
Abstract
Background
N6-methyladenosine (m6A) modification is the most abundant type of RNA modification in eukaryotic cells, playing pivotal roles in multiple plant growth and development processes. Yet the potential role of m6A in conferring the trait of male sterility in plants remains unknown.
Results
In this study, we performed RNA-sequencing (RNA-Seq) and m6A-sequencing (m6A-Seq) of RNAs obtained from the anther tissue of two wolfberry lines: ‘Ningqi No.1’ (LB1) and its natural male sterile mutant ‘Ningqi No.5’ (LB5). Based on the newly assembled transcriptome, we established transcriptome-wide m6A maps for LB1 and LB5 at the single nucleus pollen stage. We found that the gene XLOC_021201, a homolog of m6A eraser-related gene ALKBH10 in Arabidopsis thaliana, was significantly differentially expressed between LB1 and LB5. We also identified 1642 and 563 m6A-modified genes with hypermethylated and hypomethylated patterns, respectively, in LB1 compared with LB5. We found the hypermethylated genes significantly enriched in biological processes related to energy metabolism and lipid metabolism, while hypomethylation genes were mainly linked to cell cycle process, gametophyte development, and reproductive process. Among these 2205 differentially m6A methylated genes, 13.74% (303 of 2205) were differentially expressed in LB1 vis-à-vis LB5.
Conclusions
This study constructs the first m6A transcriptome map of wolfberry and establishes an association between m6A and the trait of male sterility in wolfberry.
Similar content being viewed by others


Backgrounds
N6-methyladenosine (m6A) is one of the most prevalent chemical modifications found in eukaryotic mRNAs. With recent advances in high-throughput technologies, transcriptome-wide m6A modifications have been profiled for A. thaliana and several other plant species [1,2,3,4]. These pioneering studies demonstrated that m6A modification plays important roles in multiple plant growth and development processes and is responsive to a variety of biotic and abiotic stresses [5,6,7,8,9,10]. Male sterility, which is an important agronomic trait for heterosis utilization and hybrid seed production, has been reported in several plants including maize [11], wheat [12], and tomato [13]. Its underlying molecular mechanisms have been long-time investigated, resulting in the identification of tens of male sterility-related genes in Arabidopsis thaliana and rice [11]. These male sterility-related genes are involved in multiple biological processes including transcription and lipid metabolism [14]. Recently, RNA m6A modification was also found to be associated with male infertility in human studies [15]. Despite that monumental progress, the potential association of m6A modifications in the trait of male sterility in plants is still unknown.
Lycium barbarum (Goji or wolfberry) is a famous plant with medicinal and commercial value that belongs to the Solanaceae family. In China, ‘Ningqi No.1’ (denoted as LB1) is one of the major cultivars with multiple excellent traits including high yield, large fruit size and disease resistance. ‘Ningqi No.5’ (LB5) is a natural male sterile mutant of LB1. In contrast to LB1, LB5 plants usually exhibit abortive phenotypes such as anther shrinkage, disintegration, and vacuolization, resulting in nonfunctional anthers (i.e., no pollen in the anther) [16, 17]. Hence, LB1 and LB5 are recognized as useful materials for studying pollen development, cytoplasmic and nuclear interactions, and male fertility-related gene regulations in plants [17, 18]. However, research using this important economic crop (wolfberry) to study the trait of male sterility in plants is still scarce.
Here, we performed RNA-sequencing (RNA-Seq) and m6A-sequencing (m6A-Seq) of the anther samples of LB1 and LB5, and assembled a consensus transcriptome to derive transcriptome-wide m6A maps of these two wolfberry lines. By comparing the omics data generated from LB1 vis-à-vis LB5, we identified those genes with differential expression and differential m6A methylation patterns, and then carried out their gene ontology (GO) enrichment analysis to uncover the biological processes that may be associated with the male sterility trait in wolfberry. Our research findings provide new insight into how m6A can regulate the trait of male sterility in plants.
Results
Reference-guided assembly and annotation of the wolfberry transcriptome
For both LB1 and its natural male sterile mutant LB5 (Figure S1), we respectively sequenced the transcriptomes of six anther samples from the tetrad stage (T), the single nucleus pollen stage (S), and the mature pollen stage (M) (LB1_T, LB1_S, LB1_M, LB5_T, LB5_S, LB5_M; see Methods), each with three biological replicates. Raw reads from each RNA-Seq library were pre-processed using fastp (v0.20.1) [19] to remove the adapter and low-quality sequences, resulting in a total of 908.8 million paired-end reads (63.5 Gb) (Table 1). After that pre-processing, the remaining reads were mapped onto the wolfberry reference genome sequences (accession number PRJNA640228) [20], using the HISAT2 program (v2.2.1) [21], whose alignment rate ranged from 93.39% to 96.24%. Then, using Cufflinks software (v2.2.1) [22], transcript fragments were first assembled for each RNA-Seq library, and then merged to generate a transcriptome assembly for both wolfberry lines under study. Under the criteria defined in a previous study [23], 29 324 genes and 84 709 transcript fragments were deemed high quality, that is, having a TPM ≥ 1 and read count ≥ 5 in at least one sample (Fig. 1A). Of these 29 324 genes, we found 25 841 that were expressed (i.e., TPM ≥ 1) in both LB1 and LB5 lines, with 2395 and 829 genes specifically expressed in each, respectively. In LB1, the number of specifically expressed genes decreased in the order of T stage (802) > M stage (367) > S stage (206), while in LB5, they were more abundant at the S stage (1253) than either the other two stages (T: 452 and M: 185) (Fig. 1B; Table S1).

Transcriptome assembly and annotation of the two wolfberry lines. A Overview of wolfberry transcriptome assembly pipeline. B Venn diagram of genes expressed in LB1 and LB5 at three developmental stages. C BUSCO’s assessment results. D Distribution of sources for functional annotation of assembled transcripts in wolfberry
Overall, 83.3% (24 434) of 29 324 annotated genes (Table S2) were predicted capable of encoding proteins by TransDecoder [24]. These 24 434 protein-encoding genes (PCGs) served as the input of BUSCO [25] to evaluate the completeness of the assembled transcripts. BUSCO’s assessment results revealed that of the 5950 core genes queried, 94.47% were detected (5574 complete and 49 partial) (Fig. 1C). Among these 24 434 PCGs, we found that 22 997 (94.12%) can be assigned with a function annotation label when using EggNOG-mapper (emapper) [26]. The vast majority of (90%) this annotation information came from the clade of asterids (e.g., Solanum lycopersicum, Solanum tuberosum) that are closely related taxonomically to wolfberry (Table S3; Fig. 1D). In addition, 1517 of these 24 434 PCGs (Table S4) were predicted to be transcript factors (TFs) according to PlantTFDB v5.0 [27].
Overall, the assembled transcriptome, along with its corresponding functional annotations, provided a comprehensive resource now available for the transcriptome-wide m6A analysis and other functional genomics studies in wolfberry.
Identification and expression analysis of m6A potential regulators in wolfberry
A total of 25 m6A potential regulators (7 writers, 10 erasers, and 8 readers) in wolfberry were identified from wolfberry’s assembled transcriptome by applying a specified bioinformatics pipeline (Fig. 2B; Table 2; Figures S2 and S3). These potential regulators were named according to their predicted orthologs from Arabidopsis [28] and tomato [29]. Among these 25 potential regulators, 22 were relatively highly expressed with TPM ≥ 10 in both LB1 and LB5 lines. These 22 genes exhibited dynamic expression patterns across T to M stages in either line (Figure S4). Several genes had markedly divergent expression dynamics between LB1 and LB5. For example, certain m6A writer genes, namely XLOC_003715 (LbaMTC, homologous with AtMTC) and XLOC_10786 (LbaMTB, homologous with AtMTB; Figure S3B), displayed different expression patterns when going from the T to M stages between LB1 and LB5 (Fig. 2A). There were 14 genes whose expression was greater in LB5 than LB1 at all three stages studied. One representative example is XLOC_016741 (LbaYTH5) encoding an YTH domain-containing protein. Gene LbaYTH5 is close to the experimentally demonstrated Arabidopsis m6A readers AtECT6 and AtECT7 in the phylogenetic tree, constructed with the FastTree software [30] using YTH domain-containing proteins from wolfberry, tomato, maize, A. thaliana and Physcomitrium patens (Fig. 2B). In LB1, the expression level of LbaYTH5 was 68.64, 51.97, and 56.27 at the T, S, and M stages, but higher at 76.26, 83.20, and 104.35 in LB5, respectively (Figure S5).

Identification and expression analysis of putative m6A regulators in wolfberry. A Dynamic trends in the expression levels of two example genes. B Phylogenetic tree of the YTH gene family (Ppa: Physcomitrella patens, Zma: Zea mays, Ath: Arabidopsis thaliana, Sly: Solanum lycopersicum and Lba: Lycium barbarum); the color-marked genes are annotated YTH genes in wolfberry. C Volcano plot of differentially expressed genes at each stage (p-value < 0.05, absolute fold-change > 2)
Pair-wise comparisons also revealed that genes of some m6A potential regulators differed starkly in their expression between LB1 and LB5. Using DESeq2 [31], we identified 4499, 4575, and 7491 differentially expressed genes (DEGs) between LB1 and LB5 at the T, S, and M stages, respectively (Fig. 2C; Table S5). One representative gene is XLOC_021201, which may encode an m6A eraser regulator, belonged to the same evolutionary branch as the Arabidopsis gene AtALKBH10 (Figure S3A). Compared with LB5, this gene had markedly higher values of TPM in LB1 at both S and M stages (Figure S4). Altogether, these results revealed the differential expression patterns of genes encoding m6A potential regulators between LB1 and LB5 during the anther development of wolfberry from the T to M stage.
Transcriptome-wide identification of m6A methylation in LB1 and LB5
With the DEGs encoding m6A potential regulators in mind, we then asked whether there some differences in the m6A methylome also arise between LB1 and LB5. To address this question, we firstly measured the m6A/A ratio of pollens from these two wolfberry lines at the S stage, and found that the m6A level in mRNA was slightly different between LB1 and LB5 (Figure S6). Then, we used anther samples at the S stage as an example to obtain m6A maps of LB1 and LB5 via m6A-Seq technology. Six m6A-immunoprecipitation (IP) and matched input (non-IP control) libraries were constructed and sequenced for RNAs from the anther samples of these two lines at the S stage, with three biological replicates per sample. Raw sequencing reads from each library were processed to discard adaptor sequences and low-quality bases using the fastp (v0.20.1) [19]. The resulting reads from the wolfberry LB1 and LB5 samples were aligned to the wolfberry reference genome (accession number PRJNA640228) using HISAT2 (v2.2.1) [20, 21]. Read distribution analysis showed that the reads from m6A-IP samples accumulated highly around the stop codon and within the 3’-untranslated region (3’UTR) in all samples, with the sequencing data of input and IP being highly correlated between replicates, thus confirming the high quality of m6A-Seq in this study (Fig. 3A).

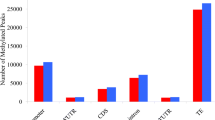
Overview of the m6A methylome in wolfberry. A The read distribution of input and IP data from m6A-Seq of LB1 and LB5. B Comparison of m6A peaks between LB1 and LB5. C Peak density in five non-overlapping transcript segments: the 5’-untranslated region (5’UTR), near start codon, coding sequence (CDS), near stop codon, and 3’-untranslated region (3’UTR). D Relative enrichment of the m6A peaks in the five non-overlapping transcript segments. E The motif on the top is the 1st-ranked enriched URUAY motif (where R denotes A/G, A is m6A and Y denotes C/U). The motif on the bottom is the canonical RRACH motif (where R denotes A/G, A is m6A, and H denotes A/C/U). F Volcano plot of differentially methylated genes (p-value < 0.05, absolute fold-change > 2)
We detected 10 389 and 9301 m6A peaks in LB1 and LB5, corresponding to 9587 and 8668 m6A-modified genes, respectively (Fig. 3B; Table S6). In both LB1 and LB5 lines, their m6A-modified genes (m6A genes) were significantly longer than genes without m6A peaks (non-m6A genes) (Student’s t-test, p-value < 0.001) (Figure S7). In addition, those m6A-modified genes exhibited greater expression than non-m6A genes (Figures S8 and S9). Moreover, m6A peaks were enriched in the following order: 3’UTR (3’-untranslated region) > near stop codon > CDS (coding sequence) > start codon > 5’UTR (5’-untranslated regions) (Fig. 3C, D). All these m6A peaks were scanned further for enriched motifs, using MEME suite (http://meme-suite.org/index.html) [32]. As expected, the URUAY (where R represents A/G and Y represents C/U; Fig. 3E) motif was significantly enriched within the m6A peaks, and in both lines the URUAY motif is the most enriched one. We next examined the canonical m6A motif RRACH (where R represents A/G, A is m6A, and H represents A/C/U; Fig. 3E), using another commonly used motif analysis program, HOMER (v4.10) [33]. Evidently, the RRACH motif could also be detected in our m6A-Seq data, it being significantly enriched in m6A peaks vis-à-vis non-m6A regions (Figure S10).
Furthermore, a differential methylation analysis was performed by RADAR [34], which uncovered 2205 genes that were differentially m6A-modified between LB1 and LB5. Among these 2205 differentially m6A-modified genes, 1642 genes (including 67 TFs) were hypermethylated in line LB1 compared with line LB5, while 563 genes (including 19 TFs) were hypomethylated (Table S7; Fig. 3F). Previous studies have linked a number of TFs from MADS, MYB, ARF, and GATA gene families to plant male sterility [35,36,37,38,39]. In our study, 86 differentially m6A-modified genes may have encoded members of those TF families (i.e., bHLH: 11, B3: 6, WRKY: 5, NAC: 5). Interestingly, we found that the gene XLOC_003715 encoding an m6A writer also had differential methylation between the LB1 and LB5 lines (Figure S3B). Further, the gene XLOC_027737 that may encode an m6A demethylase was specifically hypermethylated in LB1 (Figure S3A). These results suggested that m6A methylation at the RNA level may form a complex framework for epigenetic regulation of male sterility in wolfberry.
Functional enrichment analysis of hypermethylated genes in wolfberry
Among 1642 hypermethylated genes, 1175 were LB1-specific methylation genes while the other 467 genes featured higher m6A methylation levels in LB1 than LB5. Gene ontology (GO) enrichment analysis revealed these 1175 LB1-specific methylation genes being mainly enriched in biological processes, such as energy metabolism and biosynthesis, photosynthesis, lipid metabolism and biological component synthesis (Fig. 4A; Table S8). As an essential process in anther and pollen development, the destruction of lipid metabolism has been reported to lead to male sterility [14, 40]. Here, we found that 44 LB1-specific methylation genes, including XLOC_002359 (GATase), XLOC_002916 (Lipoxygenase), and XLOC_010833 (Cytochrome p450, CYP), were annotated to the lipid metabolic process (Tables S3 and S8).

Function enrichment analysis of the hypermethylated genes. A Scatterplot of the GO enrichment analysis of hypermethylated genes. B Volcano plot of DEGs and differentially methylated genes (p-value < 0.05, absolute fold-change > 2). C Enrichment of the m6A methylation of gene XLOC_009804 between LB1 and LB5
Similarly, the GO enrichment analysis showed that, for the 467 genes with higher methylation level in LB1 than LB5, they were also specifically enriched in biological processes, namely those associated with gametophyte development, pollen wall assembly and lipoprotein biosynthetic (Fig. 4A; Table S8). As other research has shown, pollen development is affected by pollen wall assembly in maize, wheat, and rice; abnormally developed pollens can cause male sterility [41,42,43]. We also found that 53 of these 467 genes were both differentially methylated and differentially expressed between LB1 and LB5 at the S stage (15 genes up-regulated and 38 genes down-regulated in LB1). One representative gene is XLOC_009804, encoding a bHLH TF, which showed hypermethylation and down-regulation in LB1 (Fig. 4B, C). In alfalfa plants, a tapetum-specific bHLH TF has been identified to play a regulatory role in the process conferring male sterility [44]. These results indicated that at least some of hypermethylated genes were closely linked to the trait of male sterility in wolfberry.
Functional enrichment analysis of hypomethylated genes in wolfberry
We further explored whether (or not) hypomethylated genes were associated with infertility in wolfberry. Of the 563 hypomethylated genes, 256 were LB5-specific methylation genes while the other 307 genes underwent a higher m6A methylation level in LB5 than LB1. GO enrichment analysis showed that these 256 LB5-specific methylation genes were mainly enriched in the cell cycle process, gametophyte development, and reproductive process (Fig. 5; Table S9). Being the most critical cell type for plant fertility and crop production, gametocyte development is directly linked to the formation of plant male sterility. Many of these pathways have been implicated in gametocyte development; for example, the cell cycle and reproductive processes directly affect the two pivotal stages: microsporogenesis and microgametogenesis [45]. Further, the 256 genes with a higher methylation level in LB5 were also specifically enriched in biological processes, such as cellular response to DNA damage, double-strand break repair, and DNA repair (Fig. 5; Table S9). Extensive studies of rice have shown that sustained double-strand breaks cause the programmed cell death of male gametes and complete male sterility [46,47,48,49].

Function enrichment analysis of hypomethylated genes. Network plot of GO enrichment analysis of hypomethylated genes (yellow: LB5-specific methylation genes; blue: genes with higher methylation level in LB5). The size of the circle represents the number of genes enriched in the GO terms
To gain deeper insight into the functioning of these hypomethylation genes, we performed a combinational analysis of transcriptome and m6A methylome in the anthers of LB1 and LB5 at the S stage. We found that 62 LB5-specific methylation genes are concomitantly differentially expressed (9 genes up-regulated and 53 genes down-regulated in LB1). One representative example is XLOC_020363, which encodes a TF belonging to the ERF gene family. The ERF gene family is thought to be involved in MYB regulation of pollen ablation and male sterility in canola [50]; our results suggested that at least some of hypomethylated genes may also participate in the trait of male sterility in wolfberry.
Collectively, these results suggested that m6A-modified genes figure prominently in how male sterility arises in wolfberry, and that gaining mechanistic insight into the functions of genes with both differential expression and differential methylome patterns is imperative for a better understanding of male sterility in plants.
Discussion
In the present study, we performed a series of m6A-Seq and RNA-Seq experiments on the anther samples of two wolfberry lines LB1 and LB5, and carried out in-depth bioinformatics analysis to establish the association between m6A modification and the male sterility trait in wolfberry.
Constructing the first m6A transcriptome map of wolfberry
Using the generated RNA-Seq datasets, we assembled a consensus transcriptome of wolfberry consisting of 29 324 genes and 84 709 transcript fragments (Fig. 1A; https://github.com/cma2015/Wolfberry_m6A). This addressed the problems caused by the lack of public availability of gene annotations for wolfberry. Based on the newly assembled transcriptome, we constructed the first m6A transcriptome map of wolfberry. The distribution of m6A within different gene contexts in wolfberry anther is similar to that reported in Arabidopsis [7], maize [1] and a few other under-studied plant species [2]. However, the top ranked motifs (and significance) enriched within m6A peaks in wolfberry anther (URUAY [Y = C/U] and RRACH [R = A/G, H = A/C/U]; Fig. 3E) differed from those reported in the anther of tomato (only URUAH) [9] as well as rice (WKUAH) [51]. This difference may represent the unique biological significance of m6A methylation among these species. We hope that the constructed m6A transcriptome map for the anther of wolfberry will be helpful to further understanding the regulatory mechanism of m6A in the anthers of plants [52].
Linking m6A-modified genes to the trait of male sterility in wolfberry
Plant male sterility is the outcome of a complex process that has been associated with abnormalities in growth substance metabolism, epigenetic regulation, and mitochondrial function [14, 35, 53,54,55]. So far, however, the association of m6A methylation with male infertility has only been explored in humans [15]. Our results show that an m6A potential regulator is differentially expressed between LB1 and LB5 (Figures S3A and S5). In addition, both hypermethylated and hypomethylated genes between LB1 and LB5 are involved in multiple biological processes (e.g., transcript and lipid metabolism) (Tables S8 and S9), which have been previously reported to be related to the trait of male sterility in plants. Among these differentially m6A modified genes, some are homologous to those of already annotated male sterility-related genes. Figure S11 depicts two representative genes, one encoding a lipoxygenase and the other encoding a bHLH transcription factor, with high evolutionary conservation of motif constituents between wolfberry and Arabidopsis. These observation results suggest the association between m6A and the trait of male sterility in wolfberry.
The integrative analysis of the transcriptome and m6A methylatome also enabled us to identify 4575 DEGs and 2205 differentially m6A-modificated genes (1642 hypermethylated and 563 hypomethylated genes) between LB1 and LB5. We found that 303 genes (including 9 TFs) were both differentially expressed and differentially methylated (Fig. 6; Table S10). This preliminary result shows that changes to m6A methylation could occur in genes with and without differential expression, which suggests that m6A modification may have different regulatory mechanisms in wolfberry (Fig. 7).

Venn diagram of numbers of differentially methylated genes and differentially expressed genes. A Venn diagram of numbers of hypermethylated genes and differentially expressed genes. B Venn diagram of numbers of hypomethylated genes and differentially expressed genes. Numbers in parentheses represent the number of TFs

Schematic illustration of potential m6A regulatory mechanisms in wolfberry
Limitations and future work
Despite these preliminary findings, there are still some limitations to this analysis that can be pursued as further work. Firstly, we applied the m6A-Seq technology to investigate the patterns of m6A modification peaks in the anther samples of LB1 and LB5 at the S stage. Further considering the m6A methylation changes across different developmental stages and using m6A sequencing technologies with single-nucleotide resolution will help us to improve the association between m6A modification and the trait of male sterility. Secondly, we only established the potential association between m6A modification and the trait of male sterility. More work, including ‘wet’ experiments and advanced bioinformatics analysis [56], is still needed to carefully validate whether m6A modification is a contributor to the male sterility trait or not. Thirdly and lastly, we computationally identified some m6A-modified genes of interest in wolfberry. In-depth study of the functions of these genes will contribute to field of plant genetics and breeding.
Conclusions
We show that the m6A landscape is altered in LB1 and its natural male sterile mutant LB5. Hence, changes to and from m6A methylation may be associated with the trait of male sterility in wolfberry. The modulation of m6A epitranscriptome might be a promising target for breeding male sterility lines in the future improvement of wolfberry and other crops.
Material and methods
Plant materials
The flower buds of LB1 and LB5 were harvested from the Quanxing Wolfberry Plantation (Zhongning County, Ningxia, China) at three developmental stages: (1) the tetrad (T) stage, when the flower bud length is 3.0–5.0 mm; (2) the single nucleus pollen (S) stage, when the flower bud length is 5.0–7.0 mm; (3) the mature pollen (M) stage, when the flower bud length is greater than 8.0 mm. For each developmental stage, three biological replicates were collected, each of which contained samples from at least three plants. After their collection, the samples of anthers were rapidly stripped on the dry ice, sub-packaged into centrifuge tubes, and frozen immediately in liquid nitrogen, then stored in − 80 °C freezer until used. These frozen plant materials were preserved in the sample storage room of College of Life Science, Ningxia University, China.
RNA isolation and library preparation for sequencing
Total RNA was isolated from the flower bud samples of LB1 and LB5 using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. For each sample, the NanoDrop ND-1000 (NanoDrop, Wilmington, DE, USA) was used to determine the amount and purity of total RNA. The completeness of total RNA was assessed by using the Bioanalyzer 2100 (Agilent, CA, USA), and the electrophoresis with denaturing agarose gel was used to confirm the results.
High-throughput RNA-Seq and m6A-Seq
For the RNA-Seq libraries, clustering of index-coded samples was performed on a cBot Cluster Generation System by using the TruSeq PE Cluster Kit v3-cBot-HS (Illumia) according to the manufacturer’s instructions. After cluster generation, the library preparation was sequenced on the Illumina HiSeq 2000 platform, from which the paired end reads (2 × 150 bp) were generated. For the m6A-Seq libraries, the m6A-Seq was performed as previously described [2]. Briefly, the Poly (A) RNA was purified from the 50 μg of total RNA using Dynabeads Oligo (dT)25-61005 (Thermo Fisher, CA, USA). After that, the Poly (A) RNA was truncated into small fragments using the Magnesium RNA Fragmentation Module (NEB, cat.e6150, USA). The ensuing truncated RNA small fragments were incubated with m6A-specific antibodies (No. 202003, Synaptic Systems, Germany) in an IP buffer (50 mM Tris–HCl, 750 mM NaCl, and 0.5% IgepalCA-630). And the RNA incubated with m6A-specific antibodies (IP RNA) was reverse transcribed using SuperScript™ II (Invitrogen, cat. 1896649, USA) to generate the complementary DNA (cDNA); Then, the U-labeled second-stranded DNAs was synthesized with E.coli DNA polymerase I (NEB, cat.m0209, USA), RNase H (NEB, cat.m0297, USA) and the dUTP Solution (Thermo Fisher, cat.R0133, USA).
Next, the A base, which connected to the indexed adapter, was added to the blunt end of each chain. The T base dangle on each adapter of each chain was used to connect the adapter to the A-tailed segment DNA. After that, both single- and dual-index adapters were connected to the fragments, and AMPureXP beads was used for size selection. After treatment of U-labeled second-strand DNAs with heat-labile UDG enzyme (NEB, cat.m0280, USA), the ligation products were amplified by PCR under conditions consistent with the previously described [2]. The mean insert size for the final cDNA library was 300 bp with SD 50. Finally, the 2 × 150 bp paired-end sequencing (PE150) was performed on an illumina Novaseq™ 6000 (LC-Bio Technology CO., Ltd., Hangzhou, China), this done following the vendor’s recommended protocol.
Reference-guided transcriptome assembly for wolfberry
In this study, the reference genome of L. barbarum (wolfberry) was downloaded from the GenBank (https://www.ncbi.nlm.nih.gov/genbank) repository at the National Center for Biotechnology Information (NCBI) [57] under accession number PRJNA640228 [20]. To perform the reference-guided transcriptome assembly, the quality of RNA-Seq data was first examined using MultiQC (v1.13.dev0) [58], after which any adapter and low-quality reads were removed using fastp (v0.20.1) [19]. Once the quality control was completed, the remaining clean reads were mapped to the genome sequences using the fast, splice-aware alignment program HISAT2 (v2.2.1) [21], this yielding the BAM (binary sequence alignment format) file recording read-genome alignments. Cufflinks (v2.2.1) software [22] was used to perform the transcriptome assembly. Finally, the RNA-Seq assembly results from the same line were merged using the Cuffmerge tool in Cufflinks (v2.2.1).
Construction of the consensus transcriptome for the two wolfberry lines
We screened the assembled transcript fragments on the basis of expression-level evidence [23]. The TPM (Transcripts Per Million) values were calculated using featureCounts (v2.0.1) [59] and read counts per base were calculated using bedtools genomecov (v2.30.0) [60]. Those assembled transcripts with either low expression (e.g., TPM < 1) or low read coverage (e.g., < 5) in the majority of each transcript fragments (e.g., 60%) were discarded. Finally, we merged the remaining high-confidence transcript fragments from two lines to construct a consensus transcriptome, and further calculated the TPM for the latter using featureCounts (v2.0.1) [59].
Gene expression analysis
To investigate the conditions under which certain genes are specifically expressed, their expression levels (TPM values) in samples from different developmental stages were compared. For each line, a gene was considered as expressed under a condition if it was expressed (e.g., TPM ≥ 1) in at least one of the three biological replicates. The R package ‘DESeq2’ [31] was used to search for DEGs (differentially expressed genes) based on the criteria of p-value < 0.05 and absolute fold-change > 2.
Gene structure and functional annotation
To predict the open reading frame (ORF) of assembled transcript fragments, we used the transcripts coding regions-finding software TransDecoder (https://github.com/TransDecoder/TransDecoder/releases, v5.5.0) [61]. The fast functional annotation tool EggNOG-mapper (emapper v2.1.9; eggNOG DB v5.0.2, http://eggnog5.embl.de; diamond vv2.0.1) [22, 62, 63] was used to annotate the assembled transcript fragments under these parameters: tax_scope = ‘Viridiplantae’. Further, the transcription factor information was simultaneously annotated using the Plant Transcription Factor Database (PlantTFDB v5.0, http://plantregmap.gao-lab.org) [27].
Annotation of putative m6A regulators in wolfberry
Six HMM (hidden Markov model) profiles of the m6A regulators were downloaded from the Pfam database (http://pfam-legacy.xfam.org) [64], namely eraser: ALKBH10B (PF13532), reader: YTH (PF04146), and writer: MT-A70 (PF05063), FIP37 (PF17098), VIR (PF15912), HAKAI (PF18408). These HMM profiles were respectively inputted into the hmmsearch program of HMMER software (v3.1b2) [65] to search the HMM-profile domain against known wolfberry protein sequences, following by building a new HMM profile for wolfberry, using the hmmbuild program of HMMER software. Next, the fast multiple sequence alignment software MAFFT (v7.310) [66] set to its default parameters was used to complete the multiple sequence alignment of the putative m6A regulators in five plant species (Physcomitrella patens, Zea mays, Arabidopsis thaliana, Solanum lycopersicum, and Lycium barbarum). FastTree (v2.1.10) [30] set to its default parameters was used to construct approximate maximum likelihood phylogenetic tree based on the multiple sequence alignment results (Figure S3).
Analyses of m6A-Seq data
Raw sequencing reads were cleaned using the fastp tool (v0.20.1) [19] to remove any reads containing adapter or low-quality sequences. Cleaned reads were then mapped onto the reference genome (accession number PRJNA640228) using HISAT2 (v2.2.1) [20, 21] under its default settings. To identify the m6A peak, the R package ‘exomePeak’—with a major update released (exomePeak2)—was used with default parameters [67]. The DREME (Discriminative Regular Expression Motif Elicitation) tool in MEME suite (http://meme-suite.org/tools/dreme) [32] was used to distinguish the relatively short (up to 8 bp), ungapped motifs, according the following parameters: minimum length of the motif = 5; maximum length of the motif = 7; E-value threshold = 1E-5. For a specified motif (e.g., RRACH or URUAY), to calculate the significance level of its relative enrichment, the AME tool in MEME suite (http://meme-suite.org/tools/ame) was used. The discovered m6A peaks were divided into five categories based on their positions: 5′-untranslated region (5’UTR), near start codon, coding sequence (CDS), near stop codon, and 3′-untranslated region (3’UTR). The coordinates of these genomic elements were extracted, then bedtools intersect (v2.30.0) [60] searched for overlapping between m6A peaks and each element. The R package ‘RADAR’ [34] was implemented to reveal the differentially methylated genes from the two wolfberry lines, using the criteria of p-value < 0.05 and an absolute fold-change > 2.
Quantitative analysis of mRNA m6A by LC–MS/MS
The LC–MS/MS technique was used to detect global m6A levels in wolfberry. For each sample, the RNA sample were digested into single nucleosides in a digestion buffer which contains phosphodiesterase I (0.01 U), nuclease S1 (180 U), 1 mM zinc sulfate, 280 mM sodium chloride and 30 mM sodium acetate. The digestion buffer was placed at 37℃ for 4 h at PH 6.8, and the bacterial alkaline phosphatase (30 U) was used to dephosphorylated for 2 h at 37℃. Enzymes were removed by filtration (Amicon Ultra 10 K MWCO). Then, the nucleosides samples were subjected to liquid chromatography coupled with tandem mass spectrometry (LC–MS/MS) which analysis on a QTRAP 4500 mass spectrometer (SCIEX, Framingham, MA, USA). The quantification of nucleosides was performed using the nucleoside-to-base ion mass transitions of 268.1 to 136.1 for A, 245.1 to 113.0 for U, 244.1 to 112.1 for C, 184.1 to 152.1 for G, 282.1–150.1 for RNA m6A. We determined the concentration of m6A and A by comparing with the standard curve obtained from their nucleoside standards, and analyzed the ratio of m6A to A based on the calculated concentrations. Three independent biological replicates were performed for this experiment.
Gene ontology enrichment analysis
Gene Ontology (GO) enrichment analysis was performed by implementing the R package ‘clusterProfiler’ [68].
Conservation analysis
Multiple sequence alignments of protein sequences were performed using MAFFT (v7.310) [65] with default parameters. FastTree (v2.1.10) [30] was used to construct approximate maximum likelihood phylogenetic tree with default parameters. Motif analysis was performed using MEME suite (http://meme-suite.org/tools/dreme) [32]. The conservation analysis results were displayed using TBtools (v1.120) [68].
Statistical analysis
The Student’s t test and Wilcoxon test were applied to the data, as respectively needed, using the R package ‘ggsignif’ (https://github.com/const-ae/ggsignif).
Availability of data and materials
All sequencing data have been deposited into the National Genomics Data Center (NGDC)’s Genome Sequence Archive database under the accession numbers PRJCA015572 (https://ngdc.cncb.ac.cn/search/?dbId=&q=PRJCA015572). The assembled wolfberry transcriptome and corresponding functional annotations are deposited at https://github.com/cma2015/Wolfberry_m6A. The frozen plant materials are available from corresponding authors upon reasonable request.
Abbreviations
- m6A:
-
N6-Methyladenosine
- RNA-Seq:
-
RNA-sequencing
- m6A-Seq:
-
M6A-sequencing
- LB1:
-
Ningqi No.1
- LB5:
-
Ningqi No.5
- T:
-
Tetrad stage
- S:
-
Single nucleus pollen stage
- M:
-
Mature pollen stage
- FC:
-
Fold-change
- GO:
-
Gene ontology
- bHLH:
-
Basic Helix-Loop-Helix
- ERF:
-
Ethylene-Responsive Factor
References
Miao Z, Zhang T, Qi Y, Song J, Han Z, Ma C. Evolution of the RNA N6-methyladenosine methylome mediated by genomic duplication. Plant Physiol. 2020;182:345–60.
Miao Z, Zhang T, Xie B, Qi Y, Ma C. Evolutionary Implications of the RNA N6-methyladenosine methylome in plants. Mol Biol Evol. 2022;39:msab299.
Zhu C, Zhang S, Zhou C, et al. RNA methylome reveals the m6A-mediated regulation of flavor metabolites in Tea leaves under solar-withering. Genomics Proteomics Bioinformatics. 2023;S1672-0229(23)00035-9. https://doi.org/10.1016/j.gpb.2023.02.003.
Yu Q, Liu S, Yu L, Xiao Y, Zhang S, Wang X, et al. RNA demethylation increases the yield and biomass of rice and potato plants in field trials. Nat Biotechnol. 2021;39:1581–8.
Hu J, Cai J, Xu T, Kang H. Epitranscriptomic mRNA modifications governing plant stress responses: underlying mechanism and potential application. Plant Biotechnol J. 2022;20:2245–57.
Govindan G, Sharma B, Li Y, Armstrong CD, Merum P, Rohila JS, et al. mRNA N6-methyladenosine is critical for cold tolerance in Arabidopsis. Plant J. 2022;111:1052–68.
Hu J, Cai J, Park SJ, Lee K, Li Y, Chen Y, et al. N6-methyladenosine mRNA methylation is important for salt stress tolerance in Arabidopsis. Plant J. 2021;106:1759–75.
Zhang K, Zhuang X, Dong Z, Xu K, Chen X, Liu F, et al. The dynamics of N6-methyladenine RNA modification in interactions between rice and plant viruses. Genome Biol. 2021;22:189.
Yang D, Xu H, Liu Y, Li M, Ali M, Xu X, et al. RNA N6-methyladenosine responds to low-temperature stress in tomato anthers. Front Plant Sci. 2021;12:687826.
He Y, Li L, Yao Y, Li Y, Zhang H, Fan M. Transcriptome-wide N6-methyladenosine (m6A) methylation in watermelon under CGMMV infection. BMC Plant Biol. 2021;21:516.
Wan X, Wu S, Li Z, Dong Z, An X, Ma B, et al. Maize genic male-sterility genes and their applications in hybrid breeding: progress and perspectives. Mol Plant. 2019;12:321–42.
Melonek J, Duarte J, Martin J, Beuf L, Murigneux A, Varenne P, et al. The genetic basis of cytoplasmic male sterility and fertility restoration in wheat. Nat Commun. 2021;12:1036.
Secgin Z, Uluisik S, Yıldırım K, Abdulla MF, Mostafa K, Kavas M. Genome-wide identification of the aconitase gene family in tomato (Solanum lycopersicum) and CRISPR-based functional characterization of SlACO2 on male-sterility. Int J Mol Sci. 2022;23:13963.
Wan X, Wu S, Li Z, An X, Tian Y. Lipid metabolism: critical roles in male fertility and other aspects of reproductive development in plants. Mol Plant. 2020;13:955–83.
Cai Z, Niu Y, Li H. RNA N6-methyladenosine modification, spermatogenesis, and human male infertility. Mol Hum Reprod. 2021;27:gaab020.
Shi J, Chen L, Zheng R, Guan C, Wang Y, Liang W, et al. Comparative phenotype and microRNAome in developing anthers of wild-type and male-sterile Lycium barbarum L. Plant Sci. 2018;274:349–59.
Zheng R, Yue S, Xu X, Liu J, Xu Q, Wang X, et al. Proteome analysis of the wild and yx-1 male sterile mutant anthers of wolfberry (Lycium barbarum L). PLoS One. 2012;7:e41861.
Yu J, Jiang M, Guo C. Crop Pollen development under drought: from the phenotype to the mechanism. Int J Mol Sci. 2019;20:1550.
Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–90.
Cao Y-L, Li Y, Fan Y-F, Li Z, Yoshida K, Wang J-Y, et al. Wolfberry genomes and the evolution of Lycium (Solanaceae). Commun Biol. 2021;4:671.
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907–15.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and abundance estimation from RNA-Seq reveals thousands of new transcripts and switching among isoforms. Nat Biotechnol. 2010;28:511–5.
Chen S, Ren C, Zhai J, Yu J, Zhao X, Li Z, et al. CAFU: a Galaxy framework for exploring unmapped RNA-Seq data. Brief Bioinform. 2020;21:676–86.
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity. Nat Protoc. 2013;8:1494–512. https://doi.org/10.1038/nprot.2013.084.
Seppey M, Manni M, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness. Methods Mol Biol. 2019;1962:227–45.
Cantalapiedra CP, Hernández-Plaza A, Letunic I, Bork P, Huerta-Cepas J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol Biol Evol. 2021;38:5825–9.
Tian F, Yang D-C, Meng Y-Q, Jin J, Gao G. PlantRegMap: charting functional regulatory maps in plants. Nucleic Acids Res. 2020;48:D1104–13.
Scutenaire J, Deragon J-M, Jean V, Benhamed M, Raynaud C, Favory J-J, et al. The YTH domain protein ECT2 is an m6A reader required for normal trichome branching in Arabidopsis. Plant Cell. 2018;30:986–1005.
Yin S, Ao Q, Tan C, Yang Y. Genome-wide identification and characterization of YTH domain-containing genes, encoding the m6A readers, and their expression in tomato. Plant Cell Rep. 2021;40:1229–45.
Price MN, Dehal PS, Arkin AP. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Bailey TL, Johnson J, Grant CE, Noble WS. The MEME suite. Nucleic Acids Res. 2015;43:W39-49.
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89.
Zhang Z, Zhan Q, Eckert M, Zhu A, Chryplewicz A, De Jesus DF, et al. RADAR: differential analysis of MeRIP-seq data with a random effect model. Genome Biol. 2019;20:294.
Hou Q, Zhang T, Qi Y, Dong Z, Wan X. Epigenetic dynamics and regulation of plant male reproduction. Int J Mol Sci. 2022;23:10420.
Zhou X, Liu W, Wang C, Xu Q, Wang Y, Ding S, et al. A MADS-box transcription factor MoMcm1 is required for male fertility, microconidium production and virulence in Magnaporthe oryzae: MoMCM1 and pathogenesis in M. oryzae. Mol Microbiol. 2011;80:33–53.
Yang W, Lou X, Li J, Pu M, Mirbahar AA, Liu D, et al. Cloning and functional analysis of MADS-box genes, TaAG-A and TaAG-B, from a wheat K-type cytoplasmic male sterile line. Front Plant Sci. 2017;8:1081.
Xia R, Xu J, Meyers BC. The emergence, evolution, and diversification of the miR390-TAS3-ARF pathway in land plants. Plant Cell. 2017;29:1232–47.
Li H, You C, Yoshikawa M, Yang X, Gu H, Li C, et al. A spontaneous thermo-sensitive female sterility mutation in rice enables fully mechanized hybrid breeding. Cell Res. 2022;32:931–45.
Liu S, Li Z, Wu S, Wan X. The essential roles of sugar metabolism for pollen development and male fertility in plants. Crop J. 2021;9:1223–36.
Begcy K, Nosenko T, Zhou L-Z, Fragner L, Weckwerth W, Dresselhaus T. Male sterility in maize after transient heat stress during the tetrad stage of pollen development. Plant Physiol. 2019;181:683–700.
Bu Y, Niu F, He M, Ye J, Yang X, Du Z, et al. The gene TaPG encoding a polygalacturonase is critical for pollen development and male fertility in thermo-sensitive cytoplasmic male-sterility wheat. Gene. 2022;833:146596.
Zhang L, Liu Y, Wei G, Lei T, Wu J, Zheng L, et al. POLLEN WALL ABORTION 1 is essential for pollen wall development in rice. Plant Physiol. 2022;190:2229–45.
Zheng X, He L, Liu Y, Mao Y, Wang C, Zhao B, et al. A study of male fertility control in Medicago truncatula uncovers an evolutionarily conserved recruitment of two tapetal bHLH subfamilies in plant sexual reproduction. New Phytol. 2020;228:1115–33.
Huang J, Dong J, Qu L-J. From birth to function: male gametophyte development in flowering plants. Curr Opin Plant Biol. 2021;63:102118.
Zhang Y, Chen Q, Zhu G, Zhang F, Fang X, Ren H, et al. CHR721, interacting with OsRPA1a, is essential for both male and female reproductive development in rice. Plant Mol Biol. 2020;103:473–87.
Kou Y, Chang Y, Li X, Xiao J, Wang S. The rice RAD51C gene is required for the meiosis of both female and male gametocytes and the DNA repair of somatic cells. J Exp Bot. 2012;63:5323–35.
Zhang P, Zhang Y, Sun L, Sinumporn S, Yang Z, Sun B, et al. The rice AAA-ATPase OsFIGNL1 is essential for male meiosis. Front Plant Sci. 2017;8:1639.
Lu J, Wang C, Wang H, Zheng H, Bai W, Lei D, et al. OsMFS1/OsHOP2 complex participates in rice male and female development. Front Plant Sci. 2020;11:518.
Phan HA, Li SF, Parish RW. MYB80, a regulator of tapetal and pollen development, is functionally conserved in crops. Plant Mol Biol. 2012;78:171–83.
Ma K, Han J, Zhang Z, Li H, Zhao Y, Zhu Q, et al. OsEDM2L mediates m6A of EAT1 transcript for proper alternative splicing and polyadenylation regulating rice tapetal degradation. J Integr Plant Biol. 2021;63:1982–94.
Zhou L, Gao G, Tang R, Wang W, Wang Y, Tian S, et al. m6A-mediated regulation of crop development and stress responses. Plant Biotechnol J. 2022;20:1447–55.
Khan AH, Min L, Ma Y, Zeeshan M, Jin S, Zhang X. High-temperature stress in crops: male sterility, yield loss and potential remedy approaches. Plant Biotechnol J. 2023;21(4):680–97.
Peng G, Liu Z, Zhuang C, Zhou H. Environment-sensitive genic male sterility in rice and other plants. Plant Cell Environ. 2023;46:1120–42.
Chase CD. Genetically engineered cytoplasmic male sterility. Trends Plant Sci. 2006;11:7–9.
Zhai J, Song J, Zhang T, Xie S, Ma C. deepEA: a containerized web server for interactive analysis of epitranscriptome sequencing data. Plant Physiol. 2020;185:29–33.
Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Ostell J, Pruitt KD, et al. GenBank. Nucleic Acids Res. 2018;46:D41–7.
Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–8.
Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2.
Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47:D309-14.
Buchfink B, Reuter K, Drost H-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods. 2021;18:366–8.
Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, et al. Pfam: the protein families database in 2021. Nucleic Acids Res. 2020;49:D412–9.
Potter SC, Luciani A, Eddy SR, Park Y, Lopez R, Finn RD. HMMER web server: 2018 update. Nucleic Acids Res. 2018;46:W200–4.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Meng J, Lu Z, Liu H, Zhang L, Zhang S, Chen Y, et al. A protocol for RNA methylation differential analysis with MeRIP-Seq data and exomePeak R/Bioconductor package. Methods. 2014;69:274–81.
Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2:100141.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13:1194–202.
Acknowledgements
The authors would like to thank members of the C. Ma and C. Guan Labs for their constructive feedback on this work.
Funding
This work was supported by the National Natural Science Foundation of China (32170681[CM]), Ningxia Natural Science Foundation (2022AAC05020[CG]), the Hundred Talents Program of Shaanxi Province of China (CM).
Author information
Authors and Affiliations
Contributions
C.M. and C.G. conceived and designed the study; C.G. and W.M. prepared plant materials; J.Z., C.Z., J.Y., M.Y., S.L. and Y.M. analyzed the data; C.M., C.G. and J.Z. wrote the article; All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The flower buds of LB1 and LB5 were kindly provided by Quanxing Wolfberry Plantation in Zhongning County, Ningxia, China. The LB1 and LB5 lines were identified by Ningxia Academy of Agriculture and Forestry Science. The seeds of LB1 and LB5 are deposited in National Center for Forestry and Grassland Genetic Resources, with corresponding deposition number of LYBAR6401050025 and LYBAR6401050029, respectively. All methods were performed in accordance with the relevant institutional, national and international guidelines and legislation.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Flowers of two wolfberry lines under study. (A) Flower of LB1. (B) Flower of LB5. Figure S2. Bioinformatics pipeline for the identification of putative m6A regulators in wolfberry. Figure S3. Phylogenetic analysis of m6A regulators. (A) Phylogenetic tree of ALKBH10B genes. (B) Phylogenetic tree of MT-A70 genes. (C) Phylogenetic tree of FIP37 genes. (D) Phylogenetic tree of HAKAI genes. (E) Phylogenetic tree of VIR genes. Color-marked genes are m6A regulators identified in wolfberry. Figure S4. Expression levels of 22 m6A regulators with TPM greater than 10 at three developmental stages. Figure S5. Expression levels of the gene XLOC_016741 in LB1 and LB5 at three developmental stages. Figure S6. LC-MS/MS assay showing the amount of mRNA m6A in LB1 and LB5. Data are presented as mean ± standard deviation (n = 3). Figure S7. Comparison of gene length between the m6A and non-m6A genes of two wolfberry lines. In each boxplot, the horizontal line indicates the median. Statistical analysis was conducted using the Student’s t test; *** p < 0.001. Figure S8. Comparison of expression levels between the m6A and non-m6A genes in wolfberry. (A) LB1. (B) LB5. Statistical analysis was conducted using the Student’s t test; *** p < 0.001. Figure S9. Gene expression levels based on the distributions of differential m6A peaks in wolfberry. (A) LB1. (B) LB5. Statistical analysis was conducted using the Wilcoxon test; ** p < 0.05, *** p < 0.001. Figure S10. Detection of the canonical m6A motif RRACH within the m6A peak regions. (A) LB1. (B) LB5. Figure S11. Conservation analysis of two genes between Arabidopsis, rice, maize, tomato, and wolfberry. (A) The gene encoding a lipoxygenase. (B) The gene encoding a bHLH transcription factor.
Additional file 2:
Table S1. Expression patterns of assembled genes in LB1 and LB5.
Additional file 3:
Table S2. De novo prediction of encoding capacity of assembled transcripts.
Additional file 4:
Table S3. Function annotation of assembled transcripts in wolfberry.
Additional file 5:
Table S4. Transcription factor annotation in wolfberry.
Additional file 6:
Table S5. Differential expression analysis of two wolfberry lines at three stages.
Additional file 7: Table S6.
m6A peak regions of assembled transcripts.
Additional file 8:
Table S7. Differential methylation analysis of LB1 and LB5 at S stage.
Additional file 9:
Table S8. GO enrichment results of hypermethylated genes.
Additional file 10:
Table S9. GO enrichment results of hypomethylated genes.
Additional file 11:
Table S10. List of wolfberry genes with differential expression and differential methylation patterns at the S stage.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhao, J., Zhang, C., Li, S. et al. Changes in m6A RNA methylation are associated with male sterility in wolfberry. BMC Plant Biol 23, 456 (2023). https://doi.org/10.1186/s12870-023-04458-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-023-04458-7