs (sucrose transporters), and SWEETs (sugars will eventually be exported transporters) [5, 6]. Unlike two other transporters, the SWEETs can typically act as bidirectional transmembrane transporter for sugars to cross the biomembrane system along a pH-independent concentration gradient via MtN3/saliva structural domains [7]. Members of the SWEET gene family encode membrane proteins consisting of conserved transmembrane (TM) domains linked by a PQ-loop-repeat, which are conserved across eukaryotes and prokaryotes [8, 9]. However, Eukaryotic SWEET proteins generally contain seven transmembrane helices (TMHs), obviously distinguishable from semiSWEETs consisting of 3 TMHs in prokaryotes, which may be correlated to the evolution process of plants [10, 11].
As a transporter identified about sugars recently, the SWEETs are found to be present in all kingdoms of life [5, 9]. SWEET proteins play central roles in plant growth and development such as plant defense, long-distance transport of sucrose, vegetative and reproduction growth, senescence, and stress responses [12,13,14]. Currently, SWEET proteins have been reported to form four clades (Clade I, II, III and IV) in several plants, furthermore genes belonging to the same clade hold similar structures and functions [8, 15]. Considering the physiological relevance of the fertility of sucrose, the AtSWEET13 sucrose transporter contributes to male fertility restoration [16]. Vacuolar Sugar Carrier AtSWEET16 overexpression lines promote stress tolerance under non-favorable conditions [17]. OsSWEET11 and OsSWEET15 participate in seed development in rice [18]. Clade III OsSWEETs, which are taken as interaction factors, mediate bacterial blight susceptibility by interacting with OsHMGB1 and OsHsp20L to breed broad-spectrum disease-resistant rice [19]. SlSWEET7a and SlSWEET14 negatively regulate the sugar transport and storage of fruits in tomatoes [20]. MdSWEET9b positively regulates sugar accumulation in apples, providing the foundation for exploring the metabolism between hormone and sugar pathways [21]. GmSWEET15 in soybean mediates sucrose export from the endosperm to the early embryo seed development [22]. CsSWEET17 is activated to positively regulate cold tolerance by interacting with CsLHY in tea plants [23]. Pineapple SWEET10 functions as a potential glucose transporter, and fruit crop yield and quality may be improved by manipulating the glucose transport activity of AcSWEET10 [24]. SWEET11b transports both sugar and cytokinin to maintain normal development in barley grains [25].
Cell membrane transfers chemical signals to the cytoplasm when plants receive stress signals under harsh environmental conditions. Interestingly, for most stresses, the prior exposure can strengthen plants tolerance during subsequent exposures [26]. In the long term, environmental stresses like drought and salinity can activate stress-related response genes of plants to adjust to osmotic pressure [27,28,29,30]. Therefore, innovative solutions are inevitable to develop to mitigate soil salinity and drought stress. Sucrose transport, a key physiological process, is impacted by abiotic stresses [31]. Overexpression of OsMST6 confers improved resilience to drought and salt stress in Arabidopsis thaliana [32]. Up-regulation of SUTs may be necessary to maintain phloem transport despite decreased sucrose available under salt and drought stresses in plants [33]. OsSUTs show significant importance in coping with drought and salt stress for rice plants [34]. The increase of soluble sugar contents and tolerances to both drought and salt stress are enhanced by elevating the production of trehalose in OsTPSP overexpression lines [35]. OsSWEET13 and OsSWEET15 transporters of Clade III pose critical importance of sucrose translocation and distribution in adaptive responses of rice plants to abiotic stresses [31].
Night lily (Hemerocallis citrina Borani) is one of the nutritionally, medicinally, and ornamentally important horticultural perennial crops cultivated worldwide. However, the lack of the night lily genome restrains the progression that molecular mechanisms and gene function analysis are explored. Therefore, as genomic information was released in 2021, new insights will be provided for a deeper understanding of the evolution, development, and biological functions of night lily [36]. As previous results reported, the SWEET gene family has become the research focus on responsiveness to abiotic stress. To investigate how night lily responds to drought and salt stress, we performed genome-wide identification of the SWEET members. In this study, members of the HcSWEET gene family were identified at the genomic level, followed by physicochemical properties, phylogenetic trees, conserved motifs, gene structures, chromosomal positions, cis-acting elements, and collinearity relationships were comprehensively analyzed using bioinformatics methods. Additionally, tissue-specific expression patterns of HcSWEET genes, as well as relative expression levels of genes under drought and salt treatments, were analyzed by transcriptome sequencing (RNA-seq). And that RNA-seq data from stress treatments were validated by RT-qPCR. The function of HcSWEET4a in stress response was further investigated by the Agrobacterium-mediated watermelon transformation method. The research can serve as a promising reference for offering strategies for improving tolerance to abiotic stress in night lily.
Results
Identification and characterization of the SWEET gene family in night lily
A total of 19 genes that encoded HcSWEET proteins were identified after homology alignment and conservative domain verification. These genes were named HcSWEET1 to HcSWEET16 according to their homology with dicotyledons such as Arabidopsis AtSWEETs and monocotyledons such as rice OsSWEETs. HcSWEET gene characteristics of open reading frame (ORF) size, number of amino acid (AA/aa), protein molecular weight (MW), theoretical isoelectric point (pI), instability index (II), aliphatic index (AI), grand average of hydropathicity (GRAVY), number of predicted TMHs and conserved domain position were analyzed in Table 1. As a consequence, the deduced number of amino acids in the night lily SWEET proteins was from 229 to 299, with the MW varying widely from 25.617 kDa to 32.938 kDa, and the PI index was between 5.49 and 9.66. All proteins ranged from 27.08 to 42.19 on the instability index, from 105.08 to 133.65 on the aliphatic index, and from 0.419 to 0.934 on GRAVY. The majority of predicted TMHs number was seven other than 6 TMHs of HcSWEET5 and HcSWEET13a/d. Coupled with subcellular localization, the secondary structure including α-helix, extended strand, β-turn and random coil were summarized in Supplementary Table S1. Subcellular localization predictions suggested that the majority of HcSWEET genes located in the plasma membrane, were also found in the chloroplast, vacuole or cytoplasm (Supplementary Table S1). Considering the transmembrane characteristic, we predicted 3D structural models of 19 HcSWEET proteins using a homology modeling approach, shown in Supplementary Figure S1. The results demonstrated that various properties existed in different HcSWEET members.
Phylogenetic analysis of HcSWEET proteins
To better understand the evolutionary interrelatedness among these HcSWEET members, we constructed a phylogenetic tree containing SWEETs from six species (Hemerocallis citrina, Hemerocallis fulva, Oryza sativa, Zea mays, Arabidopsis thaliana, Vitis vinifera) based on amino acid sequences using the TBtools software (Fig. 1). SWEET protein sequences of six species were listed in Supplementary Information 1. The results revealed that 19 HcSWEET members were classified into four major clades, namely Clade I, II, III, and IV respectively. Multiple HcSWEETs were tightly clustered with other SWEETs. Among these clades, the member number varied significantly. Clade III contained the most HcSWEET members (9), followed by Clade II (5), I (4), and IV (1), indicating a similar distribution pattern and gene function to these of other species.

Phylogenetic tree analysis and classification of the SWEETs in night lily and some other plants. Gene members of Hemerocallis citrina (Hc), Hemerocallis fulva (Hf), Oryza sativa (Os), Zea mays (Zm), Arabidopsis thaliana (At), Vitis vinifera (Vv) were classified into 4 clades. A phylogenetic tree was constructed via the TBtools software with 5000 bootstrap replicates
Conserved motif composition, domain, and gene structure analysis of HcSWEET genes
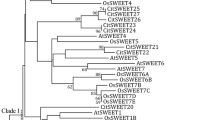
According to the results from phylogenetic analysis, we divided the identified 19 HcSWEET proteins into 4 clades (Fig. 2A). Ten different conserved motifs were identified by analyzing amino acid sequences of HcSWEET proteins using the MEME online tool (Fig. 2B). The number and distribution of these motifs showed variations in different proteins. Each protein had 5–7 motifs, in which Motif 1, 3, and 4 were found in all proteins, moreover, different clades had distinct motifs. Motif analysis suggested that the HcSWEET gene family showed both conservation and differences, which might result from the diversity of protein functions within clades. Conserved domain analysis revealed that HcSWEET proteins possessed domains consisting of MtN3_slv and MtN3_slv superfamily (Fig. 2C), implying high conservation during evolution. In addition, to understand the structural compositions of HcSWEET genes, a structural map including the untranslated region (UTR) and coding sequence (CDS) was constructed based on the genome sequence (Fig. 2D). The number of CDSs ranged from 5 to 6. Coding sequences of 19 HcSWEETs were displayed in Supplementary Information 2. Gene structure analysis suggested that members within the same evolutionary clade exhibited similar gene structures.

Constructed phylogenetic tree, conserved motif, conserved domain, and gene structure analysis of SWEETs in Hemerocallis citrina. (A) Intraspecific evolutionary tree of 19 HcSWEET members. (B) The compositions and distribution of HcSWEETs conserved motifs. (C) The conserved functional domains of HcSWEETs. (D) Gene structures of HcSWEET genes
Chromosomal localization, gene duplication, and collinearity analysis of HcSWEETs in Hemerocallis citrina, Arabidopsis thaliana and Oryza sativa
To investigate the chromosomal localization of HcSWEET genes, we analyzed the distribution of identified 19 genes on night lily chromosomes (Fig. 3). The distributions of these genes were determined via the night lily genome reported in 2021 [36]. The results showed that 19 genes were evenly dispersed across 9 of 11 chromosomes (LGs) with gene counts ranging from 1 to 4 in total. Notably, the largest number of genes were located in LG2, in contrast, LG6 and LG8 hosted only one gene and no gene was located in LG7 and LG9.

Chromosome distributions of SWEET genes on night lily (LG1-11). All HcSWEET genes named are displayed on night lily LGs, with the LG number marked at the edge of each strip. The 0-500 Mb scale represents chromosome length. The lines inside the LGs represent gene density
To set forth whether gene duplication events forced the evolution of the HcSWEET gene family, an intra-species collinear analysis was performed (Fig. 4A). Among all gene pairs of the night lily genome, four pairs of duplicated segments were identified in the HcSWEETs, implying that segmental duplication events were likely to contribute to genetic diversity. Subsequently, we constructed inter-species syntenic maps of Hemerocallis citrina with other two representative species including monocotyledon Oryza sativa and dicotyledon Arabidopsis thaliana to identify orthologous genes through utilizing TBtools (Fig. 4B). It was observed that seven genes exhibited collinearity relationships with two genes in A. thaliana and ten genes in O. sativa. These results suggest that HcSWEET genes likely share more similarities in function with OsSWEET genes in O. sativa.

Collinearity of HcSWEET gene pairs. (A) Chromosome location and inter-chromosomal relationships of SWEETs in Hemerocallis citrina. The identified duplication events are marked by red lines. The LG number is labeled within different colored rectangles. (B) Collinearity analysis between the SWEETs of Hemerocallis citrina (H. citrina), Arabidopsis thaliana (A. thaliana), and Oryza sativa (O. sativa)
Identification and distribution of cis‑regulatory elements in HcSWEET promoter
We specifically extracted 2000 base pairs upstream of the HcSWEET genes transcription start codon to explore their potential biological functions through the database named PlantCARE online. As shown in Fig. 5, a total of three types of main responsive elements were identified, hormone responses, plant growth and development, and responses to abiotic stress respectively. Among them, 6 to 26 light-responsive elements were found in 19 genes, indicating that the majority of HcSWEET genes may be regulated by light signaling. Moreover, all 19 genes contained hormone-related and development-related cis-acting elements. Hormone-related elements were referred to as MeJA, salicylic acid, abscisic acid, auxin, and gibberellin response elements. In contrast, development-related elements contained zein metabolism regulation, anaerobic induction, cell cycle regulation, seed-specific regulation, circadian control, flavonoid biosynthesis, endosperm, and meristem expression, anoxic specific inducibility and phytochrome down-regulation response elements. Additionally, HcSWEET genes other than HcSWEET4c, HcSWEET13d, and HcSWEET14b had cis-acting elements linked to abiotic stress, involving defense and stress, drought, and low-temperature responsive elements. HcSWEET13d also contained the wound-responsive element. Overall, various types of cis-elements were contained in the promoter regions of 19 HcSWEETs, indicating their possible involvement in diverse biological processes, environmental stresses, and regulatory pathways.

Distributions of cis-acting elements of the HcSWEET promoter
Expression patterns of HcWEET genes across different tissues and phenotype observations under drought and salt stress
To further analyze the possible roles of HcSWEET genes in abiotic stress, 19 genes were analyzed for relative expression profiles in different tissues and developmental stages (tender root, mature root, bud, tender leaf, mature leaf, tender scape, and mature scape) (Fig. 6A). As listed in Supplementary Table S3, RNA-seq results of tissue-specific expression revealed that most HcSWEETs in Clade II (HcSWEET4a, HcSWEET4b, HcSWEET5) and III (HcSWEET13c, HcSWEET13d, HcSWEET14a, HcSWEET14b, HcSWEET14c, HcSWEET15) exhibited the higher transcript levels in roots than the other organs. Among them, HcSWEET4a and HcSWEET13c were simultaneously in a related highly expression in the scape. Except these genes, HcSWEET2 in Clade I was expressed especially and abundantly in roots throughout the developmental process.
The transporters of the SWEET gene family in Arabidopsis thaliana have been reported to increase root proliferation by enhancing sucrose transport from shoot to root during abiotic stress [37]. Therefore, we carried out stress experiments to determine whether the case was suitable for HcSWEET genes in night lily. We treated 30 d seedling roots with drought stress for 0 h, 24 h, 48 h, 72 h, and 108 h, and identically salt stress for 0 h, 24 h, 48 h, 72 h, and 96 h. As was displayed in Fig. 6B and C, night lily plants primarily showed a slight symptom of loss of water when treated for 24 h. After treatment for 48 h, leaves presented severe yellow and rotten roots were generated.

Expression dynamics of the HcSWEET genes in different tissues of night lily and phenotypes under different abiotic stresses. (A) Expression heatmap of HcSWEETs in the tender root, mature root, bud, tender leaf, mature leaf, tender scope, and mature scope. (B, C) The phenotypic observations of night lily under drought stress (B) and salt stress (C)
Expression pattern analysis of HcSWEETs under drought and salt stress
Based on the phenotypes observed in night lily plants treated by drought and salinity, roots were collected, followed by RNA-Seq analysis (Supplementary Table S4 and S5). Depicted in Fig. 7A, expressions of genes including HcSWEET1b, HcSWEET4a, HcSWEET4b, HcSWEET5, HcSWEET13b, HcSWEET13c, and HcSWEET13d were induced compared with the control group (0 h) under drought stress. On the contrary, HcSWEET2, HcSWEET13a and HcSWEET14a expressed in a down-regulation trend. Relative expression levels of two genes (HcSWEET1a, HcSWEET14b) first increased and then decreased, while HcSWEET14c exhibited a contrary expression pattern.
We further investigated expression levels of HcSWEETs under salt treatment (Fig. 7B). Five HcSWEET genes (HcSWEET4a, HcSWEET5, HcSWEET11, HcSWEET13c, HcSWEET13d) showed significantly elevated expression levels. By contrast, the expressions of HcSWEET2, HcSWEET6, HcSWEET13b, and HcSWEET14b increased during different salt stresses. After salt treatments, there were 4 genes (HcSWEET1b, HcSWEET14a, HcSWEET14c, and HcSWEET16) initially up-regulating, subsequently decreasing again in expression levels. HcSWEET4c, HcSWEET13a, HcSWEET15 went down and then up. The various expression types showed that functional differences were likely to exist in HcSWEET genes in response to abiotic stresses.

Expression heatmaps of HcSWEETs under drought (A) and salt (B) stress based on RNA-seq data
To unveil the potential functions of HcSWEET genes in responding to different stresses including drought and salinity, nine HcSWEET genes showing significant responses were selected and RNA-Seq results were further confirmed using RT-qPCR experiments (Fig. 8). HcSWEET4a, HcSWEET4b, and HcSWEET5 showed an up-regulation in expressions under two stresses, notably, the expressions of HcSWEET4a and HcSWEET5 significant increased after salt stress, suggesting the potential involvement of HcSWEET4a and HcSWEET5 in salt stress response. HcSWEET2 exhibited a pattern of decreased expression compared with 0 h. Given whole wavelike patterns, the expressions of HcSWEET1a, HcSWEET4a, HcSWEET4b, HcSWEET5, and HcSWEET13c had a sharp increase at the time point of 24–72 h, but decreased in 96–108 h throughout stresses. Overall, our analysis revealed that expression trends of 19 genes were consistent with those of the RNA-Seq and the majority of HcSWEETs presented a dynamic response pattern.

Expression patterns of 19 HcSWEET genes after drought and salt treatment. The different lowercase letters indicate significant differences by t-test (p < 0.05)
Subcellular localization of HcSWEET4a and HcSWEET5
HcSWEET4a and HcSWEET5 were predicted to function in the plasma membrane (Supplementary Table S1). To evaluate the validity of results, we performed co-expressed experiments in N. benthamiana leaf cells (Fig. 9). The fusion constructs pSuper: HcSWEET4a-GFP and pSuper: HcSWEET5-GFP were co-expressed with the plasma membrane marker (mCherry). Excluding empty vectors pSuper-GFP, GFP fluorescent signals were all exclusively observed in the plasma membrane. Results indicated that HcSWEET4a and HcSWEET5 belonged to plasma membrane-localized proteins.

Subcellular localization of HcSWEET4a and HcSWEET5. pSuper: GFP represents the negative control. The second panel represents a positive marker for the plasma membrane. The rightmost panel indicates the fusion of green fluorescence, red fluorescence, and bright field. Bar = 25 μm
Overexpression of HcSWEET4a in watermelon and response to salt stress
Previous experiments have implied that both HcSWEET4a and HcSWEET5 exhibited significant up-regulation under salt stress. To further interpret the function of HcSWEETs in response to salt stress. We ectopically overexpressed alternative HcSWEET4a in watermelon through Agrobacterium-mediated transformation. Two transgenic lines (OE-1, OE-2) were obtained and expression levels were 3.67–4.18 times higher than the wild-type (WT) watermelon plant (Fig. 10B). Under NaCl treatment (150 mM) for 24 h, transgenic plants exhibited slightly curled leaf margins, while WT plants have been almost wilted (Fig. 10A). Phenotype observations further showed the up-regulation of HcSWEET4a might improve tolerance to salinity of plants.

Phenotype observations of HcSWEET4a overexpression lines under salt treatment. (A) Phenotype changes of transgenic lines (OE-1, OE-2) after treated for 0–24 h with NaCl (150 mM). Bar = 17 mm. (B) The relative expression levels of HcSWEET4a in OE-1 and OE-2 lines. The significance of difference is marked by two asterisks (p < 0.05)
Discussion
Hemerocallis citrina, belonging to Liliaceae Hemerocallis, is an important specialty crop widely cultivated around the world. With continuously expanded consumption patterns, night lily tends to be a model vegetable species for understanding deeply of perennial plant growth and development [38, 39]. Thereby, its tolerance to abiotic stress has become one of most key elements which influence production and quality. SWEET genes are plant-specific and taken as an essential class of sugar transporters in multiple plants [14, 40]. No studies were reported on the genome-wide identification of HcSWEETs or their function validation in perennial night lily previously. The present study investigated the response patterns of HcSWEET genes through genome-wide and expression analysis under drought and salt stress, aiming at promising new insights into enhancing stress resistances of night lily.
The study isolated 19 functional HcSWEETs successfully (Table 1), whose characteristics were similar to those reported in other plants especially HfSWEETs [41]. Secondary (Supplementary Table S1) and tertiary (Supplementary Figure S1) structure of HcSWEET proteins were predicted, revealing potential functions and interactions between proteins [42]. HcSWEETs fell into four phylogenetic clades by bioinformatics analysis, which were uniform with HfSWEETs, AtSWEETs, VvSWEETs, and LcSWEETs [5, 41, 43, 44]. There are not only similarities but also discrepancies in the members’ number of the same clade, for instance, as was shown in (Fig. 1), nine members were found in Clade III contrast sharply to only one in AtSWEET and OsSWEET [45,46,47,48]. Phylogenetic analyses were supported by analyses of conserved motifs and gene structures. Conserved motif analysis indicated that most HcSWEETs harbored seven conserved transmembrane domains (TMDs), while genes within each clade also shared more unique motifs (Fig. 2A-B). Above conclusion is consistent with Arabidopsis thaliana, Solanum lycopersicum, and Oryza sativa [5, 49, 50]. Gene structure model demonstrated that HcSWEETs within each clade were similar in an exon-intron organization (Fig. 2D). For instance, each member of Clade I, III (except HcSWEET13d), and IV harbors 6 exons, whereas genes in Clade II contain 5 exons. Previous relative report in Oryza sativa indicated the loss-of-exon rate is larger than the gain-of-exon rate after undergoing segmental duplication [51]. Therefore it is proposed that all clades except Clades II might contain the original ancestor HcSWEET genes, and genes in Clade II might result from gene duplication and divergence.
HcSWEET genes were uniformly located in 9 chromosomes (Fig. 3). Intra-species collinear analysis identified four pairs of duplicated segments (Fig. 4A), and inter-species syntenic maps identified seven homologous genes in Arabidopsis thaliana but two in Oryza sativa (Fig. 4B). Therefore, we speculated that tandem or segmental duplication might result in species evolution by gene family expansion, consistent with other plant species reported [9, 48, 52]. The promoters of HcSWEETs mainly contain stress response elements, growth and development-related elements, hormone response elements (Fig. 5), suggesting that HcSWEET genes are indispensably in the process of various biological activities and coping with abiotic stresses of night lily. At the same time, some previous reports establish a theoretical foundation for further exploring gene functions of HcSWEETs [53,54,55,56,57,58,59,60,61,62].
SWEETs in the same or different species may have different cellular localizations [5, 17, 63]. AtSWEET4 in Arabidopsis is localized in the plasma membrane, mediating sugar transport to axial sinks as a hexose facilitator [64]. OsSWEET4 and ZmSWEET4c are located in the basal endosperm transfer layer membrane, transferring hexoses across BETL to sustain the development of large endosperm in cereal grains [65]. SWEET4 in grapevine as a glucose transporter, was also identified as a plasma membrane protein [44]. Sugar transporter gene PbSWEET4 in pear, localizing in the plasma membrane, causes the reduction of sugar content and leads to early senescence of leaves [66]. In our present study, HcSWEET4a and HcSWEET5, falling in Clade II, were localized in the plasma membrane (Fig. 9). AtSWEET11 and close paralog AtSWEET12 localize to the plasma membrane of the phloem, suggesting the same localization also may exist in different clades [67].
At present, the SWEET family of transporters has been characterized as one of the key players in sugar translocation in higher plants [5, 6]. The major types of translocated sugar and their regulation in partition are necessary concerning plant development and stress responses [68, 69]. In divided four clades, genes in Clade I and II prefer hexose, Clade III sucrose, and Clade IV SWEETs likely fructose transporters in Arabidopsis [17, 67]. Expression patterns of genes are tightly correlated with functions, thus, tissue-specific analysis can be used to predict biological functions [70]. In this study, genes in the same clade tended to present the same expression pattern (Fig. 6). In Clade II, HcSWEET4a, HcSWEET4b, and HcSWEET5, highly expressing in roots except for HcSWEET4c and HcSWEET6 in buds, might play central roles in mediating stress response as reported in Arabidopsis [64]. Our finding indicated that similar biological functions are performed more possibly in the same subfamily.
SWEETs in cabbage and Medicago truncatula have been reported in response to chilling stress [59, 71]. MaSWEETs are involved in abiotic/biotic stress responses in bananas [45]. ZmSWEET gene in maize responds to abiotic stress [72]. The SWEET gene family in wheat response widely to abiotic stress [73]. Our RNA-seq data preliminary showed that expression levels of most HcSWEETs first increased after treatments for 24 h compared with 0 h, then changed towards different trends (Fig. 7), RT-qPCR assay further verified the expression levels of some differential expression genes (DEGs) in the HcSWEET gene family to ensure the reliability (Fig. 8). These experiments jointly suggested that most HcSWEETs might respond to drought and salt stress in different regulatory pathways with functional redundancy.
The members of the SWEET gene family may act as sugar transporters exposed under adverse environmental conditions [74, 75]. To elucidate precise roles of HcSWEET4a in night lily, ectopic expression of HcSWEET4a was conducted in watermelon. The results showed that up-regulated expression of HcSWEET4a in transgenic lines was distinct compared with WT plants (Fig. 10A). Phenotype observations indicated transgenic lines have greater resistance against salinity than WT plants. All data integrated in our present study, It is concluded that HcSWEET4a enhances salt tolerance by positively regulating sugar transport. However, the internal regulatory network remains poorly elaborated.
Conclusions
In summary, our present study first analyzed all extracted SWEET sequences in Hemerocallis citrina. Nineteen HcSWEET genes were selected as members comprehensively characterized, including basic properties, evolutionary relationships, conserved motifs, protein domains, gene structure analysis, chromosomal localization, gene duplication, and collinearity analysis. The phylogenetic tree classified the HcSWEET gene into four clades. Each clade had similar motif compositions and gene structures. Tissue-specific expression patterns of HcSWEETs and DEGs under drought and salt treatments suggested the function diversity and potential response mechanism to environments. Ectopic expression in watermelon further assured the response of HcSWEET4a to salt stress. Our research lays a foundation for casting light on the regulatory mechanism of HcSWEET genes in response to abiotic stress in detail.
Materials and methods
Identification and characterization of the HcSWEET genes in night lily
The whole genome sequences of night lily were downloaded from NCBI(https://www.ncbi.nlm.nih.gov/assembly/GCA_017893485.1_ASM1789348v1_genomic).To identify all members of HcSWEET genes, the protein sequence of SWEETs in Hemerocallis citrina, Zea mays and Vitis vinifera also were obtained from National Center for Biotechnology Information (NCBI), Arabidopsis thaliana from the Arabidopsis Information Resource (TAIR) [76], Oryza sativa from Ensembl Genome database [77], and Hemerocallis fulva from reported data previously [41], respectively. Functional genes with incomplete domains were supplemented and screened by searching conserved domains (CD-Search) [78]. Molecular weight (MV) and theoretical isoelectric points (PI) were detected using TBtools [79]. Transmembrane topology is predicted by Deep TMHMM Server v1.0.24. Subcellular localization analysis was preliminarily predicted via WoLF PSORT software online [80].
Secondary and tertiary structure predictions
The secondary structures of 19 HcSWEET proteins were predicted by the SOPMA tool online. The three-dimensional (3D) structures of these HcSWEET proteins were constructed using homology modeling. All 19 protein sequences were aligned with all other proteins of the Protein Data Bank (PDB), and the optimum models were finally built with a standard of relatively high GMQE and seq identity values by Swiss-Model [81].
Phylogenetic tree construction, gene structure analysis, and prediction of conserved motifs and domains
Multiple Alignment Trimming in TBtools was used to carry out protein sequence alignments in night lily and other four plants. Based on the primary results of sequence alignment, trimAL Wrapper proceeded to have a shear to remove non-conserved regions as soon as possible. After shears were completed, a phylogenetic tree was constructed by One Step Build an ML Tree with 5000 bootstrap replicates. The visualization of gene structure was performed by Visualize Gene Structure of TBtools. MEME online and NCBI conserved domain database were respectively used to predict the conserved motifs and domains of the HcSWEETs.
Chromosomal location, collinearity, and cis-acting element (CREs) analysis
The genome annotation file in GFF format of HcSWEETs was obtained from the NCBI database. Both Arabidopsis and rice genomes were downloaded from the Ensembl Plants database. Intra-species and intre-species collinear analysis about night lily were constructed via MCScanX and TBtools software [79, 82]. A 2000 bp sequence in the promoter region was extracted utilizing TBtools from the genome. The online PlantCARE database was employed to predict the CREs of interest [83].
Plant materials and stress treatments
Night lily F1 hybrid population 116 (Dongzhuang Huanghua as female parent and Chonglihua as male parent) was used as treated plant material in this study, which was grown in the experimental base of Shanxi Agricultural University in Taigu under natural conditions and managed in the same regular agricultural practice. Population 116 showed moderate resistance to abiotic stress compared with the daylily cultivar ‘Golden Doll’. The experiments were conducted in 2023, and plants used for experimental treatments were from tissue cultivation. These seedlings through domestication were cultured in the same conditions for one month, after which drought and salt treatment were carried out. Drought and salt stress were respectively simulated using PEG6000 (20%) and NaCl (250 mM). Drought stresses were designed into 5 groups (0 h, 24 h, 48 h, 72 h, 108 h), and salt treatments were divided into 5 groups (0 h, 24 h, 48 h, 72 h, 96 h). Roots of treated plants were collected and immediately stored at -80 °C for follow-up experiments.
Comparative transcriptome analysis
To analyze tissue-specific expression patterns, different tissues including tender root, mature root, bud, tender leaf, mature leaf, tender scape, and mature scape were collected. Total RNA of different tissues and treated roots was isolated using the TIANGEN RNAsimple Total RNA Kit, and RNA quality was evaluated by 1.5% (w/v) agarose gel. Samples with three biological replicates were selected and sent to BMK Biotechnology Company to conduct an RNA-seq assay. Before analyzing the RNA-Seq data, quality control checks were performed through trimmomatic software [84], followed by mapping high-quality reads to the H. citrina reference genome using HISAT2 [85]. Differentially expressed genes (DEGs) were identified and modelled using DESeq2 tools [86], and filtered with standreds of Fold Change ≥ 2 and False Discovery Rate < 0.05. The average value of FPKM (reads per kilobase per million reads) was used to measure gene expression levels in the study. The heat map was drawn with log2FPKM values to show different expression levels intuitively.
RT-qPCR analysis
Based on RNA-seq results, an RT-qPCR assay was performed for further verification of HcSWEETs expression levels with HcACTIN as the standard. Real-time quantitative PCR amplification was performed by PerfectStart® Green SuperMix (TransGen, China). RT-qPCR procedure with triplicate was completed using a Real-Time PCR System (ABI QuantStudio 5, USA), and lg2−ΔΔCt values were taken as a measure of relative expression levels. SPSS 20 software was used to calculate statistical data and different letters represented significant differences between different treatments in accordance with Duncan’s multiple test. All gene-specific primers were given in Supplementary Table S2.
Construction of HcSWEET4a transient expression vector and subcellular localization
The first-strand cDNA of night lily was synthesized using TransScript® One-Step gDNA Removal and cDNA Synthesis SuperMix (TransGen, China). Night lily WT cDNA was used as template to amplify the coding sequence without stop codon of HcSWEET4a using primers containing the XbaI/KpnI restriction sites. PCR product purified was sub-cloned into enzyme-digested pSuper1300-GFP vector by the homologous recombination method using Monclone Single Assembly Cloning Mix (Monad, China). The recombinant pSuper1300-HcSWEET4a-GFP was transformed into Agrobacterium tumefaciens strain GV3101. Tobacco leaf cells were transiently injected by the cultures. After 16–24 h, fluorescence images were captured under a fluorescence microscope (Leica STELLARIS 5, Germany). Relative primers were listed in Supplementary Table S2.
Overexpression vector construction and transformation of HcSWEET4 in watermelon
The PCR product with the KpnI and BamHI restriction sites was obtained by using binary pSuper1300-HcSWEET4a-GFP as template, then was cloned into 1305.4-GFP vector and sequenced. Constructed 1305.4-HcSWEET4a-GFP vector was transformed into watermelon YL using an optimized genetic transformation system [87]. Transgenic watermelon plants were screened by GFP fluorescence observation and PCR analysis. WT and positive plants were transplanted to the same growing conditions to cultivate. The salt tolerance of transgenic plants was analyzed with WT watermelon plants as the negative control. Primers referred to in the experiment were given in Supplementary Table S2.
Data availability
The data that support results are included in this manuscript and supplementary information files. Other relevant materials also are available from corresponding authors upon reasonable request.
References
Boyer JS. Plant productivity and environment. Science. 1982;218(4571):443–8.
EJD E, Norlyn JD, Rush DW, Kingsbury RW, Wrona AF. Saline culture of crops: a genetic approach. Science. 1980;210(4468):399–404.
Conde A, Neves A, Breia R, Pimenel D, Dinis LT, Bernardo S, Correia CM, Cunha A, Moutinho-Pereira J. Kaolin particle film application stimulates photoassimilate synthesis and modifies the primary metabolome of grape leaves. J Plant Physiol. 2018;1617(18):30026–9.
Conde A, Soares F, Breia R, Gerós H. Postharvest dehydration induces variable changes in the primary metabolism of grape berries. Food Res Int. 2018;105(03):261–70.
Chen LQ, Hou BH, Lalonde S, Takanaga H, Hartung ML, Qu XQ, Guo WJ, Kim JG, Underwood W, Chaudhuri B, et al. Sugar transporters for intercellular exchange and nutrition of pathogens. Nature. 2010;468:527–32.
Broek VD, Gompel EV, Luttik MA, Pronk JT, Leeuwen CV. Mechanism of glucose and maltose transport in plasma-membrane vesicles from the yeast Candida utilis. Biochem J. 1997;321:487–95.
Zhang XH, Wang S, Ren Y, Gan CY, Li BB, Fan YYW, Zhao XQ, Yuan ZH. Identification, analysis and gene cloning of the SWEET gene family provide insights into sugar transport in Pomegranate (Punica granatum). Int J Mol Sci. 2022;23(5):2471.
Hamada M, Wada S, Kobayashi K, Satoh N. Ci-Rga, a gene encoding an MtN3/saliva family transmembrane protein, is essential for tissue differentiation during embryogenesis of the ascidian Ciona intestinalis. Differentiation. 2005;73(7):364–76.
Jia BL, Zhu XF, Pu ZJ, Duan YX, Hao LJ, Zhang J, Chen LQ, Jeon CO, Xuan YH. 2017. Integrative view of the diversity and evolution of SWEET and semiSWEET sugar transporters. Front. Plant Sci. 2017;8:2178.
Xuan YH, Hu YB, Chen LQ, Sosso D, Ducat DC, Hou BH, Frommer WB. Functional role of oligomerization for bacterial and plant SWEET sugar transporter family. Proc NatI Acad Sci U S A. 2013;110(39):E3685–94.
Mizuno H, Kasuga S, Kawahigashi H. The sorghum SWEET gene family: stem sucrose accumulation as revealed through transcriptome profiling. Biotechnol Biofuels. 2016;9:127.
Singh J, Das S, Jagadis Gupta K, Ranjan A, Foyer H, Kumar Thakur C. Physiological implications of SWEETs in plants and their potential applications in improving source-sink relationships for enhanced yield. Plant Biotechnol J. 2023;21(8):1528–41.
Singh Jeena G, Kumar S, Kumar Shukla R. Structure, evolution and diverse physiological roles of SWEET sugar transporters in plants. Plant Mol Biol. 2019;100(4–5):351–65.
Breia R, Conde A, Badim H, Margarida Fortes A, Geros H, Granell A. Plant SWEETs: from sugar transport to plant-pathogen interaction and more unexpected physiological roles. Plant Physiol. 2021;186(2):836–52.
Yuan M, Zhao JW, Huang RY, Li XH, Xiao JH, Wang SP. Rice MtN3/saliva/SWEET gene family: evolution, expression profiling, and sugar transport. J Integr Plant Biol. 2014;56(6):559–70.
Isoda R, Palmai Z, Yoshinari A, Chen LQ, Tama F, Frommer B, Nakamura W. M. SWEET13 transport of sucrose, but not gibberellin, restores male fertility in Arabidopsis sweet13;14. Proc NatI Acad Sci U S A. 2022;119(42):e2207558119.
Klemens PAW, Patzke K, Deitmer J, Spinner L, Le Hir R, Bellini C, Bedu M, Chardon F, Krapp A, Ekkehard Neuhaus H. Overexpression of the vacuolar sugar carrier AtSWEET16 modifies germination, growth, and stress tolerance in Arabidopsis. Plant Physiol. 2013;163(3):1338–52.
Yang J, Luo DP, Yang B, Frommer B, Eom W. SWEET11 and 15 as key players in seed filling in rice. New Phytol. 2018;218(2):604–15.
Wang X, Ju YH, Wu T, Kong LG, Yuan M, Liu HF, Chen XS, Chu ZH. The clade III subfamily of OsSWEETs directly suppresses rice immunity by interacting with OsHMGB1 and OsHsp20L. Plant Biotechnol J. 2024;14338:1–15.
Zhang XS, Feng CY, Wang MN, Li TL, Liu X, Jiang J. Plasma membrane-localized SlSWEET7a and SlSWEET14 regulate sugar transport and storage in tomato fruits. Hortic Res. 2021;8(1):186.
Zhang SH, Wang H, Wang T, Zhang J, Liu WJ, Fang HC, Zhang ZY, Peng FT, Chen XS, Wang N. Abscisic acid and regulation of the sugar transporter gene MdSWEET9b promote apple sugar accumulation. 2023;192(3):2081–2101.
Wang SD, Yokosho K, Guo R, Whelan J, Ruan YL, Ma JF, Shou HX. The soybean Sugar Transporter GmSWEET15 mediates sucrose export from endosperm to early embryo. 2019;180(4):2133–41.
Wu YD, Di TM, Wu ZJ, Peng J, Wang J, Zhang KX, He MM, Li NN, Hao XY, Fang WP, Wang XC, Wang L. CsLHY positively regulates cold tolerance by activating CsSWEET17 in tea plants. Plant Physiol Biochem. 2024;207:108341.
Fakher B, Arif Ashraf M, Wang LL, Wang XM, Zheng P, Aslam M, Qin Y. Pineapple SWEET10 is a glucose transporter. Hortic Res. 2023;10(10):uhad175.
Radchuk V, Belew ZM, Gundel A, Mayer S, Hilo A, Hensel G, Sharma R, Neumann K, Ortleb S, Wagner S et al. SWEET11b transports both sugar and cytokinin in developing barley grains. 2023;35(6):2186–207.
Nishad A, Kumar Nandi A. Recent advances in plant thermomemory. Plant Cell Rep. 2021;40(1):19–27.
Forni C, Duca D, Glick BR. Mechanisms of plant response to salt and drought stress and their alteration by rhizobacteria. Plant Soil. 2017;410:335–56.
Tiwari S, Lata C, Singh Chauhan P, Prasad V, Prasad M. 2017. A functional genomic perspective on drought signalling and its crosstalk with phytohormone-mediated signaling pathways in plants. Curr Genom. 2017;18:469–82.
Yang YQ, Guo Y. Elucidating the molecular mechanisms mediating plant salt-stress responses. New Phytol. 2018;217:523–39.
Muhammad M, Waheed A, Wahab A, Majeed M, Nazim M, Liu YH, Li L, Li WJ. Soil salinity and drought tolerance: an evaluation of plant growth, productivity, microbial diversity, and amelioration strategies. Plant Stress. 2024;11:100319.
Mathan J, Singh A, Ranjan A. Sucrose transport in response to drought and salt stress involves ABA-mediated induction of OsSWEET13 and OsSWEET15 in rice. Physiol Plant. 2021;171(4):620–37.
Monfared HH, Chew JK, Azizi P, Xue GP, Ee SF, Kadkhodaei S, Hedayati P, Ismail I, Zainal Z. Overexpression of a rice monosaccharide transporter gene (OsMST6) confers enhanced tolerance to drought and salinity stress in Arabidopsis thaliana. Plant Mol Biol Rep. 2020;38:151–64.
Kaur H, Manna M, Thakur T, Gautam V, Salvi P. Imperative role of sugar signaling and transport during drought stress responses in plants. Physiol Plant. 2021;171(4):833–48.
Ibraheem O, Dealtry G, Roux S, Bradley G. The effect of drought and salinity on the expressional levels of sucrose transporters in rice (Oryza sativa nipponbare) cultivar plants. Plant Omics J. 2011;4(2):68–74.
Redillas MC, Park SH, Lee JW, Kim YS, Jeong JS, Jung H, Jung H, Bang SW, Hahn TR, Kim JK. Accumulation of trehalose increases soluble sugar contents in rice plants conferring tolerance to drought and salt stress. Plant Biotechnol Rep. 2012;6:89–96.
Qing ZX, Liu JH, Yi XX, liu XB, Hu GA, Lao J, He W, Yang ZH, Zou XY, Sun MS, et al. The chromosome-level Hemerocallis citrina Borani genome provides new insights into the rutin biosynthesis and the lack of colchicine. Hortic Res. 2021;8(1):89.
Fatima U, Anjali A, Senthil-Kumar M. AtSWEET11 and AtSWEET12: the twin traders of sucrose. Trends Plant Sci. 2022;27(10):958–60.
Li S, Ji FF, Hou FF, Shi QQ, Xing GM, Chen H, Weng YQ, Kang XP. Morphological, palynological and molecular assessment of Hemerocallis core collection. Sci Hortic. 2021;285:110181.
Zuo GY, Li K, Guo YN, Niu XR, Yin LJ, Wu ZQ, Zhang XM, Cheng XJ, Yu J, Zheng SW, et al. Development and optimization of a rapid in vitro micropropagation system for the perennial vegetable night lily. Hemerocallis citrina Baroni Agron. 2024;14:244.
Yue WH, Cai KF, Xia X, Liu L, Wang JM. Genome-wide identification, expression pattern and genetic variation analysis of SWEET gene family in barley reveal the artificial selection of HvSWEET1a during domestication and improvement. Front Plant Sci. 2023;14:1137434.
Huang DM, Chen Y, Liu X, Bai NDA, Qin L. Genome-wide identification and expression analysis of the SWEET gene family in daylily (Hemerocallis fulva) and functional analysis of HfSWEET17 in response to cold stress. BMC Plant Biol. 2022;22(1):211.
Su H, Meng LJ, Qu ZC, Zhang W, Liu N, Cao PJ, Jin JJ. Genome-wide identification of the N6-methyladenosine regulatory genes reveals NtFIP37B increases drought resistance of tobacco (Nicotiana tabacum L). BMC Plant Biol. 2024;24:134.
Xie HH, Wang D, Qin YQ, Ma AN, Fu JX, Qin YH, Hu GB, Zhao JT. Genome-wide identification and expression analysis of SWEET gene family in Litchi chinensis reveal the involvement of LcSWEET2a/3b in early seed development. BMC Plant Biol. 2019;19(1):499–512.
Chong J, Piron MC, Meyer S, Merdinoglu D, Bertsch C, Mestre P. The SWEET family of sugar transporters in grapevine: VvSWEET4 is involved in the interaction with Botrytis Cinerea. J Exp Bot. 2014;65(22):6589–601.
Miao HX, Sun PG, Liu Q, Miao YL, Liu JH, Zhang KZ, Hu W, Zhang JB, Wang JY, Wang Z, et al. Genome-wide analyses of SWEET family proteins reveal involvement in fruit development and abiotic/biotic stress responses in banana. Sci Rep. 2017;7(1):3536.
Miao LM, Lv YX, Kong LJ, Chen QZ, Chen CQ, Li J, Zeng FH, Wang SY, Li JB, Huang L. Genome-wide identification, phylogeny, evolution, and expression patterns of MtN3/saliva/SWEET genes and functional analysis of BcNS in Brassica rapa. BMC Genomics. 2018;19(1):174.
Chandran D, Reinders A, Ward JM. Substrate specificity of the Arabidopsis thaliana sucrose transporter AtSUC2. J Biol Chem. 2003;278(45):44320–5.
Patil G, Valliyodan B, Deshmukh R, Prince S, Nicander B, Zhao MZ, Sonah H, Song L, Lin L, Chaudhary J, et al. Soybean (Glycine max) SWEET gene family: insights through comparative genomics, transcriptome profiling and whole genome re-sequence analysis. BMC Genomics. 2015;16:520.
Feng L, Frommer WB. Structure and function of SemiSWEET and SWEET sugar transporters. Trends Biochem Sci. 2015;40:480–6.
Yuan M, Wang S. Rice MtN3/saliva/SWEET family genes and their homologs in cellular organisms. Mol Plant. 2013;6:665–74.
Nuruzzaman M, Manimekalai R, Sharoni AM, Satoh K, Kondoh H, Ooka H, Kikuchi S. Genome-wide analysis of NAC transcription factor family in rice. Gene. 2010;465(1–2):30–44.
Gautam T, Saripalli G, Gahlaut V, Kumar A, Sharma PK, Balyan HS, Gupta PK. Further studies on sugar transporter (SWEET) genes in wheat (Triticum aestivum L). Mol Biol Rep. 2019;46:2327–53.
Lin IW, Sosso D, Chen LQ, Gase K, Kim SG, Kessler D, et al. Nectar secretion requires sucrose phosphate synthases and the sugar transporter SWEET9. Nature. 2014;508:546–9.
Guan YF, Huang XY, Zhu J, Gao JF, Zhang HX, Yang ZN. Ruptured pollen grain1, a member of the MtN3/saliva gene family, is crucial for exine pattern formation and cell integrity of microspores in Arabidopsis. Plant Physiol. 2008;147:852–63.
Sun MX, Huang XY, Yang J, Guan YF, Yang ZN. Arabidopsis RPG1 is important for primexine deposition and functions redundantly with RPG2 for plant fertility at the late reproductive stage. Plant Reprod. 2013;26:83–91.
Kanno Y, Oikawa T, Chiba Y, Ishimaru Y, Shimizu T, Sano N, Koshiba T, Kamiya Y, Ueda M, Seo M. AtSWEET13 and AtSWEET14 regulate gibberellin-mediated physiological processes. Nat Commun. 2016;7:13245.
Zhou Y, Liu L, Huang W, Yuan M, Zhou F, Li X, et al. Overexpression of OsSWEET5 in rice causes growth retardation and precocious senescence. PLoS ONE. 2014;9(4):e94210.
Wang L, Yao LN, Hao XY, Li NN, Qian WJ, Yue C, Ding CQ, Zeng JM, Yang YJ, Wang XC. Tea plant SWEET transporters: expression profiling, sugar transport, and the involvement of CsSWEET16 in modifying cold tolerance in Arabidopsis. Plant Mol Biol. 2018;96:577–92.
Zhang W, Wang SY, Yu FW, Tang J, Shan X, Bao K, Yu L, Wang H, Fei ZJ, Li JB. Genome-wide characterization and expression profiling of SWEET genes in cabbage (Brassica oleracea var. capitata L.) reveal their roles in chilling and clubroot disease responses. BMC Genomics. 2019;20(1):93.
Sosso D, Luo D, Li QB, Sasse J, Yang J, Gendrot G, et al. Seed filling in domesticated maize and rice depends on SWEET-mediated hexose transport. Nat Genet. 2015;47:1489–93.
Guo C, Li H, Xia X, Liu X, Yang L. Functional and evolution characterization of SWEET sugar transporters in Ananas comosus. Biochem Biophys Res Commun. 2018;496:407–14.
Zhang Z, Zou LM, Ren C, Ren FR, Wang Y, Fan PG, Li SH, Liang ZC. VvSWEET10 mediates sugar accumulation in grapes. Genes. 2019;10:255.
Chardon F, Bedu M, Calenge F, Klemens PAW, Spinner L, Clement G, Chietera G, Leran S, Ferrand M, Lacombeet B. Leaf fructose content is controlled by the vacuolar transporter SWEET17 in Arabidopsis. Curr Biol. 2013;23:697–702.
Liu XZ, Zhang Y, Yang C, Tian ZH, Li JX. AtSWEET4, a hexose facilitator, mediates sugar transport to axial sinks and affects plant development. Sci Rep. 2016;6:24563.
Sosso D, Luo DP, Li QB, Sasse J, Yang JL, Gendrot G, Suzuki M, Koch KE, McCarty DR, Chourey PS, et al. Seed filling in domesticated maize and rice depends on SWEET-mediated hexose transport. Nat Genet. 2015;47(12):1489–93.
Ni JP, Li JM, Zhu RX, Zhang MY, Qi KJ, Zhang SL, Wu J. Overexpression of sugar transporter gene PbSWEET4 of pear causes sugar reduce and early senescence in leaves. Gene. 2020;743:144582.
Chen LQ, Qu XQ, Hou BH, Sosso D, Osorio S, Fernie AR, Frommer WB. Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science. 2012;335(6065):207–11.
Wind J, Smeekens S, Hanson J. Sucrose: metabolite and signaling molecule. Phytochemistry. 2010;71:1610–4.
Xie HH, Wang D, Qin YG, Ma AN, Fu JX, Qin YH, Hu GB, Zhao JT. Genome-wide identification and expression analysis of SWEET gene family in Litchi chinensis reveal the involvement of LcSWEET2a/3b in early seed development. BMC Plant Biol. 2019;19(1):499.
Gao Y, Wang ZY, Kumar V, Xu XF, Yuan DP, Zhu XF, Li TY, Jia BL, Xuan YH. Genome-wide identification of the SWEET genefamily in wheat. Gene. 2018;642:284–92.
Hu B, Wu H, Huang W, Song J, Zhou Y, Lin Y. SWEET gene family in Medicago truncatula: genome-wide identification, expression and substrate specificity analysis. Plants. 2019;8(9):338.
Zhu JL, Zhou L, Li TF, Ruan YY, Zhang A, Dong XM, Zhu YS, Li C, Fan JJ. Genome-wide investigation and characterization of SWEET gene family with focus on their evolution and expression during hormone and abiotic stress response in maize. Genes. 2022;13(10):1682.
Qin JX, Jiang YJ, Lu YZ, Zhao P, Wu BJ, Li HX, Wang Y, Xu SB, Sun QX, Liu ZS. Genome-wide identification and transcriptome profiling reveal great expansion of SWEET gene family and their wide-spread responses to abiotic stress in wheat (Triticum aestivum L). J Integr Agric. 2020;19(7):1704–20.
Valifard M, Le Hir R, Müller J, Scheuring D, Neuhaus HE, Pommerrenig B. Vacuolar fructose transporter SWEET17 is critical for root development and drought tolerance. Plant Physiol. 2021;187(4):2716–30.
Yao LN, Ding CQ, Hao XY, Zeng JM, Yang YJ, Wang XC, Wang L. CsSWEET1a and CsSWEET17 mediate growth and freezing tolerance by promoting sugar transport across the plasma membrane. Plant Cell Physiol. 2020;61(9):1669–82.
Rhee SY, Beavis W, Berardini TZ, Chen G, Dixon D, Doyle A, Garcia-Hernandez M, Huala E, Lander G, Montoya M, et al. The Arabidopsis Information Resource (TAIR): a model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res. 2003;31(1):2248.
Yates AD, Allen J, Amode RM, Azov AG, Barba M, Becerra A, Bhai J, Campbell LI, Carbajo MM, Chakiachvili M, et al. Ensembl genomes 2022: an expanding genome resource for non-vertebrates. Nucleic Acids Res. 2022;50(D1):D996–1003.
Lu S, Wang J, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Marchler GH, Song JS, et al. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 2020;48(D1):D265–8.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, Xia R. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13(8):1194–202.
Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35(1):W585–7.
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer T, Rempfer C, Bordoli L, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–303.
Wang YP, Tang HB, Debarry JD, Tan X, Li JP, Wang XY, Lee TH, Jin HZ, Marler B, Guo H, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40(7):e49.
Lescot M, Déhais P, Thijs G, Marchal K, Moreau Y, Peer YVD, Rouzé Pi, Rombautsets S, Plant CARE. A database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002;30:325–7.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60.
Love MI, Huber W, Anders S. Moderated estimation of Fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Cao LH, Wei W, Shen JJ, Li Z, Xu ZM. Study on the optimization of transformation systems in watermelon. Vegetable Res. 2022;2:12.
Acknowledgements
We thank Prof. Zheng Li from College of Horticulture in Northwest A&F University for providing watermelon materials. We also thank intermediate laboratory technician Yi-Ping Wang from Experimental Teaching Center for offering technological assistance in subcellular localization.
Funding
This study was supported by The first Shanxi postdoctoral innovation competition project, Shanxi provincial breeding joint research project (YZGG-10), Shanxi provincial key research and development project (202102140601009), Biological breeding engineering program of Shanxi Agricultural University (YZGC122).
Author information
Authors and Affiliations
Contributions
S.L. and G.X. planned and designed the research. L.C., J.W., L.W., H.L., W.W., F.H., Y.L., Y.G. and X.C. performed experiments and analyzed the data. S.L., G.X., and L.C. wrote and revised the paper.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Voucher specimen
The authors declare that there are no voucher specimen included in the study.
Competing interests
The authors declare that there are no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cao, L., Wang, J., Wang, L. et al. Genome-wide analysis of the SWEET gene family in Hemerocallis citrina and functional characterization of HcSWEET4a in response to salt stress. BMC Plant Biol 24, 661 (2024). https://doi.org/10.1186/s12870-024-05376-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-024-05376-y