
Abstract
Background
Mycoheterotrophs, acquiring organic carbon and other nutrients from mycorrhizal fungi, have evolved repeatedly with substantial plastid genome (plastome) variations. To date, the fine-scale evolution of mycoheterotrophic plastomes at the intraspecific level is not well-characterized. A few studies have revealed unexpected plastome divergence among species complex members, possibly driven by various biotic/abiotic factors. To illustrate evolutionary mechanisms underlying such divergence, we analyzed plastome features and molecular evolution of 15 plastomes of Neottia listeroides complex from different forest habitats.
Results
These 15 samples of Neottia listeroides complex split into three clades according to their habitats approximately 6 million years ago: Pine Clade, including ten samples from pine-broadleaf mixed forests, Fir Clade, including four samples from alpine fir forests and Fir-willow Clade with one sample. Compared with those of Pine Clade members, plastomes of Fir Clade members show smaller size and higher substitution rates. Plastome size, substitution rates, loss and retention of plastid-encoded genes are clade-specific. We propose to recognized six species in N. listeroides complex and slightly modify the path of plastome degradation.
Conclusions
Our results provide insight into the evolutionary dynamics and discrepancy of closely related mycoheterotrophic orchid lineages at a high phylogenetic resolution.
Similar content being viewed by others


Introduction
Mycoheterotrophs possess the ability to survive by acquiring nutrients from mycorrhizal fungi, relaxing the dependence on their own photosynthesis for carbon fixation [1,2,3,4,5,6]. Mycoheterotrophs commonly undergo loss or pseudogenization of photosynthesis-related genes due to relaxed selective constraints, further leading to dramatic plastid genome (plastome) reductions and structural rearrangements [1, 4, 7,8,9,10,11,12,13,14]. Moreover, there are repeated plastome-based phylogenetic changes during trophic transitions from autotrophy, via partial mycoheterotrophy (mixotrophy), to holomycotrophy [15, 16].
The loss of plastid-encoded genes coinciding with or following the evolution of parasitic plants has been extensively explored [1, 12, 16,17,18,19]. Recently, an increasing number of studies revealed more and more evolution details by incorporating dense sampling of plastomes across higher taxonomic levels (e.g. genera, families or tribes) containing parasitic plants, from the phylogenetic-comparative perspective [8, 15, 16, 19,20,21,22,23,24,25]. Plastomes of parasitic plants are characterized by elevated substitution rates, gene pseudogenization and loss. The path of plastome degradation in parasitic plants, proposed and revised based on syntheses of previous plastome evolution in parasitic plants, includes the following major stages: (1) loss and/or pseudogenization in the ndh genes complex; (2) loss and/or pseudogenization of photosynthesis genes; (3) loss and/or pseudogenization of photosynthesis-related genes with secondary functions, including atp genes; (4) loss and/or pseudogenization of other genes, such as accD, ycf1, ycf2; and (5) nearly complete or complete loss of the plastome [3, 19, 26, 27]. This model of plastome evolution has been observed in subsequent studies (such as [14, 19, 28,29,30,31,32]). Nevertheless, these studies may leave phylogenetic sampling or evolutionary route ‘gaps’ due to large temporal- and spatial-scale patterns [20]. By now, comparative plastome analyses at a fine scale, such as infrageneric or intraspecific levels, are rather scant [20]. These limited studies have revealed unexpectedly high plastome divergence among mycoheterotrophic species complex members, which is presumably related to mycorrhizal interactions, geographical barriers, or other biotic/abiotic factors [20]. Adaptive processes underlying such divergence, however, remain largely unknown [20]. Therefore, it is necessary to explore plastome variation at higher phylogenetic resolution and across diverse habitats to exquisitely illustrate plastome evolution of mycoheterotrophs.
Neottia Guettard (Orchidaceae, Neottieae) comprises approximately 73 species, including both autotrophic and mycoheterotrophic species and are widely distributed in northern temperate regions and alpine areas of Asian subtropical regions [2, 15, 18, 31, 33,34,35,36,37,38,39,40,41,42] (https://powo.science.kew.org/results?q=Neottia). Previous studies indicated that leafless Neottia are fully mycoheterotrophic and have evolved from leafy species only once in Neottia [15, 32, 43]. N. listeroides complex is composed of approximately six morphologically similar leafless holomycotrophic species (Fig. 1), including N. listeroides, N. megalochila, N. Microglottis, N. smithianus and N. tenii [44, 45]. These species generally occur in forests dominated by Abies or Pinus in temperate and alpine region of subtropical area of China (Yunnan, Sichuan, Shaanxi, and Tibet), Myanmar, Nepal, and neighboring regions (Table 1, Table S1) [44, 45]. In this study, we analyzed plastomes of N. listeroides complex members using densely sampling from different habitats, to explore their variation patterns and effects of environmental factors on micro-evolution of closely related mycoheterotrophic taxa.

Members of the Neottia listeroides complex collected from different sites. a N. listeroides (LCY), b N. megalochila (LGZ), c N. naungmungensis (MMD), d N. listeroides (LGS), and e N. megalochila (MLJ)
Results
Phylogenetic relationships and molecular dating of Neottia listeroides complex
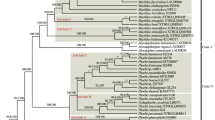
Topologies from the phylogenetic analyses of protein coding genes of mitochondrial genomes (Fig. 2) and plastome genomes (Fig. S1) are different about the phylogenetic position and the interrelationships of Neottia. We here use the phylogram from mitochondrial genomes for subsequent analyses and discussion. The N. listeroides complex, along with other mycoheterotrophic species, such as N. acuminata, N. camtschatea, and N. alternifolia, formed a monophyletic group nesting within Neottia and sister to the clade consisting of N. divaricata, N. nujiangensis, N. fugongensis, N. pinetorum, and N. ovata (Fig. 2). Mycoheterotrophic species of Neottia evolved from the autotrophic plants approximately 13.04 million years ago (Ma) (Fig. 3). The N. listeroides complex evolved approximately 8.83 Ma and was subdivided into three clades according to forest habitats: Clade I, including ten samples (i.e. HHL-1, HHL-2, HLZ, N. smithianus; LJL1, LJL2, LCY1, LCY2, LLZ1, and LLZ2, N. listeroides; MMD, N. naungmungensis) from pine-broadleaf mixed forests; Clade II, including four samples (i.e. MLJ, LGZ, LLJ, N. megalochila; LGS, N. listeroides) from alpine fir forests (Fig. 3), and Clade III, including one sample (HWQ, N. listeroides) from alpine fir-willow forests. We thus designated the three phylogenetic clusters, Clade I, Clade II and Clade III as Pine Clade, Fir Clade, and Fir-willow Clade, respectively. Fir-willow Clade diverged from other two clades approximately 6.31 Ma, Pine Clade diverged from Fir Clade approximately 4.72 Ma (Fig. 3).

Phylogenetic relationships based on maximum likelihood (ML) analysis of mitochondrial protein-coding sequences (mtCDS). Numbers above branches represent bootstrap support (* indicates 100%). N. listeroides complex members were divided into three clades: Pine Clade (indicated by blue) including samples from pine-broadleaf mixed forests; Fir Clade (indicated by the green) including samples from alpine fir forests; and Fir-willow Clade (indicated by the yellow)

Time-calibrated phylogram based on concatenated sequences of all mtCDS. Numbers at nodes are median ages in million years ago (Ma). Pine Clade indicated by blue; Fir Clade indicated by the green; and Fir-willow Clade indicated by the yellow. The schematic diagrams of record of Earth’s climate was edited according to reported references [107]
General plastome features
Plastome size of 15 members of the N. listeroides complex range from 94,499 bp to 110,855 bp, with guanosine-cytosine (GC) content varies from 37.2% to 37.8%. All these plastomes possess the typical quadripartite structure, consisting of a pair of IR regions (26,145–27,814 bp) separated by LSC (35,272–48,291 bp) and SSC (6,500–9,932 bp). The number of annotated genes is 80 to 84, including 34–38 coding sequences (CDS), eight rRNA genes, and 38 tRNA genes (Table 2). In brief, within the N. listeroides complex, Fir-willow Clade has the largest plastome size, Fir Clade has the smallest plastome size. According to nucleotide diversity (Pi > 0.1), mutation hotspots among 15 samples are largely located in intergenic spacers, including trnC-trnD, trnY-trnT, trnM-atpB, rbcL-accD, and rps15-ccsA (Fig. S2). When the sample of N. listeroides (LCY1) was selected as the reference, sequences of other members from Pine Clade showed greater synteny to it, and divergence of three samples of N. megalochila (LLJ, LGZ, MLJ) one sample of N. listeroides(LGS) from the reference mainly occurred in intergenic non-coding regions with frequent deletions (Fig. S3).
A consistent gene order, without rearrangement, is maintained among all complex members (Fig. S4). At the junction of LSC/IRa (JLA), trnK-UUU flanked the JLA for Pine Clade samples, whereas it is located completely within the LSC for Fir Clade samples (Fig. S5). Genes encoding thylakoid NAD(P)H dehydrogenase (ndh), plastid-encoded RNA polymerase (PEP; rpo), and thylakoid ATP synthase (atp), as well as photosynthesis-related genes (ccsA, cemA, pet, psa, psb, rbcL, and ycf3 and ycf4), are largely pseudogenized or deleted in all 15 samples, except retention of a few genes of photosystem II (psbJ, psbK, and psbM in LLJ and LGZ of N. megalochila, and psbZ in LGS of N. listeroides and cytochrome b6f complex (petG in LGS of N. listeroides and petL in LLJ of N. megalochila) in some Fir Clade samples (Fig. 4). Housekeeping genes have intact ORFs in almost all samples, although LJL-1 and LJL-2 (N. listeroides) functionally lost the ribosomal protein gene rps15. Overall, samples assigned to Pine Clade showed high similarity in gene content.

Protein-coding genes in N. listeroides complex. Black, grey and white boxes respectively represent intact genes, pseudogenes and gene loss. PEP = plastid-encoded RNA polymerase
Members of the N. listeroides complex are split into three subgroups, i.e., Pine Clade, Fir Clade, and Fir-willow clade approximately 6.31 and 4.72 Ma, respectively. These three clades differ greatly in gene content, substitution rates and size of plastomes. Fir-willow and Pine clades have lost or pseudogenized all genes related to photosynthesis. Fir Clade retains a few photosynthesis-related genes, such as pet and psb, displaying substantial variations in gene content among individuals. RNA polymerase gene rpoB had been deleted from MMD (N. naungmungensis) and ribosomal protein gene rps15 pseudogenized in LJL-1 and LJL-2 (N. listeroides) in Pine Clade. Fir Clade members show significantly reduced plastome size in comparison with two other clades.
Molecular evolution
Fir-willow Clade has the lowest substitution rates in N. listeroides complex, whereas Fir Clade had highest evolutionary rates (Table S2, Fig. S6). Using N. fugongensis as a reference, all N. listeroides complex members are under purifying selection, with the signature of negative selection in most genes (ω < 0.5). However, selective constraint on some of genes are relaxed in Pine Clade (e.g. rpl33) and Fir Clade (e.g. rpl22, rpl32, rps8, and clpP) (Table S3). The branch-site model detected positively selected sites (BEB probability > 0.95) in five genes (i.e. accD, matK, rpl20, rps11, and ycf2) when Pine Clade was set as the foreground branch, while in two genes (i.e. accD and rpl32) as Fir Clade was the foreground branch (Table S4). Biased codon usage (RSCU > 1) existed in most amino acids, except tryptophan (Trp) and methionine (Met). Significance of difference in RSCU values among three clades was estimated using the t-test. Eight of 32 frequently used codons were underused in Fir Clade and Fir-pillow Clade sample compared with Pine Clade samples, whereas seven codons were overused (Table S5).
Discussion
Taxonomic treatment of N. listeroides complex
The taxonomy of fully mycoheterotrophic group, such as Gastrodia and Neottia, has been notoriously difficult due to greatly reduced plant body, few specimens that was usually in poor condition, variation of floral characters and the rarity of specimens [46]. The number of identified species of Gastrodia has been triple in last two decades with the help of digital cameras, molecular systematics and botanic survey [47,48,49,50,51,52]. The taxonomy of N. listeroides complex has been confused for a long time [44]. N. smithianus and N. microglottis were even transferred to another genus, Holopogon [53, 54]. N. microglottis and N. tenii have not been discovered in field since they were described. Phylogenomics indicated that all samples of N. megalochia, N. naungmungensis and N. smithianus, were nested within N. listeroides. Two samples of N. smithianus (LJL1, LJL2) were nested within Pine clade and diverged from other members of Pine Clade about 2.2 Ma. The third sample of N. smithianus diverged from N. listeroides less than 1 Ma. Three samples of N. megalochila belong to Fir clade and diverged from N. listeroides (LGS) within Fir Clade about 2.43 Ma. N. naungmungensis diverged from N. listeroides (LLZ1) less than 1 Ma and is characterized by its ecological niches and morphological characters (Table 1, Fig. 1). N. listeroides complex was rapid diversification in icehouse period of last 5 Ma, however, there is the discrepancy between the molecular systematic tree and the morphology. N. listeroides( HWQ) is morphological stasis although it diverged very early. Instead, some recent evolved sampling (such as MMD, LGZ, LLJ) differ greatly from N. listeroides. This discrepancy might have been contributed by many factors, such as incomplete lineage sorting, hybridization or molecular evolution rates.
Based on these, we tentatively propose to recognize approximately six species in this complex, i.e., N. listeroides, N. megalochia, N. microglottis, N. naungmungensis sp. nov. (MMD from Naungmung Mountains, Chin state, Myanmar), N. smithianus and N. tenii. Neottia naungmungensis differs from its relatives by its elliptic lip with about 2.5 cm long, apex bilobed, lobelets acute, and stigma lateral with stalk. N. listeroides is characterized by narrowly obovate-oblong lip about 3-4 mm wide, apex bilobed, lobelet apex acute or obtuse; N. megalochila by obovate lip about 6-10 mm wide, apex bilobed, lobelet apex truncate-rounded; N. smithianus by terminal stigma and anther more or less with filament; N. tenii differs from other species by its lip with a pair of auricles at base [44]. N. microglottis is characterized by its entire lip not bilobed at apex, however, the taxonomic identity remains to be confirmed. Species delimitation is urgently needed in this complex based on more sampling across the distribution range.
Factors driving plastome diversification
Our results demonstrated that plastome structure and substitution rates of closely related mycoheterotrophic lineages within a species complex could diverge rapidly based on forest habitat types. Fully mycoheterotrophic orchids uptake organic carbon and essential nutrients from their mycorrhizal fungi that concurrently liaise with surrounding trees, thus forming tripartite symbiotic associations [55,56,57]. In this system, green trees are ultimate energy sources, and subsequently shape their microflora [55, 58]. As a consequence, diversity and composition of mycorrhizal symbionts at the local-scale vary substantially along with changes in forest dominant trees [56, 59, 60].
In this study, alpine fir forests are simply dominated by Abies fabri, whereas pine-broadleaf forests are characterized by more abundant ectomycorrhizal hosts, mainly composed of Quercus (e.g. Q. mongolica) and Pinus (e.g. P. densata and P. yunnanensis). Given that tree host diversity contributes to ectomycorrhizal fungi (ECM) richness, alpine fir forests are expected to possess fewer ECM fungal species than pine-broadleaf forests [61]. Together, we infer that, although the key ECM taxa colonizing N. listeroides complex members remain largely the same (possibly as ectomycorrhizal Sebacinales Clade A), the specificity of ECM associates in Fir Clade samples is potentially higher than that in Pine Clade samples, further leading to forest-type-dependent plastome divergence of complex members. As mycorrhizal symbioses are indispensable for growth and survival of mycoheterotrophic orchids, additional studies are needed to clarify tripartite vegetation-fungus-orchid associations.
From the forgoing considerations, identity and specificity of mycorrhizal associations differ considerably among orchid taxa, and profoundly affect the distribution, evolution, and diversification of mycoheterotrophic orchids [16, 62,63,64]. Two ecologically different clades of Sebacinales have been found to predominate in various putative mycorrhizal associates of Neottia [39]. In particular, Sebacinales Clade A including ectomycorrhizal fungi (ECM) mainly connects with trees and mycoheterotrophic orchids (e.g. N. nidus-avis and N. camtschatea), whereas taxa in non-ectomycorrhizal Sebacinales Clade B commonly form rhizoctonia symbionts with green orchids (e.g. N. ovata and N. cordata) [42, 56, 65, 66]. Based on limited empirical evidence, shifts of association from rhizoctonia to ECM symbionts, with increased specificity of fungal partners, are potential steps in the sequential evolution from autotrophy to holomycotrophy in Neottia [2, 39, 59]. Moreover, variation in mycorrhizal specificity within a certain orchid species complex may contribute to fine-scale phylogenetic diversification [67, 68]. Recently, Suetsugu et al. (2022) showed that the use of different symbiotic microbiont can contribute to the diversification of species in mycoheterotrophic plants [46], which suggests that a shift in symbiotic microbiont may have played a role in the ecological speciation of these plants.
The Fir Clade have highest substitution rates among three clades, likely associated with changes in codon preference resulting from mutational bias or selection, since seven of 32 frequently used codons were overused in Fir Clade samples. Moreover, selection on codon usage bias related to translation efficiency might reflect adaptation of individuals to their environments [30, 69, 70]. Selective constraints are relaxed in more genes for Fir Clade (e.g. rpl22, rpl32, rps8, and clpP) than Pine Clade (e.g. rpl33). We detected the signature of positive selection in five genes (i.e. accD, matK, rpl20, rps11, and ycf2) with Pine Clade as the foreground branch, while in two genes (i.e. accD and rpl32) with Fir Clade as the foreground branch. These genes may function importantly during adaptation of complex members to different habitats, contributing to the divergence of Pine Clade and Fir Clade [71].
Plastome evolution in parasitic plants
It is worth noting that all members of Neottia listeroides have green plants despite the degeneration of the plastomes. Previous results indicated that some leafless orchids with green stems or purple stems, such as Corallorhiza trifida [72, 73], Cymbidium macrorhizon [74,75,76], and Limodorum abortivum [77, 78], can perform photosynthesis in stems or fruits even in the absence of leaves. However, plastomes of Cymbidium macrorhizon, Corallorhiza trifida and Limodorum abortivum [79] have nearly intact photosynthesis and photosynthesis-related genes, instead, these genes were nearly lost or pseudogenized in plastomes of Neottia listeroides complex. It seems that all members of Neottia listeroides complex have lost the ability of photosynthesis and fully depend on symbiotic microbionts for organic carbon.
Recent studies on extremely reduced plastomes (minimal plastomes) of several parasitic plant groups, including Epipogium (Orchidaceae) [1], Epirixanthes (Polygalaceae) [14], Exacum (Gentianceae) [28], Gastrodieae (Orchidaceae) [12] and Sciaphilla (Triuridaceae) [80, 81], reveal little known evolutionary trends, including the formation of rrn gene block, the retention or even increase of gene copies, such as accD, clpP, ycf 1, and ycf2. Comparative genomics of nuclear genome of mycoheterotrophic Gastrodia menghaiensis and autotrophic orchids (Apostasia zhenzhenica, Dendrobium officinale, Phalaenopsis equestris) showed that genes involved synthesis and degradation of chlorophyll were absent in genome of Gastrodia menghaiensis [82]. However, nuclear encoded genes related to plastid biosynthesis of fatty acids, and hormones are intact or even increased in copies [82]. These suggest plastids play important role even in fully mycoheterotrophic species and the loss and/or pseudogenization of all “housekeeping” genes is very rare case. These minimal plastomes don’t belong to any stage proposed in previous studies [3, 19, 26, 27]. The plastome degradation in parasitic plants also display a highly lineage-specific manner in gene retentions, pseudogenization and loss even within genus or species [15, 16, 20, 73].
Therefore, we propose to slightly modify the path of plastome degradation in previous studies [3, 19, 26, 27, 81, 83,84,85]: (1) loss and/or pseudogenization in the ndh genes complex; (2) retention, pseudogenization and/or loss of photosynthesis and photosynthesis-related genes, including atp genes; (3) loss and/or pseudogenization genetic apparatus and Maturase K gene, such as rpo, matK; (4) retention even expansion of gene copies of other genes, including accD, rrn, ycf1 and ycf 2 genes; and (5) nearly complete or complete loss of the plastid genome. Plastomes of Cymbidium macrorhizon [75] fall within stage 1, plastomes of Corallorhiza trifida [27] in stage 2, Neottia listeroides complex and Rhizanthella gardneri [7] in stage 3, instead, most minimal plastomes of Epipogium [1], Thismia [10, 86] and Gastrodieae [12] are in stage 4.
Conclusions
We analyzed plastome evolution of the N. listeroides complex composed of 15 samples from different habitats via phylogenetic and comparative approaches, to explore fine-scale evolutionary dynamics and discrepancies of closely related mycoheterotrophic lineages. We detected the rapid diversification of plastomes in terms of structure, gene content, and evolutionary rates during the last 4 Ma. Unexpectedly, the observed divergence is closely related to forest habitats. We hypothesized that specificity of mycorrhizal fungal partners contributes to such divergence, which needs to be further demonstrated by empirical evidences. In addition to gene loss, plastome evolution involves intricate but coordinated nucleus-plastid interactions, such as transfer of genes to the nucleus via multiple steps [7, 26, 87, 88]. Such transfer processes are species- or lineage-specific [1]. Therefore, it is also necessary to deeply explore the nuclear genome variation in complex members to understand their diversification mechanisms.
Materials and Methods
Sampling, DNA extraction, and sequencing
Fifteen samples of N. listeroides complex were collected from thirteen sites covered with different vegetation (Table 1, Fig. 1). Total genomic DNAs from silica gel-dried materials were extracted using a modified CTAB method [89]. DNAs with a concentration higher than 100 ng/μl were sonicated into ~ 500 bp fragments (Covaris M220; Woburn, MA, USA). Libraries were prepared following the user’s manual of the NEBNext Ultra DNA Library Prep Kit (New England Biolabs, Ipswich, MA, USA). Paired-end sequencing was performed on the Illumina HiSeq 2500 platform (Illumina, Inc., San Diego, CA, USA).
Assembly and annotation
For quality control (QC), raw data were processed using the NGS QC Toolkit v.2.3.3 [90] to trim adaptors and filter low-quality reads (PHRED < 20, length < 70 bp). Clean reads were mapped to the plastome of Calanthe triplicata (NC_024544.1) using Geneious v.10.1.2 (Biomatters, Inc., Auckland, New Zealand) with medium–low sensitivity in five iterations. Plastid contigs from consensus sequences were de novo assembled in VELVET [91] over a range of k-mer values from 37 to 45 with auto-adjustment for coverage cutoffs. Subsequently, contigs were combined into scaffolds. After inverted repeat (IR) boundaries identification via BLAST [92], all reads were mapped with high stringency to the draft chloroplast genome for assembly errors correction [15]. The assembled chloroplast genomes were annotated using Geneious with 70% identity to the C. triplicate reference sequence, and then were checked and corrected manually [15]. Non-triplet frame shifts and premature stop codons were considered as pseudogenes [15].
Phylogenetic analyses and molecular dating
In addition to 15 members of the N. listeroides complex, we also included another 16 species (Table S1) to complement the phylogenetic analysis. Two species of Orchidoideae, Hemipilia cordifolia and Platanthera minor, were used as outgroup. To minimize effects of accelerated substitution rates in mycoheterotrophic plastomes on phylogenetic inference [4, 93], we used mitochondrial protein-coding sequences (mtCDS) for phylogenetic reconstruction [4, 93]. Clean reads were mapped to mitochondrial genomes of Gastrodia elata (MF070084-MF070102) and Phoenix dactylifera (NC_016740) to obtain mtCDS sequences. Thirty-eight mtCDS were assembled for each sampling (supplementary Table S6). All mtCDS extracted by Geneious were aligned in MAFFT with default settings [94], and were manually adjusted via Bioedit v.5.0.9 [95]. After examination of saturation, aligned sequences were concatenated into a single multi-gene supermatrix using PhyloSuite [96]. The phylogenetic tree was constructed based on Maximum Likelihood (ML) with 1000 standard bootstrap (BS) pseudoreplicates using IQtree v.2.1.2 [97]. The tree was visualized in FigTree v.1.4.3 [98]. The phylogenetic tree based on 26 common house-keeping genes (Table S3) of plastomes was constructed as above.
All mtCDS were selected for molecular dating. The ML tree calibrated via BEAST v.2.4.8 [99] was used as a topological constraint, with Corybas taliensis, Cheirostylis yunnanensis, and Spiranthes sinensis as outgroup. The lognormal relaxed clock was selected in MrModelTest v.2.3 [100]. One orchid fossil was set as a calibration point for crown clades: subtribe Goodyerinae, 15 Ma (mean: 0, sigma: 1.5). Priors were placed on the stem node of Neottieae (offset: 67.17 Ma, mean: 0, sigma: 1.0), and the most recent common ancestor of Neottiaeae (offset: 26.19 Ma, mean: 0, sigma: 1.0) based on previous results [93, 101]. We conducted two independent runs of Markov chain Monte Carlo (MCMC) searches, sampling every 10,000 generations over 20 million generations, with four non-independent chains used for each run. Log files were monitored in Tracer v.1.6 [102]. Convergence was determined according to distribution and effective sample size (ESS) > 200. A maximum clade credibility (MCC) chronogram was generated in TreeAnnotator v.1.7.5 [103] with median heights for node ages. The time tree was visualized using FigTree.
Plastome structure
Boundaries between single-copy regions (LSC and SSC) and inverted repeats (IR) regions (i.e. IR/SSC and IR/LSC) of each sample were visualized in Geneious. All 15 plastomes, excluding one IR in each sample, were compared by mVISTA (http://genome.lbl.gov/vista/mvista/submit.shtml) with the Shuffle-LAGAN model; the annotation for LCY (KU551272) was used as a reference. Nucleotide diversity (Pi) was calculated to identify mutation hotspots of the N. listeroides complex by a sliding window analysis via DnaSP v.6.10.04 [104]. The step size was set as 200 bp, with a 500 bp window length. Collinearity for 11 chloroplast genomes was evaluated through progressiveMauve algorithm in Geneious to identify syntenic blocks and visualize structural rearrangements.
Detection of selection
To estimate non-uniform synonymous codon usage in protein-coding genes of complex members, the relative synonymous codon usage (RSCU) was calculated using CodonW v.1.4.2 (http://codonw.sourceforge.net/). RSCU < 1 denotes lack of usage bias, whereas RSCU > 1 indicates that a codon is overrepresented. A total of 34 retained protein-coding genes from the 15 plastomes were involved in selective pressure analyses. Single-copy CDS sequences without stop codons were aligned at the codon level using MUSCLE (codon) in MEGA v.7.0.2 [105]. The tree topology was inferred as described above. Non-synonymous (dN) and synonymous (dS) substitution rates indicated by branch lengths were estimated using PAML v.4.8 [106] under the branch model (run model = 0, model = 1, NSsites = 0) in the CODEML module [15].
Pairwise comparisons between samples were conducted via the pairwise model (runmode = 2, NSsites = 0), with Neottia fungongensis as the reference, followed by calculation of the selection intensity parameter ω (i.e. dN/dS). The codon frequencies were determined by the F3 × 4 model [1]. Significance of the ω parameter was determined by likelihood ratio tests (LRT), with Bonferroni correction for multiple comparisons. Values of ω > 1, ω ≈ 1, and ω < 1 suggest positive, neutral, and negative (purifying) selection, respectively (Yang & Nielsen, 2002).
Our phylogenetic analysis revealed that N. listeroides complex members could be separated into three clades designated as Pine, Fir and Fir-willow Clade (see RESULTS). To identify positively selected genes potentially functioning in adaptation of each group, the branch-site model was performed (model = 3, NSites = 3, fixed omega = 0, omega = 2) based on 31 protein-coding genes (excluding rpl36, rps12, and rps7). Pine Clade, Fir Clade and Fir-pillow Clade were respectively set as the foreground branch. Differences between the models and the null model M0 (model = 0, NSites = 0, assuming no site-wise or branch-wise dN/dS variation) were evaluated by the LRT with a χ2 distribution at a threshold of p < 0.05. In addition, the Bayesian Empirical Bayes (BEB) method was used to identify specific sites under positive selection by calculating posterior probabilities. A gene with a high posterior probability (BEB > 0.95) and a p-value < 0.05 was considered as positively selected.
Availability of data and materials
All 15 newly sequenced and annotated plastid genomes generated in this study have been submitted to GenBase (https://ngdc.cncb.ac.cn/genbase) with accession number from C_AA002280.1 to C_AA002294.1. All sequenced data are available from GenBase (https://ngdc.cncb.ac.cn/genbase). The online resources of plastome data (NC_030712.1, NC_030711.1, NC_030710.1, KU551266.1, NC_030709.1, NC_030708.1, NC_030705.1, KU551263.1, NC_030706.1, NC_030703.1, MN416689.1) were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/).
References
Schelkunov MI, Shtratnikova VY, Nuraliev MS, Selosse MA, Penin AA, Logacheva MD. Exploring the limits for reduction of plastid genomes: a case study of the mycoheterotrophic orchids Epipogium aphyllum and Epipogium roseum. Genome Biol Evol. 2015;7(4):1179–91.
Yagame T, Ogura-Tsujita Y, Kinoshita A, Iwase K, Yukawa T. Fungal partner shifts during the evolution of mycoheterotrophy in Neottia. Am J Bot. 2016;103(9):1630–41.
Graham SW, Lam VK, Merckx VS. Plastomes on the edge: the evolutionary breakdown of mycoheterotroph plastid genomes. New Phytol. 2017;214(1):48–55.
Lin Q, Braukmann TWA, Gomez MS, Sampaio Mayer JL, Pinheiro F, Merckx VSFT, Stefanovic S, Graham SW. Mitochondrial genomic data are effective at placing mycoheterotrophic lineages in plant phylogeny. New Phytologist. 2022;236(5):1908–21.
Merckx V, Freudenstein JV. Evolution of mycoheterotrophy in plants: a phylogenetic perspective. New Phytol. 2010;185(3):605–9.
Merckx V, Bidartondo MI, Hynson NA. Myco-heterotrophy: when fungi host plants. Ann Bot. 2009;104(7):1255–61.
Delannoy E, Fujii S. des Francs-Small CC, Brundrett M, Small I: Rampant gene loss in the underground orchid Rhizanthella gardneri hghlights hvolutionary constraints on plastid genomes. Mol Biol Evol. 2011;28(7):2077–86.
Wicke S, Mueller KF, de Pamphilis CW, Quandt D, Wickett NJ, Zhang Y, Renner SS, Schneeweiss GM. Mechanisms of functional and physical genome reduction in photosynthetic and nonphotosynthetic parasitic plants of the broomrape family. Plant Cell. 2013;25(10):3711–25.
Lam VKY, Merckx VSFT, Graham SW. A few-gene plastid phylogenetic framework for mycoheterotrophic monocots. Am J Bot. 2016;103(4):692–708.
Lim GS, Barrett CF, Pang C-C, Davis JI. Drastic reduction of plastome size in the mycoheterotrophic Thismia tentaculata relative to that of its autotrophic relative Tacca chantrieri. Am J Bot. 2016;103(6):1129–37.
Bae E-K, An C, Kang M-J, Lee S-A, Lee SJ, Kim K-T, Park E-J. Chromosome-level genome assembly of the fully mycoheterotrophic orchid Gastrodia elata. G3 (Bethesda, Md). 2022;12(3):jkab433.
Wen Y, Qin Y, Shao B, Li J, Ma C, Liu Y, Yang B, Jin X. The extremely reduced, diverged and reconfigured plastomes of the largest mycoheterotrophic orchid lineage. BMC Plant Biol. 2022;22(1):448.
Xu Y, Lei Y, Su Z, Zhao M, Zhang J, Shen G, Wang L, Li J, Qi J, Wu J. A chromosome-scale Gastrodia elata genome and large-scale comparative genomic analysis indicate convergent evolution by gene loss in mycoheterotrophic and parasitic plants. Plant J. 2021;108(6):1609–23.
Petersen G, Darby H, Lam VKY, Pedersen HA, Merckx VSFT, Zervas A, Seberg O, Graham SW. Mycoheterotrophic Epirixanthes (Polygalaceae) has a typical angiosperm mitogenome but unorthodox plastid genomes. Ann Bot. 2019;124(5):791–807.
Feng YL, Wicke S, Li JW, Han Y, Lin CS, Li DZ, Zhou TT, Huang WC, Huang LQ, Jin XH. Lineage-Specific Reductions of Plastid Genomes in an Orchid Tribe with Partially and Fully Mycoheterotrophic Species. Genome Biol Evol. 2016;8(7):2164–75.
Barrett CF, Sinn BT, Kennedy AH. Unprecedented Parallel Photosynthetic Losses in a Heterotrophic Orchid Genus. Mol Biol Evol. 2019;36(9):1884–901.
Lam VKY, Darby H, Merckx V, Lim G, Yukawa T, Neubig KM, Abbott JR, Beatty GE, Provan J, Soto Gomez M, et al. Phylogenomic inference in extremis: a case study with mycoheterotroph plastomes. Am J Bot. 2018;105(3):480–94.
Kotilinek M, Tesitelova T, Jersakova J. Biological Flora of the British Isles: Neottia ovata. J Ecol. 2015;103(5):1354–66.
Wicke S, Naumann J. Molecular evolution of plastid genomes in parasitic flowering plants. In: Advances in Botanical Research. Cambridge, San Diego: Elsevier; 2018;85:315–347.
Barrett CF, Kennedy AH. Plastid Genome Degradation in the Endangered, Mycoheterotrophic, North American Orchid Hexalectris warnockii. Genome Biol Evol. 2018;10(7):1657–62.
Lin Y, Li P, Zhang Y, Akhter D, Pan R, Fu Z, Huang M, Li X, Feng Y. Unprecedented organelle genomic variations in morning glories reveal independent evolutionary scenarios of parasitic plants and the diversification of plant mitochondrial complexes. BMC Biol. 2022;20(1):49.
Ceriotti LF, Roulet ME, Sanchez-Puerta V: Plastomes in the holoparasitic family Balanophoraceae: Extremely high AT content, severe gene content reduction, and two independent genetic code changes. Mol Phylogenet Evol. 2021;162:107208.
Li Y, Zhou J-g, Chen X-l, Cui Y-x, Xu Z-c, Li Y-h, Song J-y, Duan B-z, Yao H. Gene losses and partial deletion of small single-copy regions of the chloroplast genomes of two hemiparasitic Taxillus species. Sci Rep. 2017;7:12834.
Wu C-S, Wang T-J, Wu C-W, Wang Y-N, Chaw S-M. Plastome Evolution in the Sole Hemiparasitic Genus Laurel Dodder (Cassytha) and Insights into the Plastid Phylogenomics of Lauraceae. Genome Biol Evol. 2017;9(10):2604–14.
Petersen G, Cuenca A, Seberg O. Plastome Evolution in Hemiparasitic Mistletoes. Genome Biol Evol. 2015;7(9):2520–32.
Barrett CF, Davis JI. The plastid genome of the mycoheterotrophic Corallorhiza striata (Orchidaceae) is in the relatively early stages of degradation. Am J Bot. 2012;99(9):1513–23.
Barrett CF, Freudenstein JV, Li J, Mayfield-Jones DR, Perez L, Pires JC, Santos C. Investigating the path of plastid penome degradation in an early-transitional clade of heterotrophic orchids, and implications for heterotrophic angiosperms. Mol Biol Evol. 2014;31(12):3095–112.
Li Z, Ma X, Wen Y, Chen S, Jiang Y, Jin X. Plastome of the mycoheterotrophic eudicot Exacum paucisquama (Gentianaceae) exhibits extensive gene loss and a highly expanded inverted repeat region. PeerJ. 2020;8: e9157.
Li X, Qian X, Yao G, Zhao Z, Zhang D. Plastome of mycoheterotrophic Burmannia itoana Mak. (Burmanniaceae) exhibits extensive degradation and distinct rearrangements. Peerj. 2019;7:e7787.
Li Z-H, Jiang Y, Ma X, Li J-W, Yang J-B, Wu J-Y, Jin X-H. Plastid Genome Evolution in the Subtribe Calypsoinae (Epidendroideae, Orchiaaceae). Genome Biol Evol. 2020;12(6):867–70.
Rentsch JD, Hardee LJ, Shelley CE, Williams MT. The Complete Plastid Genome of Neottia bifolia (Raf.) Baumbach (Orchidaceae): Insights Into Chlorophyllous and Achlorophyllous Plastid Genomes. Castanea. 2020;85(2):285–95.
Lallemand F, Logacheva M, Le Clainche I, Berard A, Zheleznaia E, May M, Jakalski M, Delannoy E, Le Paslier M-C, Selosse M-A. Thirteen New Plastid Genomes from Mixotrophic and Autotrophic Species Provide Insights into Heterotrophy Evolution in Neottieae Orchids. Genome Biol Evol. 2019;11(9):2457–67.
Jersakova J, Minasiewicz J, Selosse M-A. Biological flora of Britain and Ireland: Neottia nidus-avis. J Ecol. 2022;110(9):2246–63.
Zhu Z-X, Wang J-H, Sakaguchi S, Zhao K-K, Moore MJ, Wang H-F. Complete plastome sequences of two Neottia species and comparative analysis with other Neottieae species (Orchidaceae). Folia Geobot. 2019;54(3–4):257–66.
Chen B-H, Jin X-H. Neottia wuyishanensis (Orchidaceae: Neottieae), a new species from Fujian China. Plant Divers. 2021;43(5):426–31.
So J-H, Lee N-S. Phylogenetic analysis of Neottia japonica (Orchidaceae) based on ITS and matK regions. Korean J Plant Taxonomy. 2020;50(4):385–94.
Mu A-T, Aung M-H, Jin X-H. Neottia nyinyikyawii (Orchidaceae: Epidendroideae), a new species from Chin State. Myanmar Phytotaxa. 2020;446(3):205–8.
Jin X-H, Pang H-B. A new species of Neottia (Orchidaceae, Epidendroideae) from alpine border region between China and Myanmar. Phytotaxa. 2016;289(3):291–5.
Tesitelova T, Kotilinek M, Jersakova J, Joly F-X, Kosnar J, Tatarenko I, Selosse M-A. Two widespread green Neottia species (Orchidaceae) show mycorrhizal preference for Sebacinales in various habitats and ontogenetic stages. Mol Ecol. 2015;24(5):1122–34.
Jin X-H. A new species of Neottia (Orchidaceae, Epidendroideae) from southwestern China. Phytotaxa. 2014;177(3):188–90.
Raskoti BB, Wood JJ, Ale R. Neottia chandrae sp nov (Orchidaceae) from Nepal. Nord J Bot. 2012;30(2):187–9.
McKendrick SL, Leake JR, Taylor DL, Read DJ. Symbiotic germination and development of the myco-heterotrophic orchid Neottia nidus-avis in nature and its requirement for locally distributed Sebacina spp. New Phytol. 2002;154(1):233–47.
Zhou T, Jin X-H. Molecular systematics and the evolution of mycoheterotrophy of tribe Neottieae (Orchidaceae, Epidendroideae). Phytokeys. 2018;94:39–49.
Chen X-Q, Liu Z-J, Zhu G-H, Lang K-Y, Ji Z-H, Luo Y-B, Jin X-H, Cribb PJ, Wood JJ, Gale SW, et al. Flora of China, vol. 25. Beijing: Science Press; 2009.
Pearce NR, Cribb PJ. The Orchids of Bhutan. Huddersfield: Charlesworth Group; 2002.
Suetsugu K, Hirota SK, Hsu T-C, Kurogi S, Imamura A, Suyama Y. Monotropastrum kirishimense (Ericaceae), a new mycoheterotrophic plant from Japan based on multifaceted evidence. J Plant Res. 2022;136(1):3–18.
Suetsugu K. Gastrodia longiflora (Orchidaceae: Epidendroideae: Gastrodieae), a new mycoheterotrophic species from Ishigaki Island, Ryukyu Islands. Japan Phytotaxa. 2021;502(1):107–10.
Liu Q, Ya J-D, Wu X-F, Shao B-Y, Chi K-B, Zheng H-L, Li J-W, Jin X-H. New taxa of tribe Gastrodieae (Epidendroideae, Orchidaceae) from Yunnan, China and its conservation implication. Plant Divers. 2021;43(5):420–5.
Bandara C, Priyankara T, Atthanagoda AG, Lakkana T, Ediriweera S, Kumar P. Gastrodia gunatillekeorum (Gastrodieae, Epidendroideae, Orchidaceae), a new species from a lowland rainforest of Sri Lanka. Phytotaxa. 2020;436(1):55–62.
Ma L, Chen X-Y, Liu J-F, Chen S-P. Gastrodia fujianensis (Orchidaceae, Epidendroideae, Gastrodieae), a new species from China. Phytotaxa. 2019;391(4):269–72.
Aung YL, Jin X-H. Gastrodia kachinensis (Orchidaceae), a new species from Myanmar. Phytokeys. 2018;94:23–9.
Jin X-H, Kyaw M. Gastrodia putaoensis sp nov (Orchidaceae, Epidendroideae) from North Myanmar. Nord J Bot. 2017;35(6):730–2.
Chen SC: Tribe Neottieae. In: Flora Reipublicase Popularis Sinicae. Edited by Lang KY, Beijing: Science Press; 1999; 18: 74–120.
Chen S-C. The Consideration of Archineottia as Congeneric With Holopogon (Orchidaceae). J Syst Evol. 1997;35(2):178–80.
Tedersoo L, Pellet P, Koljalg U, Selosse M-A. Parallel evolutionary paths to mycoheterotrophy in understorey Ericaceae and Orchidaceae: ecological evidence for mixotrophy in Pyroleae. Oecologia. 2007;151(2):206–17.
Jacquemyn H, Waud M, Merckx VSFT, Brys R, Tyteca D, Hedren M, Lievens B. Habitat-driven variation in mycorrhizal communities in the terrestrial orchid genus Dactylorhiza. Sci Rep. 2016;6:37182.
Ogura-Tsujita Y, Yokoyama J, Miyoshi K, Yukawa T. Shifts in Mycorrhizal fungi during the evolution of autotrophy to mycoheterotrophy in Cymbidium (Orchidaceae). Am J Bot. 2012;99(7):1158–76.
Zhang X, Xing J, Zhu X, Zhao B, Liu C, Dong J, Hong L, Liu Y, Chen Y, Wen Z. Diversity and community structure of ectomycorrhizal fungi in Pinus thunbergii coastal forests bordering the Yellow Sea of China. Braz J Microbiol. 2021;52(2):801–9.
Tedersoo L, Bahram M, Toots M, Diedhiou AG, Henkel TW, Kjoller R, Morris MH, Nara K, Nouhra E, Peay KG, et al. Towards global patterns in the diversity and community structure of ectomycorrhizal fungi. Mol Ecol. 2012;21(17):4160–70.
Hui N, Liu X, Kotze DJ, Jumpponen A, Francini G, Setala H. Ectomycorrhizal Fungal Communities in Urban Parks Are Similar to Those in Natural Forests but Shaped by Vegetation and Park Age. Appl Environ Microbiol. 2017;83(23):e01797–17. https://doi.org/10.1128/AEM.01797-17.
Ishida TA, Nara K, Hogetsu T. Host effects on ectomycorrhizal fungal communities: insight from eight host species in mixed conifer-broadleaf forests. New Phytol. 2007;174(2):430–40.
Swarts ND, Sinclair EA, Francis A, Dixon KW. Ecological specialization in mycorrhizal symbiosis leads to rarity in an endangered orchid. Mol Ecol. 2010;19(15):3226–42.
Dong S, Zhao C, Chen F, Liu Y, Zhang S, Wu H, Zhang L, Liu Y. The complete mitochondrial genome of the early flowering plant Nymphaea colorata is highly repetitive with low recombination. Bmc Genomics. 2018;19:614.
McCormick M, Burnett R, Whigham D. Protocorm-Supporting Fungi Are Retained in Roots of Mature Tipularia discolor Orchids as Mycorrhizal Fungal Diversity Increases. Plants-Basel. 2021;10(6):1251. https://doi.org/10.3390/plants10061251.
Bidartondo MI, Burghardt B, Gebauer G, Bruns TD, Read DJ. Changing partners in the dark: isotopic and molecular evidence of ectomycorrhizal liaisons between forest orchids and trees. Proc Royal Soc B-Biol Sci. 2004;271(1550):1799–806.
Tesitelova T, Tesitel J, Jersakova J, Rihova G, Selosse M-A. Symbotic germination capbility of four Epipactis species (Orchidaceae) is broader than expected from adult ecology. Am J Bot. 2012;99(6):1020–32.
Taylor DL. Myco-heterotroph-fungus marriages - is fidelity over-rated? New Phytol. 2004;163(2):217–21.
Taylor DL, Bruns TD, Szaro TM, Hodges SA. Divergence in mycorrhizal specialization within Hexalectris spicata (Orchidaceae), a nonphotosynthetic desert orchid. Am J Bot. 2003;90(8):1168–79.
Albalat R, Canestro C. Evolution by gene loss. Nat Rev Genet. 2016;17(7):379–91.
Li Z-H, Ma X, Wang D-Y, Li Y-X, Wang C-W, Jin X-H. Evolution of plastid genomes of Holcoglossum (Orchidaceae) with recent radiation. BMC Evol Biol. 2019;19:63.
Dong W-L, Wang R-N, Zhang N-Y, Fan W-B, Fang M-F, Li Z-H: Molecular Evolution of Chloroplast Genomes of Orchid Species: Insights into Phylogenetic Relationship and Adaptive Evolution. Int J Mol Sci. 2018;19(3):716. https://doi.org/10.3390/ijms19030716.
Zimmer K, Meyer C, Gebauer G. The ectomycorrhizal specialist orchid Corallorhiza trifida is a partial myco-heterotroph. New Phytol. 2008;178(2):395–400.
Barrett CF, Wicke S, Sass C. Dense infraspecific sampling reveals rapid and independent trajectories of plastome degradation in a heterotrophic orchid complex. New Phytol. 2018;218(3):1192–204.
Suetsugu K, Ohta T, Tayasu I. Partial mycoheterotrophy in the leafless orchid Cymbidium macrorhizon. Am J Bot. 2018;105(9):1595–600.
Kim HT, Shin C-H, Sun H, Kim J-H. Sequencing of the plastome in the leafless green mycoheterotroph Cymbidium macrorhizon helps us to understand an early stage of fully mycoheterotrophic plastome structure. Plant Syst Evol. 2018;304(2):245–58.
Kobayashi K, Suetsugu K, Wada H. The Leafless Orchid Cymbidium macrorhizon Performs Photosynthesis in the Pericarp during the Fruiting Season. Plant Cell Physiol. 2021;62(3):472–81.
Bellino A, Alfani A, Selosse M-A, Guerrieri R, Borghetti M, Baldantoni D. Nutritional regulation in mixotrophic plants: new insights from Limodorum abortivum. Oecologia. 2014;175(3):875–85.
Girlanda M, Selosse MA, Cafasso D, Brilli F, Delfine S, Fabbian R, Ghignone S, Pinelli P, Segreto R, Loreto F, et al. Inefficient photosynthesis in the Mediterranean orchid Limodorum abortivum is mirrored by specific association to ectomycorrhizal Russulaceae. Mol Ecol. 2006;15(2):491–504.
Shevtsov S, Murik O, Zer H, Weinstein O, Keren N, Fragman-Sapir O, Ostersetzer-Biran O. The complete plastid genome sequence and the photosynthetic activity of the putative mycoheterotrophic orchid Limodorum abortivum. Israel J Plant Sci. 2019;66(1–2):69–88.
Petersen G, Zervas A, Pedersen HA, Seberg O. Genome Reports: Contracted Genes and Dwarfed Plastome in Mycoheterotrophic Sciaphila thaidanica (Triuridaceae, Pandanales). Genome Biol Evol. 2018;10(3):976–81.
Lam VKY, Gomez MS, Graham SW. The Highly Reduced Plastome of Mycoheterotrophic Sciaphila (Triuridaceae) Is Colinear with Its Green Relatives and Is under Strong Purifying Selection. Genome Biol Evol. 2015;7(8):2220–36.
Jiang Y, Hu XD, Yuan Y, Guo XL, Chase MW, Ge S, Li JW, Fu JL, Li K, Hao M, et al. The Gastrodia menghaiensis (Orchidaceae) genome provides new insights of orchid mycorrhizal interactions. BMC Plant Biol. 2022;22(1):179.
Wicke S, Müller KF, Quandt D, Bellot S, Schneeweiss GM. Mechanistic model of evolutionary rate variation en route to a nonphotosynthetic lifestyle in plants. Proc Natl Acad Sci. 2016;113(32):9045–50.
Molina J, Hazzouri KM, Nickrent D, Geisler M, Meyer RS, Pentony MM, Flowers JM, Pelser P, Barcelona J, Inovejas SA, et al. Possible Loss of the Chloroplast Genome in the Parasitic Flowering Plant Rafflesia lagascae (Rafflesiaceae). Mol Biol Evol. 2014;31(4):793–803.
Lin C-S, Chen JJ, Huang Y-T, Chan M-T, Daniell H, Chang W-J, Hsu C-T, Liao D-C, Wu F-H, Lin S-Y. The location and translocation of ndh genes of chloroplast origin in the Orchidaceae family. Sci Rep. 2015;5:9040.
Yudina SV, Schelkunov MI, Nauheimer L, Crayn D, Chantanaorrapint S, Hrones M, Sochor M, Dancak M, Mar S-S, Luu HT, et al. Comparative Analysis of Plastid Genomes in the Non-photosynthetic Genus Thismia Reveals Ongoing Gene Set Reduction. Front Plant Sci. 2021;12:602598. https://doi.org/10.3389/fpls.2021.602598.
Naumann J, Der JP, Wafula EK, Jones SS, Wagner ST, Honaas LA, Ralph PE, Bolin JF, Maass E, Neinhuis C, et al. Detecting and Characterizing the Highly Divergent Plastid Genome of the Nonphotosynthetic Parasitic Plant Hydnora visseri (Hydnoraceae). Genome Biol Evol. 2016;8(2):345–63.
Li Q, Ren Y, Shi X, Peng L, Zhao J, Song Y, Zhao G. Comparative Mitochondrial Genome Analysis of Two Ectomycorrhizal Fungi (Rhizopogon) Reveals Dynamic Changes of Intron and Phylogenetic Relationships of the Subphylum Agaricomycotina. Int J Mol Sci. 2019;20(20):5167. https://doi.org/10.3390/ijms20205167.
Jl Li. Wang S, Jing Y, Wang L, Zhou Sl: A modified CTAB protocol for plant DNA extraction. Chin Bull Botany. 2013;48(1):72–8.
Patel RK, Jain M. NGS QC Toolkit. A toolkit for quality control of next generation sequencing data. Plos One. 2012;7(2):e30619. https://doi.org/10.1371/journal.pone.0030619.
Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18(5):821–9.
McGinnis S, Madden TL. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004;32(suppl_2):W20–5.
Li YX, Li ZH, Schuiteman A, Chase MW, Li JW, Huang WC, Hidayat A, Wu SS, Jin XH. Phylogenomics of Orchidaceae based on plastid and mitochondrial genomes. Mol Phylogenet Evol. 2019;139:106540.
Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059–66.
Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In: Nucleic acids symposium series. London: Information Retrieval Ltd., c1979-c2000; 1999. p. 95–8.
Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX, Wang GT. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Res. 2018;20(1):348–55.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32(1):268–74.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29(8):1969–73.
Bouckaert R, Heled J, Kuehnert D, Vaughan T, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput Biol. 2014;10(4):e1003537. https://doi.org/10.1371/journal.pcbi.1003537.
Nylander J. MrModeltest v2. Program distributed by the author. Evolutionary Biology Centre: Uppsala University; 2004. p. 2.
Kim Y-K, Jo S, Cheon S-H, Joo M-J, Hong J-R, Kwak M, Kim K-J. Plastome Evolution and Phylogeny of Orchidaceae, With 24 New Sequences (vol 11, 22, 2020). Front Plant Sci. 2020;11:22.
Rambaut A, Suchard M, Xie D, Drummond A. Tracer v1. 6. 2014. http://tree.bio.ed.ac.uk/software/tracer/.
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior Summarization in Bayesian Phylogenetics Using Tracer 17. Syst Biol. 2018;67(5):901–4.
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol Biol Evol. 2017;34(12):3299–302.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7.
Yang Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–91.
Westerhold T, Marwan N, Drury AJ, Liebrand D, Agnini C, Anagnostou E, Barnet JSK, Bohaty SM, De Vleeschouwer D, Florindo F, et al. An astronomically dated record of Earth’s climate and its predictability over the last 66 million years. Science. 2020;369(6509):1383.
Acknowledgements
The authors thank Miss Yingying Wen, Mr. Chengwang Wang and Dr. Xiao Ma for their help on data analyses. We are also grateful to Mr. Hancheng Wang and Dr. Xueliang Guo for their advices on this paper, Zhoudong Han, Yulan Peng and Mung Htoi Aung for their help with field work.
Funding
This research was supported by the National Natural Science Foundation of China (32270214 to XJ) and the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA19050201).
Author information
Authors and Affiliations
Contributions
XH designed the research; XH and YJ collected the samples; SB and SS conducted laboratory work, and analyzed the data; XH, MZ wrote the manuscript; and all authors contributed to its final version. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Plant materials used in this study were collected in field with necessary permissions from local authorities. Voucher specimens have been deposited in publicly available herbaria, Herbarium (PE), Institute of Botany, Chinese Academy of Sciences and Herbarium (KUN), Kunming Institute of Botany, Chinese Academy of Sciences (Table 1). All methods were carried out in accordance with relevant guidelines and regulations. All specimens of Neottia listeroides complex were identified by Xiaohua Jin in Institute of Botany, Chinese Academy of Sciences and Jidong Ya in Kunming Institute of Botany, Chinese Academy of Sciences.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Taxon sampling and GenBank accession numbers. Table S2. Synonymous (dS) and non-synonymous (dN) substitution rates of N. listeroides complex members, relative to the reference N.fugongensis. Table S3. Selection pressure on 26 “housekeeping genes” of each sample relative to the reference N.fugongensis. Table S4. Positively selected sites detected by the branch-sitesmodel. Table S5. Relative synonymous codon usage of N. listeroides complex plastomes. Table S6. Mitochondrial protein-coding genes in N.listeroides complex. Fig. S1. Phylogenetic relationships based on maximum likelihood (ML) analysis of protein-coding sequences of plastid genome (ptCDS). Numbers above branches represent bootstrap support. N. listeroides complex members were divided into three clades: Pine Clade (blue branch) including samples from pine-broadleaf mixed forests; Fir Clade (green branch) including samples from alpine fir forests and For Clade (yellow branch) including sample from alpine fir--broadleaf mixed forest. Fig. S2. Mutation hotspots in plastomes of the N. listreoides complex. Fig. S3. Sequence identity of plastomes of N. listeroides complex members (LCY-1 as the reference). The vertical scale represents the percentage of identity between 50% and 100%. The horizonal axis indicates the coordinates within the plastomes. Fig. S4. Colinear analysis of plastomes of the N. listeroides complex. Color bands are locally-collinear blocks, representing homologous gene clusters. Within each block, similarity profiles of sequences corresponding to the average conservative level was shown. Fig. S5. Comparison of LSC, SSC, and IR border regions among 15 N. listeroides complex plastomes. Colored boxes for genes represent the gene position. Gene and region lengths are not to scale. Fig. S6. Branch length of non-synonymous (dN) and synonymous (dS) substitution rates of N. listeroides complex members.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shao, BY., Wang, MZ., Chen, SS. et al. Habitat-related plastome evolution in the mycoheterotrophic Neottia listeroides complex (Orchidaceae, Neottieae). BMC Plant Biol 23, 282 (2023). https://doi.org/10.1186/s12870-023-04302-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-023-04302-y