
Abstract
Background
Cucurbita ficifolia is one of the squash species most resistant to fungal pathogens, and has especially high resistance to melon Fusarium wilt. This species is therefore an important germplasm resource for the breeding of squash and melon cultivars.
Results
Whole-genome resequencing of 223 individuals from 32 populations in Yunnan Province, the main cucurbit production area in China, was performed and 3,855,120 single-nucleotide polymorphisms (SNPs) and 1,361,000 InDels were obtained. SNP analysis suggested that levels of genetic diversity in C. ficifolia were high, but that different populations showed no significant genetic differentiation or geographical structure, and that individual C. ficifolia plants with fruit rinds of a similar color did not form independent clusters. A Mantel test conducted in combination with geographical distance and environmental factors suggested that genetic distance was not correlated with geographical distance, but had a significant correlation with environmental distance. Further associations between the genetic data and five environmental factors were analyzed using whole-genome association analysis. SNPs associated with each environmental factor were investigated and genes 250 kb upstream and downstream from associated SNPs were annotated. Overall, 15 marker-trait-associated SNPs (MTAs) and 293 genes under environmental selection were identified. The identified genes were involved in cell membrane lipid metabolism, macromolecular complexes, catalytic activity and other related aspects. Ecological niche modeling was used to simulate the distribution of C. ficifolia across time, from the present and into the future. We found that the area suitable for C. ficifolia changed with the changing climate in different periods.
Conclusions
Resequencing of the C. ficifolia accessions has allowed identification of genetic markers, such as SNPs and InDels. The SNPs identified in this study suggest that environmental factors mediated the formation of the population structure of C. ficifolia in China. These SNPs and Indels might also contribute to the variation in important pathways of genes for important agronomic traits such as yield, disease resistance and stress tolerance. Moreover, the genome resequencing data and the genetic markers identified from 223 accessions provide insight into the genetic variation of the C. ficifolia germplasm and will facilitate a broad range of genetic studies.
Similar content being viewed by others

Background
A common topic in ecological genetics is the effect of a changing environment on the distribution and composition of genetically variable populations [1], as well as the mechanisms by which the genetic structure of populations may evolve and diverge [2]. Climatic factors are known to be important for the distribution of plants and are important drivers of evolution [3]. Plant populations do not remain genetically identical over time, and different populations of a species in different habitats or areas may be genetically distinct, adapting genetically and physiologically to local conditions [4]. Small-scale patterns in genetic structure are thus often driven by environmental factors [5, 6]. The effects of the environment can sometimes even be observed in populations of a species with a high potential for gene flow [2, 7].
Curcubita ficifolia Bouché (Cucurbitaceae) is a short-day plant, is sensitive to temperature and is not heat-resistant. The plant is monoecious and has unisexual flowers. It is known as “black seed squash” in English after its black seeds, and the Chinese name translates "rice noodle squash" or "Black Seeded’ figleaf squash" in Chinese [8] as the fruit pulp resembles rice noodles or vermicelli. C. ficifolia is a trailing herb with perennial roots. In sunny, warm environments, the plant grows as a perennial, but in areas that get severe winter frosts, the roots die off over winter and the plant grows as an annual. New vines can grow as much as 20 m in a year, and more than 50 high-yield squashes can be produced per plant in this time [9]. A single fruit generally weighs 2 to 5 kg and produces 50 to 150 g of seeds. The younger, tender fruits can be eaten by humans as a vegetable, whereas the slightly older, larger fruits are lignified, and are generally fed to livestock. The shells of older squashes are hard, and the fruits store well. The rind is found in three color patterns: green (dark green alternating with light green), white and “decorative” (green alternating with white) [10]. The ripe seeds are black or dark brown and also have important edible value. Compared with other species of Cucurbitaceae, C. ficifolia has strong resistance to a number of environmental stresses, including cold and drought, it grows well on barren soils and is also resistant to several diseases. The species also shows a particularly high resistance to melon Fusarium wilt and is therefore an important germplasm resource for the breeding of new cucurbit cultivars.
To date, there have been few studies investigating C. ficifolia. Some studies have focused on its pharmacological effects, and C. ficifolia extract has been found to increase the secretion of insulin, playing an important role in lowering blood sugar and can therefore be helpful in the treatment of diabetes [11,12,13,14]. C. ficifolia extracts can also modulate systemic chronic inflammation in obese mice, where they may have beneficial effects on the adaptive immune system [15]. Other studies concern the use of C. ficifolia as a rootstock onto which other species of Cucurbita can be grafted, and have found that C. ficifolia rootstocks confer improved yields and enhanced resistances to cold, salt and diseases including Fusarium wilt [10, 16,17,18,19]. Cucumbers grafted onto C. ficifolia rootstock and grown in plastic greenhouses have increased foreign income from fresh produce in Liaoning, Shandong and several other provinces in China, by virtue of the early market supply and high quality of the grafted plants. Moreover, C. ficifolia has become an excellent germplasm resource for improving the disease resistance in new cultivars. Brazil exports the largest volume of C. ficifolia worldwide, and Japan imports the most [8].
C. ficifolia originated in Central and South America [20], and the wild plant is common in mountainous areas from northern Mexico to northern Argentina and Chile. However, it is now cultivated in many countries worldwide, and has been popular for many centuries in Asia [21]. There is no data on the history of introduction and cultivation of C. ficifolia in China, but the species has a long history of cultivation in Yunnan, Sichuan, Guizhou and other higher altitude regions [22]. C. ficifolia grows well at high altitudes, and much of Yunnan Province is thus suitable for its cultivation, with this province becoming the main production area in China. C. ficifolia is an important breeding germplasm resource for cucurbits, however, to date there have been only a few genetic studies investigating this species. In order to more effectively utilize and develop this species as a genetic resource, a better understanding of its genetic diversity, the extent by which its populations differ genetically and the factors underlying these patterns are required.
Whole genome resequencing and genome-wide association studies (GWAS) have been used to investigate the genomes of several widely grown cucurbit crops, including Cucurbita pepos, pumpkins, cucumbers, watermelons and melons, and many SNPs controlling horticulturally important phenotypes have been discovered [23,24,25,26,27,28]. However, despite the utility and potential of C. ficifolia in agriculture, the C. ficifolia genome has not to date been studied in depth. In this project, we investigated genetic variation in different C. ficifolia populations in Yunnan using whole-genome resequencing. The main aims of the study were: (1) to use whole-genome resequencing to obtain a library of SNP markers, and to evaluate genetic diversity in C. ficifolia in Yunnan, (2) to use population genetic approaches to elucidate the genetic structure of C. ficifolia within and among populations, as well as to investigate the evolutionary patterns behind rind of different colors, and (3) to analyze how geographical and environmental distances affect the genetic structure of C. ficifolia. The data will improve the utilization and development of C. ficifolia germplasm resources.
Results
Plant materials and phenotypic statistics
Two hundred twenty-three individuals were collected from the field in Yunnan province of China. In order to avoid the homogenization of research materials, phenotypic traits were recorded and analyzed for all the samples. The length, breadth, and thickness of the seeds, the thousand seed weight were analyzed for each sample (Fig. 1 and Table S1). The average length of seed is 17.09 mm, beath is 11.07 mm, thickness is 2.51 mm and thousand weight is 241.31. All four phenotypic indices conform to a normal distribution. We also measured the squash rind color and showed that 109 individuals of which were ‘decorative’; 57 were white and 30 were green. We can see that the phenotypic diversity of our samples was high and suitable for genetic analyses as natural populations.SNP Calling and Analyses of Genetic Diversity and Linkage Disequilibrium Decay.

Normal distribution of the length, breath, thickness and thousand weight of Cucurbita ficifolia seeds
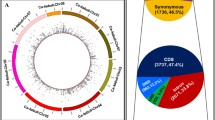
Resequencing the 223 C. ficifolia individuals on an Illumina NovaSeq6000-sequencer generated 758.08 Gbp of clean data, with a total of 2.2 million 100 bp paired-end reads (332 Gb of sequence data), 93.63% of which had Q values ≥ 30. The average alignment rate of samples with the reference genome was 93.17%, the average depth of coverage was 9 × and the genome coverage was 66.11% (with at least one base coverage).There is, to date, no published genome information available for C. ficifolia. We mapped our resequencing data to whole genomes of four species of Cucurbita (C. argyrosperma subsp. Sororia; C. pepo subsp. pepo; C. moschata; C. maxima). The mapping rate to the C. moschata genome was found to be the highest (93.88%), and the C. moschata genome was therefore selected as the reference genome for this study. After mapping to the C. moschata reference genome [29], we identified 3,855,120 SNPs and 1,361,000 InDels shorter than or equal to 6 bp (Table S2). The genome-wide variations among the 223 C. ficifolia samples are summarized in Fig. 2. We constructed 20 pseudochromosomes, using the C. moschata genome as a reference. The densities of the genes, SNPs and InDels were evenly distributed over all 20 pseudochromosomes (Fig. 2A).

Genome-wide variations among 223 Cucurbita ficifolia samples. A Circus diagram of the 20 predicted pseudomolecules of Cucurbita ficifolia illustrating the density distributions of InDels (blue inner circle), SNPs (orange middle layer) and genes (green outer circle) within the 223 Cucurbita ficifolia samples. B Plots of squared allele frequency correlations (r2) by physical distance between sites in Cucurbita ficifolia. C SNP annotation classification results. D InDels annotation classification results
We calculated r2 between pairs of SNPs to investigate patterns of linkage disequilibrium between the different C. ficifolia chromosomes. At 250 kb, the linkage disequilibrium across all samples dropped to half its maximum (r2 = 0.08, Fig. 2B). Of the 3.86 million SNPs, 47.56% were present in intergenic regions and 52.44% were located intragenically (CDS: 20.49%; intron: 31.95) (Fig. 2C). Of the 1.36 million InDels, most (63.85%) were present in intergenic regions and the intragenic InDels were predominantly introns (CDS: 3.09%; intron: 33.06%) (Fig. 2D).
To understand the extent of genetic diversity in C. ficifolia, we calculated the number of polymorphic markers and tested five common genetic diversity indices. The average number of polymorphic sites in each population was 26,376 (between 17,151 and 33,012), and the genetic diversity of C. ficifolia was high, with a π of 0.354 (0.252–0.418). The expected heterozygosity (HE) was 0.319 (0.221–0.404), the observed heterozygosity (HO) was 0.392 (0.315–0.466), the polymorphism information content (PIC) was 0.251 (0.173–0.316) and the Shannon Wiener index (I) was 0.465 (0.320–0.587) (Table 1). Of the 32 populations sampled, JS had the highest genetic diversity (π = 0.418), followed by DL and DY (π = 0.417). This high genetic diversity could provide abundant genetic information for cucurbit crop breeding.
Genetic differentiation, phylogeny construction and population structure analysis
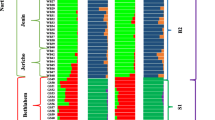
Our Arlequin analysis suggested that the Wright's FST value among populations was 0.03, indicating that the degree of genetic differentiation among different C. ficifolia populations was very small. The pairwise FST values of each population ranged from 0 to 0.841 (Table S3), showing that genetic distances among populations varied greatly. We then used a combined approach (PCA, maximum likelihood estimation of individual ancestries, and an NJ phylogenetic tree) to analyze the phylogenetic and population genetic patterns present in C. ficifolia populations. The PCA analysis reflected the larger trend, and suggested that the genetic makeup of individuals in the same population were usually relatively similar, however, the population did not form distinct groups (Fig. 3A). We then applied ADMIXTURE to make a maximum likelihood estimation of the individual ancestries of the samples. In this analysis, the range of K = 1-10 was analyzed, and the cross-validation (CV) errors value reached the lowest value at K = 3, indicating that the optimal grouping of C. ficifolia was 3 (Fig. 3B). These assignments were not completely separated by geography, but there was a geographical trend. Most of the individuals in Cluster I were from the populations in eastern Yunnan, those in Cluster II were mostly from the populations in central Yunnan, and those in Cluster III were from populations in western and northeastern Yunnan or other regions. There were also some populations whose individuals were divided between three clusters, such as ZX, YA and others (Fig. 3C). The reconstructed phylogenetic tree suggested that most individuals from the same populations were closely related, for example in populations NH, JS and SP. However, no clear clustering by geographic distribution was apparent (Fig. 3D).

Analysis of phylogeny and population structure in C. ficifolia. A Population structure within the diversity panel assessed using principal component analysis. B Plot of different number of subpopulations (K) from 1 to 10 versus cross validation error (CV error). The cross validation procedure as implemented in ADMIXTURE was used to initially find the optimum number of subpopulations (K) by minimizing the cross validation error. C Population structure based on K = 3. The different samples are shown on the x-axis. The y-axis quantifies the membership probability of samples belonging to different groups. Colors in each row represent structural components. D Neighbor-joining (NJ) phylogenetic tree
The rind of C. ficifolia fruits can be one of three colors: green (dark green alternating with light green, -G); white (-W); and decorative (green alternating with white, -H). The three rind colors did not form independent clusters in either the reconstructed phylogenetic tree or in our genetic structure analysis. However, individuals from the same local population, regardless of the color of the rind, tended to cluster in the same branch, indicating that individuals of different colors within the local population were closely related, while individuals with the same rind color from different regions were less related.
Potential correlations between genetic distance and geographic and environmental distance were tested using Mantel tests. No significant correlation between genetic and geographic distances was found (r = 0.007; P = 0.176), although there was a more obvious relationship between genetic and environmental distances (r = 0.125; P = 0.047). In order to exclude interference from environmental or geographical factors, partial Mantel tests were conducted. These tests agreed with the simple Mantel tests, and showed no correlation between genetic and geographic distance (r = 0.220; P = 0.386); when we controlled for geographical distance, genetic distance and environmental distance were still correlated (r = 0.106; P = 0.043) (Table 2). The above analyses strongly indicate that the genetic structure of C. ficifolia is driven by environmental factors.
Marker-trait association analyses
We used 3.85 million SNPs together with climate data (including annual mean temperature, annual precipitation, altitude, frost-free season and annual average sunshine) from the 32 sampling regions in an association analysis in order to identify any SNP sites associated with climate traits. We used several different statistical programs and simulations for this analysis, and performed comparisons in GEMMA (LM and LMM models), FaST-lmm, and EMMAX. The results varied to some extent with the different statistical models. The LM model of GEMMA found the greatest number of relevant marker-trait associated SNPs. Taking log10(p) = 5 as the reference point, the five environmental factors were associated with a total of 15 MTAs (Fig. 4), using the all the four models. Two MTAs were associated with altitude (T/C in chromosome NW_527.1 and C/A in NW_531.1); two MTAs were associated with annual average temperature (G/T in chromosome NW-531.1 and C/T in chromosome 517.1); one MTAs was associated with annual precipitation (A/G in chromosome NW-528.1); eight MTAs were associated with duration of annual sunshine (T/G in chromosome NW_521.1; A/T in chromosome NW-534.1; five SNP in chromosome NW_528.1) and two MTAs were associated with frost-free days (C/T and G/A in NW_532.1) (Table 3). The number of MTAs identified showed that duration of annual sunshine had the greatest impact on genetic variation in C. ficifolia.

Manhattan and QQ plots showing results from genome-wide association mapping of marker-trait association analyses. A GEMMA model Manhattan maps. The x-axis of each plot represents the physical position of each SNP, and the y-axis displays the negative log10 of p-values for each SNP included in the GWAS. B GEMMA model quantile–quantile plots (QQ plots)
A total of 293 genes within 250 kb of these MTAs (including both up- and downstream genes) were identified (Table S4). An enrichment analysis of the annotated genes showed that the up- and downstream genes of MTAs were involved in the following three functions: “cellular component”, “molecular function” and “biological process” (Fig. S1).These genes reflected the changes in C. ficifolia cell activity, molecular function and biological processes in response to different environmental conditions.Some genes, including “gene control cellular component: integral component of membrane” (GO: 0016021), “mitochondrial inner membrane” (GO: 0005743), and “spindle microtubule” (GO: 0005876) were revealed when we annotated the MTAs with duration of annual sunshine. This indicated that different durations of annual sunshine have an effect on the cell structure of C. ficifolia in different populations.
Present and future distributions of C. ficifolia
In order to further confirm the influence of environmental factors on the distribution of C. ficifolia, we performed ecological niche modeling to simulate the distribution of C. ficifolia during the present distribution, and that in the future (2080). All models performed well, with AUC values > 0.9 (n = 10 replicate model runs) [30] (Fig. 5A). C. ficifolia prefers a cooler environment, with a long growth period and fruit maturation in between November and January period. If it is too cold in winter, the fruit does not normally produce seed. Several years of data verification and field investigations suggest that the C. ficifolia populations in China are currently found mainly distributed in Yunnan and the surrounding areas. The distribution of C. ficifolia simulated by the ecological niche modeling based on environmental factors (Fig. 5B) was consistent with the current observed “real” distribution of cultivated C. ficifolia in Yunnan, which further illustrates the reliability of the model and also suggests that the distribution of this species in Yunnan is indeed limited by environmental factors.

A Reliability analysis of model prediction; B present and (C) predicted future distributions of Cucurbita ficifolia. Each cell on the grid map has a suitability index between 0 and 1. Low values (white or pale red) indicate that conditions are unsuitable for the species to occur, whereas high values (dark red) indicate that conditions are suitable. Note: The MAP is taken from CGIAR-CSI (https://srtm.csi.cgiar.org)
C. ficifolia is an excellent germplasm resource for the improvement of melon, squash and gourd crops. From the distribution map predicted for 2080, it can be seen that the suitable cultivation distribution area of C. ficifolia will migrate to the northwest, and the most suitable area will then be concentrated in the Hengduan Mountain Range (Fig. 5C). However, most areas in the southwestern regions of Yunnan will no longer be suitable for the cultivation of C. ficifolia, so it is particularly vital to collect and protect germplasm resources currently present in these regions.
Discussion
Genetic diversity of C. ficifolia
The study of genetic diversity provides insight into the origins and evolution, particularly the micro-evolution, of biodiversity. The information generated from these studies can inform animal and plant breeding programs [31]. the genetic structure of populations can be influenced by a number of factors, including the evolutionary history of the populations, their distribution, life form, breeding system and the mechanism of seed dispersal. The combined effect of various factors influences the levels of genetic variation seen within plant populations. Analysis of the spacial structure and genetic variation within plant populations is therefore important when investigating evolutionary factors [31, 32]. High genetic diversity is beneficial for the breeding and improvement of horticultural plants; however, domestication of plants can often lead to reductions in their genetic variability through anthropomorphic selection. This leads to the potential loss of valuable genes, including those conferring resistance and tolerance to biotic and abiotic factors. Although cucurbit crops are playing an increasingly important role in global nutrition, many valuable genes have been lost during domestication and manual selection, and diseases, such as Fusarium wilt, occur frequently [33]. Breeding technology can improve crop yields, disease resistance and adaptability to the environment, and closely related species with high resistances are important genetic resources for this genetic improvement. With the development of high-throughput sequencing technologies, there have been increasing numbers of studies using resequencing to analyze the genetic diversity of agricultural plants. For example, resequenced groups of chickpea resources from around the world were found to have π values ranging from 0.85 to 0.105 [34]. Significant differences in genetic diversity (π) were also found in different groups of soybean (Glycine soja, soybean landraces and soybean cultivars) in resequencing studies (G. soja: 2.94 × 10−3; landraces: 1.40 × 10−3; cultivars: 1.05 × 10−3) [35]. Similarly, resequencing of gourd resources suggested that wild gourd (Citrullus colocynthis, π = 6.75 × 10−3) and citron melon (Citrullus amarus, π = 2.28 × 10−3) had much greater nucleotide diversity than did the watermelons Citrullus mucosospermus (π = 0.792 × 10−3) and C. lanatus (π = 0.56 × 10−3) [26, 27]. C. ficifolia has relatively high genetic variation, which could provide good germplasm resources for the breeding of novel cucurbit cultivars and the improvement of races. This resequencing study of different local Chinese populations of C. ficifolia will provide a good basis for subsequent investigations using GWAS and other means to explore the excellent genetic resources of this species.
The main factors responsible for the observed genetic structure in C. ficifolia
Isolation brought about by geographical distance (IBD) and isolation from environmental distance (IBE) are recognized as important drivers of genetic differentiation [36,37,38,39]. Genetic differentiation tends to increase with increasing geographic distances between populations [1]. However, in order to adapt to different environments, the genetic diversity of neighboring populations may be reduced due to natural selection [38], which will promote or maintain genetic differentiation between different environments [40]. The large-scale genetic structure of populations may therefore be influenced by geographic processes, while over smaller scales, the population structure may be influenced more by ecological processes [41]. Climate change over time may also be an important driver of population differentiation [42]. A thorough understanding of the relative contributions of all of these factors is important to fully understand the formation of genetic structure [1, 43, 44].
The Wright's FST value among Chinese populations of C. ficifolia was 0.03, indicating that the degree of genetic differentiation among different C. ficifolia populations was very small. However, the pairwise FST values between certain populations was extremely high, for example. YA and JD: 0.841 (Table S3). Wright's FST is usually calculated for a large random-breeding population [45]. We therefore think the abnormal value of the FST is likely to be the result of the small numbers of individuals sampled: the JD site comprised only three individuals, and the YA site, six individuals. Furthermore, these nine individuals fell into different clusters in the constructed phylogenetic tree, which is also likely to contribute to this abnormal FST value.
We found that individuals from the same population have a close genetic relationship, however, C. ficifolia individuals from different, but nearby, populations are not more closely related than those from far populations. This was confirmed with Mantel and Partial Mantel tests, which also showed that the genetic distance between C. ficifolia subpopulations in Yunnan had no correlation to geographical distance. In puzzling contrast to the other species of Cucurbita, fruit shape and size in C. ficifolia are uniform, and only the rind color differs, with rinds known in three color forms: green, white and “decorative” [10]. Individuals with the same phenotype (rind color) did not cluster together in our genetic diversity or phylogenetic analyses, indicating that the C. ficifolia has not formed three distinct subspecies or varieties. Instead, individuals with all three rind colors could be found clustering together according to geographical distribution. This is consistent with Nee et al. (1990) [21], who say a single field anywhere in the range of this species may contain essentially all the variation in fruit morphology known for C. ficifolia. Interestingly, the plants with green-rinded fruits have been shown to have greater resistance to Fusarium wilt than those with rinds of other colors [46].
From Mantel and partial Mantel tests, a correlation between genetic and environmental distance was revealed. We speculate that the patterns of genetic structure in C. ficifolia occurred mostly due to environmental factors. C. ficifolia is a perennial plant, but grows as an annual in cold areas. In Yunnan, China, C. ficifolia is perennial, with a long annual growth cycle. The squash matures in December, and in the mild climate of Yunnan, the seeds mature. However, other parts of China are generally too cold in December for natural seed maturity. In other words, the growth and spread of C. ficifolia is sensitive to climate. The genetic distances observed between the C. ficifolia subpopulations in our study were obviously correlated with environmental distance, which supports our speculation. In order to verify this speculation, ecological niche modeling was used to simulate the distribution of C. ficifolia across time, from the present and into the future. The results from the ecological niche modeling suggest that the distribution of C. ficifolia will change again in the future. The analysis also suggested that environmental factors mediated the formation of the population structure in this species. As the global climate warms, the range of C. ficifolia will move northwards. In addition, because this species is a horticultural plant, human interference and the agricultural use of selected germplasms should also be considered to explain the observed population structure of C. ficifolia.
Materials and methods
Sampling, DNA extraction and sequencing
The sampling locations were chosen to cover the main C. ficifolia production areas in China. Between 3–19 individual plants were sampled from each of 32 locations (Fig. 6 and Table S1), giving a total of 223 C. ficifolia samples. Official permits for collection of these native plants were not required because these plants are not included in the list of national key protected plants, and the materials were collected in artificial planting bases or from wild plants in the field. The formal identification of the plant material was performed by Yongjie Guo of Kunming Institute of Botany based on morphological characters. The specimens of C. ficifolia has been deposited at Herbarium, Institute of Botany, Chinese Academy of Sciences (voucher # KUN 1580438). All 223 individuals were from landraces and samples were taken from plants at least 50 m apart.The length, breadth, and thickness of the seeds, the thousand seed weight and the squash rind color were recorded for each sample. Fresh leaf samples were stored at -80 °C and total DNA was extracted using the CTAB method [47]. DNA was quantified using a fluorometer with a fluorescent dye (Qubit3.0, Thermo Fisher Scientific, Waltham, MA, United States). DNA quality was assessed using agarose gel electrophoresis (Omega Bio-Tek, Norcross, GA, United States). Each qualified DNA sample was standardized to the same volume (10 µl) and quantity (200 µg) of DNA.

Locations of the 32 sampled Cucurbita ficifolia populations in Yunnan. Note: The MAP is taken from CGIAR-CSI (https://srtm.csi.cgiar.org)
The Illumina NovaSeq6000 sequencing platform was used to randomly fragment the genomic DNA, and the fragments were used to construct libraries of ~ 450 base-pair (bp) inserts and to produce paired-end reads of 150 bp. The raw sequencing data were then filtered using fastp 0.21.0 with the parameters “fastp -q 10 -u 50 -y -g -Y 10 -e 20 -l 100 -b 150 -B 150”. Low-quality reads (where > 50% of the bases had a quality score < 30) and poly-Ns (regions where > 10% of the bases were Ns) were removed. Adapter sequences were trimmed from both ends of the reads and low-quality bases (Q ≤ 13) were removed. The clean reads were then compared with the reference genome Cucurbita moschata [29] using the software bwa-mem2 v2.2 [48]. Samtools v1.9 [49] was used to sort the results, after which the sequencing depth, genome coverage and other information from each sample was taken.
SNP calling, genetic diversity and differentiation analysis
SNPs and InDels were identified using GATK v3.8 [50] with joint calling. Briefly, the genomic variant call format (GVCF) for each sample was obtained using ERC mode from reads where the mapping quality > 20 (parameters were set to “-T Haplotype Caller -ERC GVCF -variant index type LINEAR -variant index parameter 128,000 -mq 20”). Joint variant calling was then conducted using “CombineGVCFs” based on GATK HaplotypeCaller. The variants identified were then hard-filtered to remove low quality calls, with the main filtering parameters set to Q ≥ 30, QD ≥ 2, MQ ≥ 40 and FS ≥ 60. The program vcfutils.pl (varFilter-w 5-W 10) in BCFtools was used to identify SNPs within 10 bp of an adjacent indel, which were then rejected.
The populations program in the Stacks1.0 pipeline [51, 52] was used to calculate population genetic statistics (number of private alleles; observed heterozygoisty (HO); expected heterozygosity (HE); Nei’s genetic diversity (π)) for each SNP. The pairwise fixation index (FST) was calculated using Arlequin [53] to examine the distribution of genetic diversity.
Linkage disequilibrium (LD) decay was calculated between pairs of polymorphic sites using PopLDdecay v3.41 [54] with 3.8 million SNPs. The parameters were set to “-InVCF -OutStat -MaxDist 1000 -MAF 0.0001 -Miss 0.5 -OutType 2”. In order to assess the decay of LD with physical distance, the LD between pairs of polymorphic sites was plotted against genetic distance (bp) between sites in a nonlinear regression. An r2 value = 1 was assumed for a marker distance of 0 kb, and LD decay for each population was evaluated at a cut-off of r2 = 0.1.
Reconstruction of phylogeny and analysis of population structure
PLINK51 v1.90 [55] was used with the parameters “-indep-pairphase 100 10 0.2” for all individuals to generate a pruned SNP set (3,855,120 SNPs) for use in the phylogenetic analysis of population structure. Based on the distance matrix, a neighbor-joining tree was then constructed in MEGA X [56] with the parameters “-a *.mao -d *.meg -f mega -o” and the outgroup Cucurbita moschata. EIGENSOFT [57] was used with the default parameters to run a principle components analysis and the three components explaining the most variation in the data were identified. ADMIXTURE60 v 1.3.0 [58], (“-C 0.01 -s time -cv -j4” for multiple repeats with different random seeds) was used to make a maximum likelihood estimation of the individual ancestries of the samples.
Correlations between different genetic, geographical and environmental factors
To investigate any potential relationships between environmental factors and genetic differences in different populations, we performed correlation analyses between the genetic and geographical distances and environmental factors. Environmental distances were calculated in NTSYSPC v2.11c [59] and environmental data were downloaded from http://www.wheata.cn/ [60]. Five key environmental factors affecting the growth and development of C. ficifolia were applied in the analyses: annual mean temperature, annual precipitation, altitude, number of frost-free days and annual average sunshine (Table S1). To examine whether genetic distances between pairs of populations increased linearly as a function of geographic or environmental distances, we employed Mantel tests [61] against pairs of distance matrices using the program zt [62], with 10,000 permutations to test for significance.
To determine whether or not the explanation of genetic differentiation by environmental factors was due to isolation, partial Mantel tests were performed using zt [62], again with 10,000 permutations. Matrices of genetic distance, geographical and genetic distances and environmental distance were compared, where appropriate controlling for the effects of environmental or physical distance.
Marker-trait association analyses
In the IBD test described above, we found that genetic distance was significantly correlated with environmental distance. Based on this, we investigated which of the SNPs responded to environmental factors. We used our resequenced SNPs data and the five key environmental factors mentioned above (annual mean temperature, annual precipitation, latitude, frost-free season and annual average sunshine) from the 32 sampling regions in an association analysis in order to identify any SNP sites associated with climate traits. Each individual in the same population (location) was given the same environmental data, and geographical location, phenotypic information and meteorological factors for each individual were added to the analysis and are shown in Table S4. Association mapping of SNPs with genes was performed in the GEMMA software with the following models: LM and LMM, the generalized linear model (GLM) [63]; factored spectrally transformed linear mixed model (FaST-LMM) [64], and the efficient mixed model association expedited (EMMAX) [63,64,65]. SNPs were chosen for the association analysis only if their allele frequencies were > 5%. The upper and lower thresholds for screening candidate marker sites were calculated as 0.1 and 0.01. Values within -log10(p) = 5 were also fixed as candidate regions. The candidate genes screened using the above thresholds were then functionally annotated. Quantile–quantile plots were used to determine the optimum model, and the linear models were tested by plotting P values obtained from the association tests against an expected probability distribution. A cut-off of 0.05 was used to determine significance.
Ecological niche modeling
In order to further determine the impact of environmental factors on the distribution of C. ficifolia, we applied ecological niche modeling (ENM) to simulate the distribution of C. ficifolia during the the present and in the future. ENM was performed in MAXENT v3.3.3 [30, 66], as MAXENT can calculate probability distributions using presence-only data from herbarium records [30]. The 32 sampled populations were included in this study (Table S1), and the ENM was performed using the default parameters, except for the following: application of random seed and random test percentage 70%, replicates of 10 and replicated run type bootstrap. Model predictions were visualized in ARCMAP 9.3 (ESRI, Redlands, CA, USA). A general circulation model, CCSM, was used to model the suitability of the future climate for the growth of C. ficifolia. In this experiment, we projected the model of climate suitability generated with ecological niche modeling using current climate data onto the global circulation model for the year 2080. The prediction performance analyses were evaluated in MAXENT by calculating the area under the receiver operation characteristic curve.
Conclusion
The availability of reference genomes and the continuous improvements of genetic data analysis methods are fostering resequencing studies. In this study, we performed the resequencing of C. ficifolia accessions, which has allowed identification of genetic markers, such as SNPs and InDels. The SNPs discovered in this study suggest that environmental factors mediated the formation of the population structure of C. ficifolia. These SNPs and Indels might contribute to the variation in important pathways of genes for important agronomic traits such as yield, disease resistance and stress tolerance. Moreover, the genome resequencing data and the genetic markers identified from 223 accessions provide insight into the genetic variation of the C. ficifolia germplasm and facilitate a broad range of genetic studies.
Availability of data and materials
The sequencing data generated in this study for the 223 samples is currently being submitted to the NCBI Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) under the BioProject accession PRJNA924019 with run accession numbers from SRX19037395 to SRX19037617.
References
Lee CR, Mitchell-Olds T. Quantifying effects of environmental and geographical factors on patterns of genetic differentiation. Mol Ecol. 2011;20(22):4631–42.
Ortego J, Riordan EC, Gugger PF, Sork VL. Influence of environmental heterogeneity on genetic diversity and structure in an endemic southern Californian oak. Mol Ecol. 2012;21(13):3210–23.
Pearson RG, Dawson TP, Berry PM, Harrison PA. SPECIES: A spatial evaluation of climate impact on the envelope of species. Ecol Model. 2002;154(3):289–300.
Merrell DJ. Ecological Genetics. London: Longman; 1981.
Davis FW, Westfall R, Flint A, Ikegami M, Wang HF, Grivet D. Gene movement and genetic association with regional climate gradients in California valley oak (Quercus lobata Née) in the face of climate change. Mol Ecol. 2010;19(17):3806–23.
Sacks BN, Brown SK, Ernest HB. Population structure of California coyotes corresponds to habitat-specific breaks and illuminates species history. Mol Ecol. 2004;13(5):1265–75.
Freeland JR, Biss P, Conrad KF, Silvertown J. Selection pressures have caused genome-wide population differentiation of Anthoxanthum odoratum despite the potential for high gene flow. J Evol Biol. 2010;23(4):776–82.
Zhongming L. Cucurbita ficifolia, excellent germplasm resources. Yunnan Agri Sci Techn. 1991;4(4):40–2.
Lira R, Rodríguez JC, Alvarado JL, Rodríguez I, Castrejón J, Domínguez Mariani A. Diversidad e importancia de la familia Cucurbitaceae en México. Acta Bot Mex. 1998;42:43–7.
Moya-Hernández A, Bosquez-Molina E, Serrato-Díaz A, Blancas-Flores G, Alarcón-Aguilar FJ. Analysis of genetic diversity of Cucurbita ficifolia Bouché from different regions of Mexico, using AFLP markers and study of its hypoglycemic effect in mice. S Afr J Bot. 2018;116:110–5.
Banderas-Dorantes TR, Roman-Ramos R, Zamilpa A, Garcia-Macedo R, Diaz M, Campos MG, Tortoriello J, Alarcon-Aguilar FJ. Influence of two hypoglycemic Cucurbitaceae (Cucurbita ficifolia Bouche and Ibervillea sonorae Greene) on ATP-sensitive potassium channels in rat aortic rings. B Latinoam Caribe Pl. 2012;11(6):510–9.
Roman-Ramos R, Almanza-Perez JC, Fortis-Barrera A, Angeles-Mejia S, Banderas-Dorantes TR, Zamilpa-Alvarez A, Diaz-Flores M, Jasso I, Blancas-Flores G, Gomez J, et al. Antioxidant and anti-inflammatory effects of a hypoglycemic fraction from Cucurbita ficifolia Bouche in streptozotocin-induced diabetes mice. Am J Chinese Med. 2012;40(1):97–110.
Zambrowicz A, Eckert E, Pokora M, Bobak L, Dabrowska A, Szoltysik M, Trziszka T, Chrzanowska J. Antioxidant and antidiabetic activities of peptides isolated from a hydrolysate of an egg-yolk protein by-product prepared with a proteinase from Asian pumpkin (Cucurbita ficifolia). Rsc Adv. 2015;5(14):10460–7.
Miranda-Perez ME, Ortega-Camarillo C, Escobar-Villanueva MD, Blancas-Flores G, Alarcon-Aguilar FJ. Bouche increases insulin secretion in RINm5F cells through an influx of Ca2+ from the endoplasmic reticulum. J Ethnopharmacol. 2016;188:159–66.
Fortis-Barrera A, Garcia-Macedo R, Almanza-Perez JC, Blancas-Flores G, Zamilpa-Alvarez A, Flores-Saenz JL, Cruz M, Roman-Ramos R, Alarcon-Aguilar FJ. Cucurbita ficifolia (Cucurbitaceae) modulates inflammatory cytokines and IFN-gamma in obese mice. Can J Physiol Pharm. 2017;95(2):170–7.
Zhou YH, Zhou J, Huang LF, Ding XT, Shi K, Yu JQ. Grafting of Cucumis sativus onto Cucurbita ficifolia leads to improved plant growth, increased light utilization and reduced accumulation of reactive oxygen species in chilled plants. J Plant Res. 2009;122(5):529–40.
Huang Y, Bie ZL, He SP, Hua B, Zhen A, Liu ZX. Improving cucumber tolerance to major nutrients induced salinity by grafting onto Cucurbita ficifolia. Environ Exp Bot. 2010;69(1):32–8.
Zhao LL, Liu AQ, Song TF, Jin YZ, Xu X, Gao Y, Ye XL, Qi HY. Transcriptome analysis reveals the effects of grafting on sugar and α-linolenic acid metabolisms in fruits of cucumber with two different rootstocks. Plant Physiol Bioch. 2018;130:289–302.
Luan H, Niu C, Nie X, Li Y, Wei M. Transcriptome and physiological analysis of rootstock types and silicon affecting cold tolerance of cucumber seedlings. Plants (Basel). 2022;11:445.
Dámasol MP, Carlos DL. Caracterización de frutos, semillas y plántulas de portainjertos de cítricos. Réanimation Urgences. 1999;2(3):259–66.
Nee M. The domestication of Cucurbita (Cucurbitaceae). Econ Bot. 1990;44(3 Supplement):56–68.
Kehua C. Cultivation techniques of Cucurbita ficifolia. Changjiang veget. 1994;6:9–11.
Xu Q, Shi Y, Yu T, Xu X, Yan Y, Qi X, Chen X. Whole-genome resequencing of a cucumber chromosome segment substitution line and its recurrent parent to identify candidate genes governing powdery mildew resistance. PLoS ONE. 2016;11(10):e0164469.
Xanthopoulou A, Ganopoulos I, Psomopoulos F, Manioudaki M, Moysiadis T, Kapazoglou A, Osathanunkul M, Michailidou S, Kalivas A, Tsaftaris A. De novo comparative transcriptome analysis of genes involved in fruit morphology of pumpkin cultivars with extreme size difference and development of EST-SSR markers. Gene. 2017;622:50–66.
Galpaz N, Gonda I, Shem-Tov D, Barad O, Katzir N. Deciphering genetic factors that determine melon fruit-quality traits using RNA-seq-based high-resolution QTL and eQTL mapping. Plant J. 2018;94(1):169–91.
Guo S, Zhao S, Sun H, Wang X, Wu S, Lin T, Ren Y, Gao L, Deng Y, Zhang J, et al. Resequencing of 414 cultivated and wild watermelon accessions identifies selection for fruit quality traits. Nat Genet. 2019;51(11):1616–23.
Xanthopoulou A, Montero-Pau J, Mellidou I, Kissoudis C, Blanca J, Pico B, Tsaballa A, Tsaliki E, Dalakouras A, Paris HS, et al. Whole-genome resequencing of Cucurbita pepo morphotypes to discover genomic variants associated with morphology and horticulturally valuable traits. Hortic Res. 2019;6:94.
Liu S, Gao P, Zhu Q, Zhu Z, Liu H, Wang X, Weng Y, Gao M, Luan F. Resequencing of 297 melon accessions reveals the genomic history of improvement and loci related to fruit traits in melon. Plant Biotech J. 2020;18(12):2545–58.
Sun H, Wu S, Zhang G, Jiao C, Guo S, Ren Y, Zhang J, Zhang H, Gong G, Jia Z, et al. Karyotype Stability and Unbiased Fractionation in the Paleo-Allotetraploid Cucurbita Genomes. Mol Plant. 2017;10(10):1293–306.
Phillips SJ, Anderson RP, Schapire RE. Maximum entropy modeling of species geographic distributions. Ecol Modell. 2006;190(3–4):231–59.
Meffe GK, R. CC: Principle of conservation biology. Sunderland, Massachusetts; 1994.
Song G, Deyuan H. Genetic diversity and its detection method. In: Qian Q, Deping M, editors. Principles of biodiversity research and methods. Beijing: China Science and Technology; 1994.
Ma L, Wang Q, Zheng Y, Guo J, Yuan S, Fu A, Bai C, Zhao X, Zheng S, Wen C, et al. Cucurbitaceae genome evolution, gene function, and molecular breeding. Hortic res. 2022;9:uhab057.
Varshney RK, Thudi M, Roorkiwal M, He W, Upadhyaya HD, Yang W, Bajaj P, Cubry P, Rathore A, Jian J, et al. Resequencing of 429 chickpea accessions from 45 countries provides insights into genome diversity, domestication and agronomic traits. Nat Genet. 2019;51(5):857–64.
Zhou Z, Jiang Y, Wang Z, Gou Z, Lyu J, Li W, Yu Y, Shu L, Zhao Y, Ma Y, et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat Biotechnol. 2015;33(4):408–14.
Wright S. Isolation by distance. Genetics. 1943;28(2):114.
Slatkin M. Gene flow and the geographic structure of natural populations. Science. 1987;236(4803):787–92.
Nosil P, Vines TH, Funk DJ. Reproductive isolation caused by natural selection against immigrants from divergent habitats. Evolution. 2005;59(4):705–19.
Defaveri J, Jonsson PR. Meril? J: Heterogeneous genomic differentiation between walking-stick ecotypes:“isolation by adaptation” and multiple roles for divergent selection. Evolution. 2013;67(9):1425–6.
Thibert-Plante X, Hendry AP. When can ecological speciation be detected with neutral loci? Mol Ecol. 2010;19(11):2301–14.
Scheiner SM. genetic and evolution of phenotypic plasticity. Annu Rev Ecol S. 1993;24:35–68.
He Q, Edwards DL, Knowles LL. Integrative testing of how environments from the past to the present shape genetic structure across landscapes. Evolution. 2013;67(12):3386–402.
Freedman AH, Thomassen HA, Buermann W, Smith TB. Genomic signals of diversification along ecological gradients in a tropical lizard. Mol Ecol. 2010;19(17):3773–88.
Pease KM, Freedman AH, Pollinger JP, McCormack JE, Buermann W, Rodzen J, Banks J, Meredith E, Bleich VC, Schaefer RJ. Landscape genetics of California mule deer (Odocoileus hemionus): the roles of ecological and historical factors in generating differentiation. Mol Ecol. 2009;18(9):1848–62.
Wright S. The genetical structure of populations. Ann Eugenic. 1950;15:323–54.
Za Y, Ly Z. Fu Z, Gao T, Zhao K, Zhang J, Ding Ym: Response of three cultivated varieties of Cucurbita ficifotia to fusarium wilt. Chin J Trop Crops. 2017;38(1):144–9.
Porebski S, Bailey L. Grant: Modification of a CTAB DNA extraction protocol for plants. Plant Mol Biol Rep. 1997;15(1):8–8.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10(2):giab008.
Mckenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M. The genome analysis Toolkit: A map reduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Stacks: an analysis tool set for population genomics. Mol Ecol. 2013;22(11):3124–40.
Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH. Stacks: Building and Genotyping Loci De Novo From Short-Read Sequences. G3- Genes Genom Genet. 2011;1(3):171–82.
Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10(3):564–7.
Zhang C, Dong SS, Xu JY, He WM, Yang TL. PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics. 2019;35(10):1786–8.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira M, Bender D, Maller J, Sklar P, Bakker P, Daly MJ. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–9.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–9.
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 1997;19(9):1655–64.
Jensen RJ. Ntsys-Pc-numerical taxonomy and multivariate-analysis system-version 1.40. Q Rev Biol. 1989;64(2):250–2.
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A. Very high resolution interpolated climate surfaces for global land areas. Int J Climatol. 2005;25(15):1965–78.
Mantel N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967;27:209–20.
Bonnet E, Van de Peer Y. Zt: a software tool for simple and partial Mantel tests. J Stat Softw. 2002;7:1–12.
Tang Y, Liu Xl, Wang Jb, Li M, Feng Qs. GAPIT Version 2: An enhanced integrated tool for genomic association and prediction. Plant Genome. 2016;9(2):1–9.
Staff PG. Correction: Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS genet. 2018;12(3):e1005957.
Zhang Z, Ersoz E, Lai CQ, Todhunter RJ, Tiwari HK, Gore MA, Bradbury PJ, Yu J, Arnett DK, Ordovas JM, et al. Mixed linear model approach adapted for genome-wide association studies. Nat Genet. 2010;42(4).
Phillips SJ, Dudik M. Modeling of species distributions with Maxent: new extensions and a comprehensive evaluation. Ecography. 2008;31(2):161–75.
Acknowledgements
We would like to thank Yongjie Guo, Mingjian Feng, and Yan Zhao for help with sampling, and Junheng L. V. for help with data analysis.
Funding
The project was supported by the National Natural Science Foundation of China (Project numbers 31500459), Yunnan Province agricultural basic research project (202301BD070001-027), Expert Workstation of Xiaolan Zhang (202205AF150021), and the Grant of Young Talents of Yunnan Xingdian Talents.
Author information
Authors and Affiliations
Contributions
ZY and SH developed the research concepts. SH directed most of the experimental and analytical work and wrote the manuscript. JZ, GL and YD revised the manuscript. JZ, HZW, JX and HWcollected the leaf material and participated in the experimental work. ZY and SH acquired the funding. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The experiments did not involve endangered or protected species. The data collection of plants was carried out with the permission from related institution, and complied with national or international guidelines and legislation.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Geographical location, phenotypic information and meteorological factors of sample sites.
Additional file 2: Table S2.
SNP and InDel information.
Additional file 3: Table S3.
Pairwise fixation index (FST) values among 32 populations.
Additional file 4: Table S4.
Gene number annoated from different databases.
Additional file 5: Figure S1.
GO Enrichment analysis of marker-trait association related genes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
He, S., Li, G., Zhang, J. et al. The effect of environmental factors on the genetic differentiation of Cucurbita ficifolia populations based on whole-genome resequencing. BMC Plant Biol 23, 647 (2023). https://doi.org/10.1186/s12870-023-04602-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-023-04602-3