
Abstract
Background
The Polygonaceae is a family well-known for its weeds, and edible plants, Fagopyrum (buckwheat) and Rheum (rhubarb), which are primarily herbaceous and temperate in distribution. Yet, the family also contains a number of lineages that are principally distributed in the tropics and subtropics. Notably, these lineages are woody, unlike their temperate relatives. To date, full-genome sequencing has focused on the temperate and herbaceous taxa. In an effort to increase breadth of genetic knowledge of the Polygonaceae, we here present six fully assembled and annotated chloroplast genomes from six of the tropical, woody genera: Coccoloba rugosa (a narrow and endangered Puerto Rican endemic), Gymnopodium floribundum, Neomillspaughia emarginata, Podopterus mexicanus, Ruprechtia coriacea, and Triplaris cumingiana.
Results
These assemblies represent the first publicly-available assembled and annotated plastomes for the genera Podopterus, Gymnopodium, and Neomillspaughia, and the first assembled and annotated plastomes for the species Coccoloba rugosa, Ruprechtia coriacea, and Triplaris cumingiana. We found the assembled chloroplast genomes to be above the median size of Polygonaceae plastomes, but otherwise exhibit features typical of the family. The features of greatest sequence variation are found among the ndh genes and in the small single copy (SSC) region of the plastome. The inverted repeats show high GC content and little sequence variation across genera. When placed in a phylogenetic context, our sequences were resolved within the Eriogonoideae.
Conclusions
These six plastomes from among the tropical woody Polygonaceae appear typical within the family. The plastome assembly of Ruprechtia coriacea presented here calls into question the sequence identity of a previously published plastome assembly of R. albida.
Similar content being viewed by others


Background
The Polygonaceae family of plants is well-known for its weedy taxa, such as docks and sorrels (Rumex L.), Japanese knotweed (Reynoutria Houtt.), and Persicaria (L.) Mill. / Polygonum L. (knotweeds) [1, 2]. The family is also well-known for its edible taxa such as Fagopyrum Mill. (buckwheat) and Rheum L. (rhubarb) [3]. All of these taxa are herbaceous and primarily temperate in distribution [1, 2]. Within the Polygonaceae, there also exist several clades of primarily woody and exclusively tropical taxa [1, 2]. These groups include large genera such as Coccoloba P.Browne (ca. 200 species), as well as more moderately-sized genera such as Triplaris Loefl. and Ruprechtia C.A.Mey. (a few dozen species each) [1, 2].
The temperate and herbaceous taxa of the Polygonaceae are not only more readily called to mind, they have also been the subject of much of the plant science work in the family. This is true particularly of genetic work, much of it motivated by systematics research [4,5,6]. The six assembled nuclear genomes of the Polygonaceae listed on GenBank [7] as of November 2023, are all temperate in distribution and herbaceous in habit. Similarly, of the 462 assembled chloroplast genomes (“plastomes”) of the Polygonaceae listed on GenBank [7], as of November 2023, 426 (92%) of them are of temperate and herbaceous taxa in the Polygonaceae. As a result of this distribution of sequences, there exists latent diversity not represented in Polygonaceae nuclear genomes, plastomes, and mitogenomes in genera of the tropical, woody Polygonaceae, such as Coccoloba, Gymnopodium Rolfe, Neomillspaughia S.F.Blake, Podopterus Bonpl., Ruprechtia, and Triplaris.
Some genera of the tropical woody Polygonaceae are relatively species-poor: Neomillspaughia contains two species of large shrubs, both endemic to Central America. The genus is closely allied with the genus Podopterus [1] and with the genus Coccoloba [1, 8]. Podopterus contains three species of large shrubs, all endemic to Central America. Gymnopodium contains three species of large shrubs also all endemic to Central America [1]. Coccoloba, Triplaris, and Ruprechtia, are more species-rich. Ruprechtia is a genus containing approximately 20 species of small trees and large shrubs, present mostly in tropical dry forests from Central America to northern Argentina. Triplaris is a genus also containing approximately 20 species of medium-sized trees and lianas. In contrast to Ruprechtia, species of Triplaris are typically present in low elevation rain forests [9].
Coccoloba (Polygonaceae) is the largest of these genera, with some 150–200 species [1, 8]. The genus is composed of trees, shrubs, and lianas native throughout the tropics of the New World, but mostly confined to low elevations. Some species of Coccoloba, such as Coccoloba uvifera (L.) L., are extremely widespread, occurring along the coasts of North, Central, and South America, as well as nearly all of the islands of the Caribbean [8, 10, 11]. Other species, such as Coccoloba rugosa Desf., are endemic to a single island in the Caribbean (Puerto Rico; [10]). This species has been recognized as endangered since the 1990s [12].
We here improve the understanding of genetics in the tropical, woody Polygonaceae by providing the assembled and annotated chloroplast genomes of six species (in six genera) in this group of plants: Coccoloba rugosa, Gymnopodium floribundum Rolfe, Neomillspaughia emarginata S.F.Blake, Podopterus mexicanus Bonpl., Ruprechtia coriacea S.F.Blake, and Triplaris cumingiana Fisch. & C.A.Mey. ex C.A.Mey. (Table 1). We also compare the genomes of these six species, highlight areas of genetic divergence, and place them in a phylogenetic context.
Methods
The authors and their collaborators collected leaf material from living specimens of each of the six species included in this study. These collections are vouchered through herbarium specimens (Supplement 1). All identifications of the specimens were verified by the authors.
Leaf material destined for DNA extraction was preserved in silica gel and then frozen at -20C. The remaining material was used to generate a voucher specimen (Table 1). Whole genomic DNA was extracted using protocols outlined by Koenemann and Burke [8]. The DNA sample was cleaned with the Clean and Concentrator kit (Zymo Research, Irvine, CA). Whole genomic libraries were prepared using the NEBNext Ultra II DNA PCR-free Library Prep kit (New England BioLabs, Ipswich, MA). Whole genomic shotgun sequencing was conducted on an Illumina NovaSeq 6000, using a 500 bp insert size and 150 bp paired-end reads (University of South Carolina Functional Genomics Core Facility, Columbia, SC). Sequencing was scaled to generate 15 million reads per sample. These reads have been uploaded to the Sequence Read Archive [14] (Table 1).
We checked the reads for anomalies with FastQC v.0.11.8 [15] and did not find any. We then used the reads to generate a primary assembly for the chloroplast genome using GetOrganelle v.1.6.2d [16]. We did not clean the reads, as requested by GetOrganelle, so as not to interfere with the internal read cleaning of GetOrganelle. We did not provide a seed plastome to GetOrganelle as there did not exist an assembled plastome from a closely related taxon at the time we were making our assemblies (GetOrganelle uses an internal database as it's default when no seed is provided). We provided the following additional flags to GetOrganelle: -R 15 -k 21,45,65,85,105 -F embplant_pt. We annotated the assembled genome using GeSeq in the CHLOROBOX web platform [17], utilizing the added functionality of tRNAscan-SE v2.0.7 [18], but otherwise accepting the default settings.
Using the GeSeq annotation, we extracted the sequences of each feature for each species. The GeSeq annotation was returned in GFF3 format. We converted this to BED format using a custom script (see Supplement 2 for code). We then used BEDTools [19] to extract the sequence of each feature in FASTA format. We then aligned the sequences of each feature for all species using MAFFT v7.505 [20]. For each aligned feature, we then calculated the average, pairwise, per-site nucleotide diversity (π) as a measure of sequence divergence across the six plastomes. We used the pegas v.1.1 [21] package in R [22] to calculate the π values.
During the course of this study, we became aware of a possible misidentification of an existing GenBank accession. As part of our efforts to investigate this misidentification, we reconstructed a phylogeny of the Polygonaceae. The sampling for this phylogeny generally followed that of Zhang et al. [5] with the addition of the six plastomes assembled by us in this paper. We aligned all the plastomes using MAFFT with the additional flag “—adjustdirectionaccurately”. We examined the alignment using the NCBI Multiple Alignment Viewer v.1.25.0 [23] and Geneious Prime v2023.0.1 (https://www.geneious.com, [24]). We did not discover any anomalies. We removed one of the two inverted repeats from the aligned plastomes prior to phylogenetic analysis in order not to bias the contribution of the sequences in these regions.
Following alignment, we assessed the likely nucleotide substitution model using IQ-TREE v.2.1.3 [25]. The model selected by IQ-TREE was GTR + F + R5. We subsequently conducted a (maximum likelihood) phylogenetic analysis in IQ-TREE using the GTR + F + R5 model. The analysis utilized 1000 search replicates to assess topology and 1000 rapid bootstraps to assess support (Code: iqtree -s InFile.phy –alrt 1000 -B 1000 -lmap 2000) (See Supplement 3 and Supplement 4).
Results
We were able to successfully assemble a complete, circular chloroplast genome (“plastome”) for each of the six species (Supplement 5). The sizes of the six plastomes ranged from 168,651 bp – 171,221 bp, with the GC content varying between 36.3 –36.8% (Table 1). These plastome sizes are larger than has been reported for other genera of the Polygonaceae. For example, chloroplast genome size in Persicaria has been reported at 160,585 bp [26], in Rumex at 159,087 bp [6], and in Rhuem at 161,563 bp [27].
We were able to successfully annotate all six of the assembled plastomes (Supplement 6). For all six species the annotation identified 164 features: 37 tRNAs, 10 rRNAs, 103 exons, and 14 introns. This is similar to what has been documented elsewhere (e.g. [26]). These features are located in a large single copy region (LSC) (94 features), a small single copy region (SSC) (14 features), and two inverted repeat regions (IR) (28 features each).
The overall mean value of π across all features was 0.004262, and the overall median value was 0.002875. The upper quartile of π values was 0.006333. Among the different types of features, the most variable were the introns (mean: 0.0084) and the least variable the rRNAs (mean: 0.000555) (Table 2).
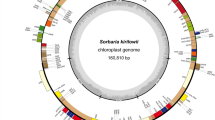
The features in the upper quartile, in order from lowest to highest π value, are: trnY-GUA, pafI, infA, ndhK, petL, petB, pafI, rpoC2, petN, ndhG, atpF, rpl20, rps16, atpF, psbE, clpP1, rpoC1, ndhC, rpl14, ndhE, ndhA (exon), psbM, rps8, ndhH, psbK, ndhA (exon), rpl22, rpl32, rps11, ndhF, pafI, accD, matK, rbcL, ndhD, ccsA, rps15, rps16, clpP1, trnW-CCA, ndhA (intron) (Fig. 1, Supplement 7).

The values of π for each annotated feature, as calculated across the six assembled plastomes. The features are placed within their proper chloroplast region and in syntenic order according to the annotation. The colors represent the feature type (exon, intron, tRNA, rRNA). The variation is to scale (values of π range from 0.0 to 0.029). Absolute π values for each annotated feature are available in Supplement 7
Of the features that have π values in the top quartile, 11 are in the SSC (of 14 total features in the SSC, 79%), and the other 30 are in the LSC (of 94 total features in the LSC, 32%). None of the features in the upper quartile of π values were found in either of the IRs. In fact, only a single feature in the IRs (ycf1, 0.00307) has a π value above the median π value. With respect to feature type, 2 of the features in the upper quartile of π values are tRNA (of 37 total tRNAs, 5%), 8 are introns (of 14 total introns, 57%), and 31 are exons (of 103 total exons, 30%).
The GC content among the six species and genera is nearly identical, varying only half a percent. GC content is notably highest in the IRs and adjacent regions of all six species, rising above 50%. This is the only location in the plastome where this is the case.
The phylogeny was fully resolved, with all nodes representing lineage bifurcations (Fig. 2). Moreover bootstrap support was above 70 for all nodes. The topology of our phylogeny is broadly reflective of those found in other phylogenetic studies in the Polygonaceae. In particular, our results are largely congruent with the phylogeny of Zhang et al. [5]. Differences include some of the relationships among species in Rumex, and a different placement of Afrobrunnichia (sister to Persicarieae in Zhang et al. [5] but sister to Eriogonoideae in ours). Importantly, the phylogeny of Zhang et al. [5] resolved Ruprechtia as sister to the entire Polygonaceae whereas ours resolved Ruprechtia as sister to Triplaris and within the Eriogonoideae.

A plastome phylogeny of the Polygonaceae reconstructed using previously assembled whole plastomes and the plastomes assembled in this study. The maximum likelihood phylogeny was generated using IQ-TREE. Branch lengths are not to scale. All nodes have bootstrap support above 70. Outgroups are highlighted with gray branches. Taxa represented by sequences generated in this study are highlighted in blue. The Ruprechtia albida specimen from Zhang et al. [5]. is highlighted in red
Discussion
Comparisons with existing assemblies
We here present six successfully assembled and annotated chloroplast genomes from six genera of the tropical woody Polygonaceae: Coccoloba rugosa, Gymnopodium floribundum, Neomillspaughia emarginata, Podopterus mexicanus, Ruprechtia coriacea, and Triplaris cumingiana. To our knowledge, these represent the first assembled and annotated plastomes for the genera Podopterus, Gymnopodium, and Neomillspaughia. Additionally, to our knowledge, these represent the first assembled and annotated plastomes for the species Coccoloba rugosa, Ruprechtia coriacea, and Triplaris cumingiana.
The sizes of the plastomes assembled here are above the median value of those reported for genera of the Polygonaceae. Our plastomes ranged in size from 168,651 bp to 171,221 bp. Of the assembled plastomes of the Polygonaceae available on GenBank [7], as of November 2023, the sizes range from 179,064 bp to 128,371 bp, with a mean size of 160,633 bp and a median size of 161,093 bp. As a result, all six of the plastomes presented here are above the average size of plastomes in the family.
For three of the genera, Coccoloba, Ruprechtia, and Triplaris, there exist recent assemblies to which we can compare our own. A previously assembled C. uvifera plastome (GenBank: NC_068873.1) reports a size of 169,369 bp, similar to the one we recovered here for C. rugosa (168,901 bp). Likewise, an existing assembly of the T. americana L. plastome (GenBank: NC_068874.1) is listed as 171,340 bp, similar in size to the one we report here for T. cumingiana (171,221 bp). We do see major differences between the existing assembly of R. albida Pendry (GenBank: NC_068875.1) and the one we present here for R. coriacea. Ruprechtia albida is reported to have a plastome size of 157,255 bp and we here report the R. coriacea plastome to have a size of 170,640 bp. Additionally, aligning the sequences of R. albida and R. coriacea shows very poor sequence identity (76.1%).
One possible explanation for this sequence divergence is a difference in assembler. The GenBank record and associated publication [5] indicate that the Ruprechtia albida sequence was assembled using NOVOPlasty and Geneious (NC_068875.1). We assembled our plastomes using GetOrganelle. Yet, in our opinion, this explanation seems unlikely. Others [28] have conducted studies comparing plastome assemblers, using both simulated and real data. What was found is that some plastid assemblers work better than others. GetOrganelle generally performed the best but both GetOrganelle and NOVOPlasty were recommended as reliable assemblers. Differences between the assemblies were slight and both had strengths and weaknesses in different situations. Moreover, the amount of divergence between the sequences, in our experience, is consistent with a generic or familial separation in taxa, not a specific separation [6].
Another possible explanation for the sequence divergence is that one of the assemblies has been generated from a misidentified voucher specimen or is the result of contamination [29]. An NCBI BLAST [30] search of the Ruprechtia albida assembly using its rbcL sequence (the land plant barcoding gene, [31]) reveals a high sequence affinity with Hydrangea L. (Hydrangeaceae) and Philadelphus L. (Hydrangeaceae) (Table 3). Using BLAST for the same feature from our (R. coriacea) assembly reveals affinities to Triplaris (Polygonaceae), Afrobrunnichia Hutch. & Dalziel (Polygonaceae), Antigonon Endl. (Polygonaceae), and Coccoloba (Polygonaceae) sequences.
We have not been able to inspect the voucher listed on GenBank for the Ruprechtia albida specimen. It is listed simply as “voucher 19693518” with no institutional affiliation indicated. Poor voucher metadata in GenBank has been written about by others [32]. Moreover, while rare, there have been documented cases of GenBank sequences having been assigned an incorrect taxonomy [33]. And while we have been unable to verify the voucher provided by Zhang et al. [5], we are confident in our own voucher and determination, which are derived from a living specimen accessioned at the Fairchild Tropical Botanic Garden and vouchered in their herbarium (Table 1, Supplement 1).
The Ruprechtia albida plastome was published as part of a study examining phylogenomics in the Polygonaceae. Another avenue for examining the identity of the sequence was to add our sequences to their phylogeny and examine the placement of taxa. Zhang et al. [5] reconstructed Ruprechtia albida as sister to the Polygonaceae as a whole. This placement is unexpected given the previous literature placing Ruprechtia as sister to Triplaris and within the Eriogonoideae [8, 34,35,36,37]. The phylogeny we reconstructed here, including the plastomes we assembled for this study, verifies the position of their Ruprechtia albida sequence, but places our Ruprechtia coriacea plastome sequence as sister to Triplaris and within the Eriogonoideae. The placement of our Ruprechtia coriacea sequence is consistent with the placement of Ruprechtia species in previous studies.
Multiple lines of evidence (voucher identification, sequence affinity in BLAST search, and phylogenetic placement) all suggest that the sequence we assembled for this study is correctly connected to the taxon Ruprechtia coriacea, but that the sequence presented in Zhang et al. [5] and currently accessioned on GenBank is likely not correctly connected to the taxon Ruprechtia albida.
Comparisons among assemblies
Among the plastomes of the six species presented in this paper, the most variable regions tended to fall within the SSC. Furthermore, among the features in the SSC, the ndh series of genes were the most variable. Moreover, the ndh genes located outside of the SSC were also within the top quartile of π values. The ndh genes, both those located within the SSC and those outside of it, code for protein elements of the NADH dehydrogenase-like complex. This complex is a membrane-embedded electron transport protein, very similar in structure to, and proposed to be homologous with respiratory complex I in the mitochondria [38]. Though its function was initially somewhat mysterious, it is now thought to be involved in the photosynthetic process, primarily in an optimizing role by helping to reduce the oxidative stress produced by processes such as photolysis [38, 39].
As the sequencing of chloroplast features and genomes has increased, variability in the ndh genes has become a known phenomenon among the land plants [39]. Additionally, certain groups of plants, notably epiphytes and parasitic plants, may lack some or all of the ndh genes [39]. As a result, our finding of variability in the SSC and ndh genes among six genera of the tropical woody Polygonaceae is unsurprising. Other studies using similar metrics (π) to quantify sequence divergence in the Polygonaceae have also found high variation in the SSC and among the ndh genes in both Rumex [6] and Rheum [40]. The list of genes in the top quartile of π values in these studies is nearly identical to the list of genes in the top quartile of π values in this study.
Two other patterns of variability that we noticed in our sequences were a strikingly low sequence variation in the IRs (only a single feature above the median π value), and a high GC content (above 50%) in these same regions. Further investigation revealed that this pattern was also observed in Rumex [6]. While this is not enough evidence to say that these patterns are common, it is at least consistent with the otherwise ordinary characterization of the plastomes assembled in this paper.
Conclusion
These six plastomes from among the tropical woody Polygonaceae appear more or less typical within the family (462 assembled Polygonaceae plastomes on GenBank as of November 2023). They are above the median size of Polygonaceae plastomes but otherwise exhibit characteristics common in the family: the features of greatest sequence variation are found among the ndh genes and in the SSC, and the IRs show little sequence variation and high GC content. The plastome assembly of Ruprechtia coriacea presented here calls into question the sequence identity of a previously published plastome assembly of R. albida.
Availability of data and materials
Data generated or analyzed during this study are included in this published article [and its supplementary information files]. The raw Illumina reads used to generate the chloroplast genome assemblies are available on the Sequence Read Archive (BioProject: PRJNA1109728 [https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1109728]; Samples: SRX22117935 [https://www.ncbi.nlm.nih.gov/sra/SRX22117935], SRX22117901 [https://www.ncbi.nlm.nih.gov/sra/SRX22117901], SRX22117878 [https://www.ncbi.nlm.nih.gov/sra/SRX22117878], SRX22117879 [https://www.ncbi.nlm.nih.gov/sra/SRX22117879], SRX22117893 [https://www.ncbi.nlm.nih.gov/sra/SRX22117893], SRX22117894 [https://www.ncbi.nlm.nih.gov/sra/SRX22117894]) (Table 1).
Abbreviations
- SSC:
-
Small single copy region of the typical green plant chloroplast genome
- IR:
-
Inverted repeat region of the typical green plant chloroplast genome
- LSC:
-
Large single copy region of the typical green plant chloroplast genome
- Plastome:
-
The complete, circular genome of the chloroplast
- Mitogenome:
-
The complete genome of the mitochondria
References
Brandbyge J. Polygonaceae. In: Bittrich V, Kubitzki K, Rohwer JG, editors. Flowering plants dicotyledons: magnoliid, hamamelid and caryophyllid families. Berlin, Heidelberg: Springer Berlin Heidelberg; 1993. p. 531–44.
Plants of the World: Polygonceae. 2023. https://powo.science.kew.org. Accessed 4 Dec 2023.
Mabberley DJ. Mabberley’s Plant-book: a portable dictionary of plants, their classification and uses. 3rd ed. Cambridge: Cambridge University Press; 2008.
Schuster TM, Reveal JL, Kron KA. Phylogeny of Polygoneae (Polygonaceae: Polygonoideae). Taxon. 2011;60:1653–66.
Zhang H, Zhang X, Sun Y, Landis JB, Li L, Hu G, Sun J, Tiamiyu BB, Kuang T, Deng T, Sun H, Wang H. Plastome phylogenomics and biogeography of the subfam. Polygonoideae (Polygonaceae) Front Plant Sci. 2022;13:893201.
Koenemann DM, Kistler L, Burke JM. A plastome phylogeny of Rumex (Polygonaceae) illuminates the divergent evolutionary histories of docks and sorrels. Mol Phylogenet Evol. 2023;182:107755.
Sayers EW, Bolton EE, Brister JR, Canese K, Chan J, Comeau DC, Connor R, Funk K, Kelly C, Kim S, Madej T, Marchler-Bauer A, Lanczycki C, Lathrop S, Lu Z, Thibaud-Nissen F, Murphy T, Phan L, Skripchenko Y, Tse T, Wang J, Williams R, Trawick BW, Pruitt KD, Sherry ST. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022;50(D1):D20–6.
Koenemann DM, Burke JM. A molecular phylogeny for the genus Coccoloba (Polygonaceae) with an assessment of biogeographic patterns. Syst Bot. 2020;45:567–75.
Pendry CA. Monograph of Ruprechtia (Polygonaceae). Sys Bot Monogr. 2004;67:1–113.
Howard RA. Studies in the genus Coccoloba, IV. The species from Puerto Rico and the Virgin Islands and from the Bahama Islands. J Arnold Arb. 1957;38:211–42.
Melo E. As espécies de Coccoloba P. Browne (Polygonaceae) da Amazonia brasileira. Âcta Amazonica. 2004;34:525–51.
Anadón-Irizarry V, Wege DC, Upgren A, Young R, Boom B, León YM, Arias Y, Koenig K, Morales AL, Burke W, Perez-Leroux A, Levy C, Koenig S, Gape L, Moore P. Sites for priority biodiversity conservation in the Caribbean Islands Biodiversity Hotspot. J Threat Tax. 2012;4:2806–44.
Thiers BM (updated continuously). Index Herbariorum. https://sweetgum.nybg.org/science/ih/.
Leinonen R, Sugawara H, Shumway M, on behalf of the International Nucleotide Sequence Database Collaboration. The sequence read archive. Nucleic Acids Res. 2011;39:D19-21.
Wingett SW, Andrews S. FastQ screen: a tool for multi-genome mapping and quality control. F1000Research. 2018;7:1338.
Jin JJ, Yu WB, Yang JB, Song Y, dePamphilis CW, Yi T-S, Li DZ. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020;21:241.
Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017;45:W6–11.
Chan PP, Lowe TM. tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol Biol. 2019;1962:1–14.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinf. 2010;26:841–2.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Paradis E. pegas: an R package for population genetics with an integrated modular approach. Bioinformatics. 2010;26:419–20.
R Core Team. 2023. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.Rproject.org/. Accessed 1 Dec 2023.
Multiple Alignment Viewer. 2023. National Center for Biotechnology Information. https://www.ncbi.nlm.nih.gov/projects/msaviewer/. Accessed 13 Dec 2023.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinf. 2012;28:1647–9.
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, Lanfear R. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37:1530–4.
Yang H, Yang D, Yang X, Li L, Zhou Q, Cheng H, Liu D. The complete chloroplast genome sequence of Persicaria perfoliata (L.) H. Gross: a medicinal plant. Mitochondrial DNA Part B. 2022;7:1961–3.
Li Y, Li H, Hei X, Li Y, Li H, Gao J, Yan Y, Liu M, Zhang G. Characterization of the complete chloroplast genome of medicinal plant Rheum officinale (Polygonaceae). Mitochondrial DNA Part B. 2019;4:2144–5.
Freudenthal JA, Pfaff S, Terhoeven N, Korte A, Ankenbrand MJ, Förster F. A systematic comparison of chloroplast genome assembly tools. Genome Biol. 2020;21:254.
Pentinsaari M, Ratnasingham S, Miller SE, Hebert PDN. BOLD and GenBank revisited – Do identification errors arise in the lab or in the sequence libraries? PLoS ONE. 2020;15(4):e0231814.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Newmaster SG, Fazekas AJ, Ragupathy S. DNA barcoding in land plants: evaluation of rbcL in a multigene tiered approach. Can J Bot. 2006;84:335–41.
Wu HY, Chan KT, But GWC, Shaw PC. Assessing the reliability of medicinal Dendrobium sequences in GenBank for botanical species identification. Scien Rep. 2021;11:3439.
Leray M, Knowlton N, Ho SL, Nguyen BN, Machada RJ. GenBank is a reliable resource for 21st century biodiversity research. PNAS. 2019;116(45):22651–6.
Sanchez A, Kron KA. Phylogenetic relationships of Afrobrunnichia Hutch. & Dalziel (Polygonaceae) based on three chloroplast genes and ITS. Taxon. 2009;58(3):781–92.
Sanchez A, Schuster TM, Kron KA. A large-scale phylogeny of the Polygonaceae based on molecular data. Int J Plant Sci. 2009;170(8):1044–55.
Burke JM, Sanchez A, Kron K, Luckow M. Placing the woody tropical genera of the Polygonaceae: a hypothesis of character evolution and phylogeny. Am J Bot. 2010;97(8):1377–90.
Sanchez A, Kron KA. Phylogenetic relationships of Triplaris and Ruprechtia: re-delimitation of the recognized genera and two new genera for tribe Triplarideae (Polygonaceae). Sys Bot. 2011;36(3):702–10.
Shikanai T. Chloroplast NDH: A different enzyme with a structure similar to that of respiratory NADH dehydrogenase. Biochim Biophys Acta Bioenerg. 2016;1857:1015–22.
Sabater B. On the edge of dispensability, the chloroplast ndh genes. Int J Mol Sci. 2021;22:12505.
Zhang HJ, Zhang X, Landis JB, Sun YX, Sun J, Kuang TH, Li LJ, Tiamiyu BB, Deng T, Sun H, Wang HC. Phylogenomic and comparative analyses of Rheum (Polygonaceae, Polygonoideae). J Syst Evol. 2021;60:1229–40.
Acknowledgements
Some of the computations in this paper were conducted on the Smithsonian High Performance Cluster (SI/HPC), Smithsonian Institution. Doi: 10.25572/SIHPC. The authors would like to thank M. Kweskin for logistical support and advice in the use of the Smithsonian HPC. The sequencing for this project was performed by the Functional Genomics Core of the USC COBRE Center for Targeted Therapeutics. The authors would like to especially thank D. Altomare and M. Shtutman of the University of South Carolina Functional Genomics Core for logistical support and troubleshooting during sequencing. The authors would like to thank B. Jestrow and B. Milne of the Fairchild Tropical Botanic Garden (Miami, Florida) for logistical support, access to living collections, and scans of voucher specimens. The authors would like to thank A. Stalter of the Bailey Hortorium at Cornell University for a digital loan of voucher specimens. The authors would like to thank L. Coykendall of the United States Botanic Garden for access to the living collection of Coccoloba rugosa, and K. Hale of Howard University for imaging the C. rugosa voucher in the Howard University herbarium.
Funding
This work was supported by Claflin University SEED Summer Research Funds, awarded to DK.
Author information
Authors and Affiliations
Contributions
JB collected some plant material and contributed to the writing of the manuscript. DK collected some plant material, conducted the data analysis, and contributed to the writing of the manuscript. Both authors read and have approved of the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All plant material utilized in this study was collected by the authors or their collaborators. Some materials were collected on private land and some on government land. In the case of collections made on private property, the permission of the landowner was obtained. In the case of collections made on government land, permission from the appropriate governing body was obtained.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Burke, J.M., Koenemann, D.M. The complete annotated plastome sequences of six genera in the tropical woody Polygonaceae. BMC Plant Biol 24, 417 (2024). https://doi.org/10.1186/s12870-024-05144-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-024-05144-y