
Abstract
Background
Waterlogging is one of the major abiotic stresses in barley and greatly reduces grain yield and quality. To explore the mechanism controlling waterlogging tolerance in barley, physiological, anatomical and transcriptional analyses were performed in two contrasting barley varieties, viz. Franklin (susceptible) and TX9425 (tolerant).
Results
Compared to Franklin, TX9425 had more adventitious roots and aerenchymas and higher antioxidant enzyme activities. A total of 3064 and 5693 differentially expressed genes (DEGs) were identified in TX9425 after 24 h and 72 h of waterlogging treatment, respectively, while 2297 and 8462 DEGs were identified in Franklin. The results suggested that TX9425 was less affected by waterlogging stress after 72 h of treatment. The DEGs were enriched mainly in energy metabolism, hormone regulation, reactive oxygen species (ROS) scavenging, and cell wall-modifying enzymes. Alcohol dehydrogenase (ADH) plays an important role in response to waterlogging stress. We found that HvADH4 was significantly upregulated under waterlogging stress in TX9425. Transgenic Arabidopsis overexpressing HvADH4 displayed higher activity of antioxidant enzymes and was more tolerant to waterlogging than the wild type (WT).
Conclusions
The current results provide valuable information that will be of great value for the exploration of new candidate genes for molecular breeding of waterlogging tolerance in barley.
Similar content being viewed by others


Background
Waterlogging stress, one of the major abiotic stresses affecting crop growth, has become more frequent, severe, and unpredictable due to the excessive water that also decreases the oxygen content in the soil and the nutrient absorption ability of the plant [1,2,3]. In general, barley is sensitive to waterlogging, which causes 40%-79% irreversible yield loss, depending on the genotype, growth stage and duration of waterlogging stress [4].
Root is the first organ responding to oxygen shortage, and it is critical for the maintenance of normal physiological activities in plants [5, 6]. As the one of the key features in waterlogging condition, the formation of adventitious roots exists widely in different plant species [7, 8]. New adventitious roots contain more aerenchymas, which can help maintain a hypoxia-tolerant pathway, store and exchange of gases within the different tissues, meanwhile reduce the number of oxygen-consuming cells [9]. This formation of aerenchyma requires ethylene, Ca2+ and reactive oxygen species (ROS) signaling [10, 11].
Antioxidant metabolism is one of the fundamental metabolic pathways under waterlogging stress [12]. The production of reactive oxygen species (ROS) is inevitable with plant’s exposure to waterlogging, this includes the superoxide radical (O2.−), hydroxyl radical (•OH) and hydrogen peroxide (H2O2), which readily attack leaf chloroplasts and ultimately lead to leaf chlorosis and senescence [13]. To avoid this, a series of antioxidant enzymes will be synthesized/activated to scavenge ROS, such as superoxide dismutase (SOD), peroxidase (POD) and catalase (CAT) [14]. The membrane structure and the activity of the cells would be destroyed by malondialdehyde (MDA), which has been utilized as a reliable indicator for waterlogging tolerance [15]. Thus, high levels of SOD, POD and CAT enzyme activity are critical for the survival of crop under waterlogging conditions [9, 15].
In addition, the energy metabolic pathway will be affected by oxygen deficiency. ATP is produced through glycolysis instead of oxidative phosphorylation [16]. Meanwhile, genes associated with ATP and carbohydrate catabolism, such as pyruvate decarboxylase 1 (PDC1), alcohol dehydrogenase 1 (ADH1) and sucrose phosphate synthase (SPS), were significantly upregulated in the process of anaerobic fermentation [17, 18]. The ADH genes of plants play an important role in the response to waterlogging. Some studies on the ADH function have been performed by transgenic assays, such as in Arabidopsis [19], soybeans [20], and kiwifruit [21]. Overexpression of the GmAdh2 gene in transgenic soybean enhanced glycolysis and alcohol fermentation, and significantly increased the germination of transgenic lines under waterlogging [20]. Two genes in kiwifruit roots were also significantly induced after waterlogging treatment. The overexpression of AdADH1 and AdADH2 in kiwifruit enhanced waterlogging tolerance in transgenic Arabidopsis [21]. However, the function of ADH genes in response to waterlogging is different in various species.
Although numerous quantitative trait loci (QTL) studies have been conducted on waterlogging in barley, the genes responsible remain unidentified [22]. RNA-sequencing (RNA-seq) technology can identify key genes involved in various biological processes, and has been successfully used to reveal waterlogging responses in cucumber [23], wild soybean [24] and wheat [25]. In this study, physiological and dynamic RNA-seq analyses of on the roots of two barley cultivars exposed to waterlogging stress were conducted. The results obtained provide insights into the physiology and molecular mechanisms underlying the response of barley to waterlogging stress, which will facilitate barley genetic study and breeding applications.
Results
Phenotypic analysis of different barley varieties under waterlogging treatment
The phenotypes of the two genotypes (TX9425 and Franklin) after 21 days of waterlogging treatment are shown in Fig. 1. No significant difference between Franklin and TX9425 under control conditions was observed in adventitious root parameters. However, after three weeks of waterlogging treatment, the length, surface area, volume and number of adventitious root of TX9425 significantly increased and the fold change value ranged from 2.36 to 4.06 compared to the control, while there was no significant difference in Franklin except adventitious root number (Table 1). The adventitious root number of TX9425 increased 4.06 times and that of Franklin increased more than 2.4 times. The adventitious roots of TX9425 became more developed than Franklin roots under waterlogging stress. Franklin leaves became more wilted and chlorotic than TX9425 leaves under waterlogging treatment. The plant height, tiller number, leaf area, shoot fresh weight and dry weight of Franklin significantly decreased. Compared with a small decline, values were detected in TX9425 leaves (Table 2). Therefore, the performance of Franklin and TX9425 displayed significant differences after three weeks of waterlogging treatment.

Morphological traits between waterlogging-sensitive Franklin and waterlogging-tolerant TX9425. (A) (C) TX9425; (B) (D) Franklin
Physiological and anatomical analysis of different barley varieties under waterlogging treatment
As shown in Fig. 2 A-C, a significant genotype difference in the activities of SOD, CAT and POD in leaves was found. The antioxidant enzyme activity in both varieties decreased under waterlogging, while the decrease in the tolerance of TX9425 was lower. Under waterlogging treatment, the increase in MDA content of Franklin was approximately 2.1-fold, but the MDA content of TX9425 increased by only 1.3-fold (Fig. 2D). The root SOD activity of Franklin and TX9425 increased by 1.2- and 1.5-fold, respectively (Fig. 2E). The CAT activity of Franklin roots increased by 1.6-fold, and the CAT activity of TX9425 increased by 2.1-fold (Fig. 2G). Moreover, the POD enzyme activity of TX9425 increased by 1.4-fold, and no significant difference was observed in the roots of Franklin between the waterlogging treatment and the control (Fig. 2F). In contrast, the MDA content of Franklin increased by 2.1-fold compared with the MDA content of the control in roots, but the change in TX9425 was not significant (Fig. 2H). Therefore, TX9425 showed higher activity of antioxidant enzymes in leaves and roots after waterlogging, suggesting that TX9425 suffered less membrane damage than Franklin.

Effect of antioxidant enzymes activity and MDA content in leaf and root between TX9425 and Franklin. (A) SOD activity in leaf. (B) POD activity in leaf. (C) CAT activity in leaf. (D) MDA content in leaf. (E) SOD activity in root. (F) POD activity in root. (G) CAT activity in root. (H) MDA content in root. Results are the mean ± SD. *and** represent the significant differences at p < 0.05 and p < 0.01, respectively
Barley leaf anatomy is a typical monocotyledonous type consisting of epidermis, mesophyll and vascular tissue. Intercellular spaces existed among the mesophyll cells in the control. Under waterlogging, mesophyll cells of Franklin were severely damaged; in contrast, the leaves of TX9425 developed more lysigenous aerenchyma under waterlogging compared with the control (Fig. 3A). The adventitious root of barley was composed of the epidermis, cortex and cylinder of vascular tissues. Cortex parenchyma cells of adventitious roots formed a larger number of lysigenous aerenchyma under waterlogging conditions, compared with small intercellular space under control conditions. Remarkably, the proportion of TX9425 aerenchyma was significantly higher than Franklin after three weeks of treatment (Fig. 3B). Under waterlogging, adventitious roots were formed in the section of the shoot base in both lines, and more adventitious root primordia were observed in TX9425 than Franklin. Otherwise, in the absence of waterlogging, few adventitious roots were found in either accession (Fig. 3C, Table 2).

Transverse of leaf, adventitious root and root nodal between waterlogging-sensitive Franklin and waterlogging-tolerant TX9425. A Transverse of leaf in two genotypes; (B) Transverse of adventitious root in two genotypes; (C) Transverse of root nodal in two genotypes. Note: (A) Me, mesophyll cells; Ep, epidermis cell; Ae, aerenchyma. (B) C, Cortex; Pc, Parenchyma Cell; Pp, Primary Phloem; Px, Primary Xylem; Ae, Aerenchyma. C Arp, adventitious root primordia. Scale bar: 50 μm
Analysis of barley root transcriptome under waterlogging stress
To reveal the molecular mechanisms of barley in response to waterlogging stress, roots were collected from TX9425 and Franklin after 0 h, 24 h and 72 h waterlogging treatments. Each sample was subjected to three replicate treatments, and a total of 18 libraries were constructed. A high-throughput Illumina sequencing platform was used to sequence the transcriptome of barley. After removing adaptor sequences, low-quality reads, and reads with more than 10% ambiguous “N” bases, 2.87–7.58 GB data were obtained from each sample. The Q20 values of all transcriptomes were all above 96.42%, and the Q30 values were at least 92.41%, indicating high-quality sequencing data in the RNA-seq experiments (Table S1). On average, more than 63% of the valid reads were mapped into the reference barley genome. Principal component analysis (PCA) was conducted on the RNA-seq dataset of 18 samples. The control and treatment samples of the two genotypes were clearly separated by the first principal component (PC1), which accounted for 98.53% of the total variation (Fig. 4A).

Transcriptome analysis in roots of TX9425 and Franklin under control and waterlogging conditions. (A) Principal component analysis (PCA) of transcript changes separates the samples under control and waterlogging (24 h and 72 h waterlogging treatment) conditions. (B) Venn diagram depicting the number of all DEGs expressed in root tissues of TX9425 and Franklin genotypes under stress and control conditions. (C) The correlation between the log2 (fold change) of 10 DEGs in the qRT-PCR experiment (x axis) and in the RNA-seq experiments (y axis). Gene relative expression was calculated by 2−△△Ct method. Actin was used as the reference gene. Data used were means of three replicates
Identification of DEGs in two barley varieties in response to waterlogging stress
We further compared the DEGs in the two barley varieties subjected to waterlogging stress. We found a total of 3064 DEGs in TX9425 and 2297 DEGs in Franklin after 24 h of waterlogging stress compared to the control, by using the parameters of log2 fold change ≥ 1 and P value ≤ 0.05. A total of 1335 DEGs were upregulated and 1729 DEGs were downregulated in TX9425, while there were 967 upregulated genes and 1330 downregulated genes in Franklin (Fig. 4B). By comparing the transcriptome profiles of TX9425 and Franklin, a total of 2183 DEGs were observed to be uniquely expressed in TX9425 only, whereas 1416 DEGs were distinctively found in Franklin under the 24 h waterlogging treatment. In addition, 881 DEGs were common between the two genotypes (Fig. 4C).
A total of 5693 DEGs and 8462 DEGs were identified under waterlogging treatment (72 h) vs the control in TX9425 and Franklin, respectively. A total of 2012 DEGs were upregulated and 3681 DEGs were downregulated in TX9425, while there were 3314 upregulated genes and 5148 downregulated genes in Franklin. There were more DEGs after 72 h of waterlogging stress than after 24 h of waterlogging stress. The number of DEGs was significantly different between TX9425 and Franklin (Fig. 4B). A total of 1664 DEGs were uniquely expressed in TX9425 only, whereas a total of 4083 DEGs were distinctively found in Franklin under 72 h of waterlogging treatment. In addition, 4029 DEGs were common between the two genotypes (Fig. 4C).
In addition, to verify the reliability of the RNA-seq data, 10 DEGs were randomly selected for qRT-PCR analysis. Significantly positive correlations were observed between qRT-PCR and RNA sequencing data (r2 = 0.82). These results suggested that the RNA-seq data were credible (Fig. 4D).
Functional annotation of waterlogging-responsive DEGs
Gene Ontology (GO) functional classification analysis was performed to categorize the functions of DEGs during waterlogging stress (Table S2). As determined through a GO enrichment analysis of these DEGs, the DEGs in TX9425 and Franklin under 24 h waterlogging stress functioned mostly in biological processes, metabolic processes, transferase activity and catalytic activity (Fig. 5 A, B). After 72 h of waterlogging, the DEGs of TX9425 functioned mainly in metabolic processes, biological processes, organic cyclic compound binding, heterocyclic compound binding and catalytic activity. However, the DEGs in Franklin mostly showed localization, oxidation–reduction process, protein binding and catalytic activity (Fig. 5 C, D, Table S2).

Gene ontology (GO) enrichment analysis of differentially expressed genes (DEGs) in roots of TX9425 and Franklin under waterlogging stress. (A) GO enrichment analysis of TX9425 at 24 h waterlogging stress. (B) GO enrichment analysis of TX9425 at 72 h waterlogging stress. (C) GO enrichment analysis of Franklin at 24 h waterlogging stress. (D) GO enrichment analysis of Franklin at 72 h waterlogging stress
For KEGG pathway enrichment analysis, these DEGs were significantly (p < 0.01) enriched into 27 KEGG pathways (Table S3). Under 24 h of waterlogging stress, the DEGs of TX9425 were enriched mostly in metabolic pathways and biosynthesis of secondary metabolites. However, the DEGs in Franklin were associated primarily with biosynthesis of secondary metabolites and phenylpropanoid biosynthesis. Under 72 h of waterlogging stress, the DEGs of TX9425 were enriched mostly in the biosynthesis of secondary metabolites, MAPK signalling pathway, toll-like receptor signalling pathway. However, the DEGs in Franklin were associated primarily with biosynthesis of secondary metabolites, biosynthesis of antibiotics and phenylpropanoid biosynthesis.
Analysis of DEGs related to energy metabolism, hormone regulation, ROS scavenging and cell wall modifying enzymes
Energy deprivation is one of the major factors affecting survival of waterlogged plants. The KEGG enrichment analysis showed that many DEGs were involved in starch and sucrose metabolism and the glycolysis/fermentation pathway. As expected, we found that several DEGs, such as sucrose synthase, pyruvate kinase family protein, ATP-dependent 6-phosphofructokinase, alpha-amylase/subtilisin inhibitor, and fructose-bisphosphate aldolase 2, were significantly accumulated in both TX9425 and Franklin.
In addition, some DEGs involved in the glycolysis/fermentation pathway such as alanine aminotransferase, glyceraldehyde-3-phosphate dehydrogenase C2, alcohol dehydrogenase 1, L-lactate dehydrogenase A, and pyruvate decarboxylase-2, were also significantly induced by waterlogging stress in the two genotypes. In this study, we found that some genes had different expression levels in the two varieties. For example, the pyruvate kinase family protein (HORVU2Hr1G040570) and fructose-bisphosphate aldolase 2 (HORVU3Hr1G088500) were induced at higher levels in TX9425 than in Franklin after 24 or 72 h of waterlogging treatment. The expression levels of ATP-dependent 6-phosphofructokinase (HORVU5Hr1G019030), alpha-amylase/trypsin inhibitor (HORVU7Hr1G035020), and alcohol dehydrogenase 1 (HORVU1Hr1G082250, HORVU4Hr1G016810) first increased and then decreased in TX9425, while they continuously increased in Franklin. Consequently, TX9425 had a greater energy state than Franklin under waterlogging stress (Table 3).
Hormones play an important role in the plant response to environmental stress. Here, we identified some DEGs related to hormones that are involved mainly in the biosynthesis of ethylene and auxin. Ethylene is biosynthesized by the activation of 1-aminocyclopropane-1-carboxylicacid synthase (ACS) and ACC oxidase (ACO). Two ACSs and 6 ACOs were identified in TX9425 and Franklin. Two ACO genes (HORVU5Hr1G067490 and HORVU5Hr1G067530) were significantly accumulated in both varieties, but the genes inductions in TX9425 were greater. Thirty-one DEGs involved in auxin metabolism were identified in the two genotypes, including 23 downregulated and 8 upregulated genes. After 72 h of waterlogging treatment, the expression levels of HORVU1Hr1G025670, HORVU3Hr1G064590 and HORVU3Hr1G084840 in TX9425 were significantly higher than the levels in Franklin (Table 3).
Reactive oxygen species (ROS), which are produced when plants experience adverse stresses, can damage normal functions in plant cells. To survive, plants have evolved multiple strategies such as activating antioxidant systems to remove excess ROS. A total of 124 DEGs involved in ROS scavenging were found in our study, and most of them were downregulated. These DEGs are involved in the synthesis of glutathione S-transferase, peroxidase, catalase, and L-ascorbate oxidase, most of which (82 genes, 66.12% of 124) were related to peroxidase. Eight genes related to glutathione S-transferase and 8 genes related to peroxidase were upregulated in both genotypes, and the fold changes of these genes in TX9425 were significantly higher than the fold changes in Franklin (Table 3).
To adapt to waterlogging stress, plants also have evolved many mechanisms, such as the formation of adventitious roots and aerenchyma. The formation of aerenchyma was related to cell wall biosynthesis and loosening. As expected, we found that 34 DEGs were involved in cell wall modifying enzymes, such as xyloglucan galactosyltransferase, pectinesterase, and respiratory burst oxidase homologue. Eight DEGs were significantly upregulated in both genotypes. Under waterlogging stress, the genes HORVU2Hr1G101150 and HORVU4Hr1G081670 in TX9425 had significantly higher expression levels than those genes in Franklin (Table 3).
Overexpression of HvADH4 enhanced waterlogging tolerance by the increasing ROS scavenging capacity
A total of 44 ADH genes were identified in the barley genome based on the BLAST program. These genes were named HvADH1- HvADH44 according to their order of distribution on the chromosomes (Table S4). In the HvADH gene family, the length of coding sequences ranged from 99 bp (HvADH17) to 1524 bp (HvADH37). The size of the corresponding amino acids varied between 32 and 507.The theoretical isoelectric point (PI) of these genes ranged from 4.51 to 9.66, and the molecular weight (Mw) varied from 3.47 to 48.11 kDa.
In this study, 17 ADH genes were found to have differential expression between the waterlogging treatment and the control, except for HvADH25 in Franklin (Fig. 6). The highest differential expression was found for HvADH4 in TX9425, and there was an approximately 50- fold difference between 24 h and the control. We thus performed a standard method to isolate HvADH4 from TX9425. Sequencing of HvADH4 showed that the full-length gene was 1158 bp in length and encoded 385 amino acids. Multiple amino acid alignment showed that the HvADH4 protein shared two highly conserved ADH GroES-like (amino acids 36–156) and zinc-binding dehydrogenase domains (amino acids 205–336) (Fig. 7A). The phylogenetic tree indicated that HvADH4 has relatively high homology with proteins from Triticum turgidum, and relatively distant sequence homology with the proteins from Setaria italica (Fig. 7B).

The expression difference levels of DEGs involved in alcohol dehydrogenase between waterlogging treatment and control in TX9425 and Franklin

Amino-acid sequence alignment and phylogenetic tree analysis of ADHs from barley and other plant species. (A) Conserved domain alignment of ADHs from different plant species by Clustal W. (B) Phylogenetic tree analysis of ADHs from different plant species by MEGA 6.0 with Neighbor-Joining method
To further verify the function of barley HvADH4 (HORVU1Hr1G082250), transgenic Arabidopsis plants overexpressing the HvADH4 gene from TX9425 were generated. Five-week-old plants of the WT and three homozygous T3 transgenic lines were selected for waterlogging stress experiments. Transgenic plants that expressed HvADH4 were confirmed by RT-PCR (Fig. 8A). Under normal growth conditions, the transgenic Line-2 and Line-3 grew better than the WT, while the differences were found to be statistically insignificant (Fig. 8B). Under waterlogging conditions, plant height was reduced by 49.1% in the WT, and 31.2, 36.1 and 40.3% in the transgenic lines (Fig. 8C). Compared to the control, the SPAD value was 61.6% lower in the WT, and 41.4, 51.4, 48.8% lower in the transgenic lines (Fig. 8D). The shoot fresh weights of the transgenic lines were 29.2, 37.2 and 36.5%, respectively, which were lower than those weights in the control, and 65.8% smaller than those weights in the WT (Fig. 8E). The shoot dry weight decreased by 51.0% in the WT, and by 29.7, 13.3 and 22.9% in the transgenic lines (Fig. 8F). In addition, the root lengths of the WT plants decreased more than the root lengths of the transgenic lines during waterlogging stress (Fig. 8G). Furthermore, the average survival rate of the transgenic lines after waterlogging was 81.8%, but the average survival rate of the WT was only 37.4% (Fig. 8H). Taken together, these data indicate that the overexpression of HvADH4 in Arabidopsis significantly enhances plant waterlogging tolerance.

Waterlogging tolerance assay of HvADH4 overexpression lines (Line1, Line2, Line 3) and wild-type (WT). (A) RT-PCR analysis of transgenic and wild-type plants. AtACT8 was chosen as control gene. (B) Five-week-old plants were subjected to waterlogging stress for further 2 weeks. (C) Plant height. (D) Soil–plant analysis development (SPAD) value (based on chlorophyll meter reading). (E) Shoot fresh weight. (F) Shoot dry weight. (G) Root length. (H) Surival rate in the wild-type and HvADH4 transgenic lines were measured under control and waterlogging stress. Values are the means ± SD. Means were generated from three independent measurements. Asterisks indicate significant differences between transgenic plants and WT according to Student’s t-test (* p < 0.05; ** p < 0.01)
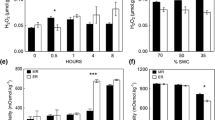
To investigate the difference in the physiological response to waterlogging stress between the WT plants and the transgenic plants, the activities of antioxidant enzymes (SOD, CAT, and POD), ADH activity and MDA content were examined under normal and waterlogging conditions. The transgenic lines showed higher ADH activity than the WT plants even when they were under control conditions, and ADH activity remained significantly higher at subsequent times (Fig. 9D). There were no significant differences in the activity of antioxidant enzymes between transgenic lines and WT under normal growth conditions. After waterlogging, the major antioxidant enzyme activity increased markedly in both WT and transgenic plants, reaching peak levels at 6 days of treatment and then decreasing after 9 days of treatment. However, the fold changes were significantly greater in the transgenic lines than in the WT (Fig. 9 A, B, C). MDA content is an important indicator to measure the level of lipid peroxidation. As shown in Fig. 9E, the MDA content in WT plants was significantly higher than the MDA content in transgenic lines, and this difference was more pronounced in the 6 d samples. Therefore, these results suggest that the overexpression of HvADH4 enhanced the scavenging ability of ROS in the plants and reduced the oxidative damage of plants under waterlogging stress.

Analysis of SOD, CAT, POD, ADH activities and proline content were carried out in transgenic lines and WT under waterlogging stressed conditions. SOD, CAT, POD, ADH and proline levels. (A–E) were measured in the leaves of plants subjected to waterlogging stress 3 days, 6 days, 9 days. The mean value and standard deviation were obtained from three independent experiments. The data represent mean ± SD of three biological repeats with three measurements per sample. Asterisks indicate significant differences from WT as determined using Student’s t-test (* p < 0.05; ** p < 0.01)
Discussion
Morpho-anatomical responses to waterlogging stress in barley
Waterlogging tolerance is a complicated trait, both physiologically and genetically [26, 27]. Waterlogging-induced plant growth inhibition is pervasive, including decreased dry weight (DW) in shoots/roots, leaf area, plant height, and chlorophyll content, and this inhibition also causes yield penalty [4]. Franklin was severely affected after 21 days of waterlogging treatment compared with TX9425, including plant height, tiller number, leaf area, shoot fresh weight and dry weight. Franklin shoots appeared wilted and presented more yellow leaves under waterlogging than the control. Remarkably, there are more morphological adaptations in waterlogging-tolerant TX9425 than the susceptible Franklin.
The root is the first organ affected by waterlogging stress. The formation of new adventitious roots and aerenchyma is one of the most important characteristics that adapts to hypoxic environments [5, 28]. Newly formed adventitious roots contain more aerenchyma, which not only provides a gas diffusion space with increased O2 transport from shoots to roots but also reduces the number of oxygen-consuming cells [29, 30]. In the present study, TX9425 had significantly more adventitious roots and aerenchyma than Franklin under waterlogging stress. This phenomenon has been reported in cucumber [23] and maize [31].
Waterlogging affects energy metabolism in barley
The energy metabolism pathway is critical for plant survival under low-oxygen stress and is related to starch and sucrose metabolism and glycolysis fermentation. As expected, we found that some DEGs, such as sucrose synthase 4, pyruvate kinase family protein, ATP-dependent 6-phosphofructokinase, fructose-bisphosphate aldolase 2, alcohol dehydrogenase, and pyruvate decarboxylase-2, were all upregulated after waterlogging. Compared to Franklin, the transcription levels of these genes were higher in TX9425 under both control and waterlogging conditions (Table 3).
Sucrose synthase (SUS) and sucrose phosphate synthase are key enzymes for the hydrolysis of sucrose, which play crucial roles in providing adequate sugar supply under waterlogging stress [32]. In low oxygen environments, the genes related to sucrose synthase in Arabidopsis [33], cucumber [23], and P. arundinacea [34], were all significantly upregulated. In addition, knockout of SUS1 and SUS4 induced less tolerance to oxygen deficits compared to wild-type in maize [35] and Arabidopsis [36]. The overexpression of sucrose synthase genes in cucumber confers tolerance against hypoxia stress [37]. Here, the expression of sucrose synthase 4 and sucrose phosphate synthase 1F were higher in TX9425 than in Franklin under 24 h of waterlogging stress. Pyruvate decarboxylase (PDC) is one of the key enzymes of ethanolic fermentation. PDC1 and PDC2 play an important role in waterlogging stress tolerance in Arabidopsis and Actinidia deliciosa [38]. In this study, only PDC2 was significantly upregulated under waterlogging stress (Table 3). This result suggests that TX9425 can accumulate more energy by decomposing more carbohydrates and amino acids, making it more readily adaptable to hypoxia stress.
Responses of ROS and hormones to waterlogging
Ethylene is an important hormone in response to waterlogging stress in plants, and can facilitate adventitious root and aerenchyma formation [5, 39]. In our previous study, we found that the ethylene content distinctly increased in TX9425 after waterlogging stress [40]. The RNA-Seq analysis showed that the expression of two ACO (HORVU5Hr1G067490 and HORVU5Hr1G067530) accumulated in both lines, while the gene expression in TX9425 was much greater. The results were consistent with published proteomic studies [41]. Auxin regulates the development of the lateral roots and plays a role in root growth [42]. Thirty-one genes involved in auxin metabolism were identified in this research, and most of them were downregulated. Interestingly, auxin storage was negatively associated with adventitious root initiation in cucumber [23].
Hypoxia stress can cause plants to overproduce ROS, which can cause progressive oxidative damage. To respond to oxidative stress plants have developed antioxidant defence systems, including SOD, CAT, POD, and GST [43]. Wang et al. [44] observed that the activities of antioxidant enzyme activity increased in soybean under waterlogging conditions. In contrast, Wang et al. [34] found that the activities of SOD, CAT, and POD were significantly decreased in Phalaris arundinacea in response to waterlogging stress. The reason for these two different results may be due to different treatment times and genotypes [45]. In this study, we identified 124 DEGs associated with the antioxidant system, in which most genes were downregulated. Similar to POD enzyme activity, 8 genes related to POD were upregulated in both genotypes, and the fold changes of these genes in TX9425 were significantly higher than in Franklin. In addition, one CAT gene (CAT2) in both lines was also upregulated after waterlogging treatment, indicating that this gene is a key regulator of CAT enzyme activity.
Xyloglucan endotransglycosylase/hydrolase (XTH) enzymes play a role in the loosening of cell walls and affect cell proliferation. XTH enzymes are involved in plant growth and resistance to stress [46]. We found that 24 DEGs were involved in XTH, and XTH 13 was significantly upregulated in TX9425 under waterlogging stress (Table 3). The present study suggests that XTH 13 plays an important role in waterlogging tolerance of barley.
Importance of the ADH gene in the response to waterlogging stress of barley
ADH is a major fermentative enzyme for oxidizing ethanol to acetaldehyde, which play a key role in resistance to waterlogging [21]. ADH family genes from tomato [47], rice [48], Pyrus bretschneideri [49] and wheat [50] have been detected at the whole genome level. Twenty-two ADH genes have been identified in the wheat genome database [50], and we identified 44 ADH genes in the barley genome.
The expression of ADH genes has been observed to be significantly elevated in soybean roots under hypoxia stress [51]. Shen et al. [50] found that TaADH1/2, TaADH3 and TaADH9 play an important role in the waterlogging tolerance of wheat, which was significantly induced by waterlogging. To further validate ADH gene function, some transgenic assays were conducted. Overexpression of ADH genes of soybean and kiwifruit increased waterlogging tolerance in transgenic plants [20, 21]. In contrast, overexpression of the Arabidopsis ADH1 gene and increased ADH activity do not affect ethanol levels and flooding survival tolerance under hypoxic conditions compared to wild-type cells [19]. Thus, the function of ADH genes varies with the different plants and stages. In this study, 17 ADH genes were differentially expressed, of which 7 genes were significantly upregulated after waterlogging stress. HvADH4 of TX9425 showed the highest level of differential expression. Overexpression of HvADH4 in transgenic Arabidopsis enhanced plant waterlogging tolerance, which could be caused by increased activity of fermentation and antioxidant enzymes.
Waterlogging is a complex trait controlled by numerous QTLs. So far, many QTLs associated with waterlogging tolerance have been successfully mapped using bi-parental linkage mapping based on various waterlogging related traits [7, 52,53,54,55]. The results of QTL mapping largely depend upon the two parents, population size, type of markers, and density of markers, and so on. For example, it has been reported that two major QTLs were mapped on 2H and 4H [52, 54]. However, Broughton et al. found that 10 QTLs associated with waterlogging-tolerant were mapped on 1H [7]. Cloning these genes have not been reported up to now. RNA-Seq mainly used to analyze gene expression with high-throughput sequencing. HvADH4 might be one of the downstream target gene under waterlogging stress. Candidate genes related to waterlogging stress in barley will be analyzed by GWAS and QTL.
In the present study, only wild-type Arabidopsis was used as a control, and transgenic line contains empty vector transgene were not used. Thus, the waterlogging tolerance of transgenic Arabidopsis lines might be attributed to the insertion of vectors in the Arabidopsis genome rather than the overexpression of HvADH4 gene. In addition, ectopic overexpression of a gene might not reflect its intrinsic function. In the future, the functions of the related genes will be further verified by over-expression, RNAi and gene editing in barley.
Conclusions
In this study, two barley varieties with different waterlogging tolerances were subjected to waterlogging treatment. Analyses of the morphological and physiological indicators revealed that TX9425 exhibited stronger waterlogging tolerance than Franklin. To further explore the mechanism controlling waterlogging tolerance in barley, RNA-seq analysis was performed. We identified 3064, 5693 and 2297, 8462 DEGs in TX9425 at 24 h, 72 h and Franklin at 24 h, 72 h, respectively. GO and KEGG analyses showed that the waterlogging tolerance of barley was closely related to energy metabolism, hormone regulation, ROS scavenging, and cell wall-modifying enzymes. Seventeen ADH genes were identified to be waterlogging responsive in barley. Among these genes, the expression level of HvADH4 was significantly different between the control and waterlogging groups. In addition, transgenic Arabidopsis with HvADH4 has improved waterlogging tolerance by deterring ROS accumulation. This work demonstrated that the HvADH4 gene plays an important role in waterlogging stress response.
Methods
Plant materials and treatments
TX9425 is waterlogging-tolerant feed barley originating from China, while Franklin is waterlogging-sensitive Australian malting barley [56]. The two accessions were obtained from the National Crop Genebank of China (NCGC, Beijing). The seeds of two genotypes were sown in plastic pots (22 cm × 25 cm) filled with the mixture of nutritional substance and vermiculite. Plants were grown in a greenhouse at a temperature of 20 °C/day and 15 °C/night. Waterlogging treatments started at the four-leaf stage and lasted for three weeks. The waterlogged plants were irrigated with tap water to 2–3 cm above the nutritional substance surface. The control plants were irrigated as needed to avoid drought stress or waterlogging stress. The experiment was carried out with three biological replications. After 21-day treatment, roots and leaves were collected and carefully washed with water for further analysis.
Morphological characteristic investigation
The leaf area and physiological traits of leaf were measured on the first fully expanded leaf below the shoot apex. Leaf chlorosis is the proportion of each plant that loses its green color (yellow) [56].
The roots were carefully rinsed with hydropneumatic elutriation device and detached from their nodal bases. Adventitious roots were arranged and floated on shallow water in a glass tray (30 cm × 30 cm), then scanned using Epson Expression 1680 scanner (Seiko Epson Corp, Japan), finally analyzed using WinRHIZO Root Analyzer System (Regent Instruments Inc., Canada) to measure length, diameter, surface area and root volume [57]. The parameters related to adventitious roots were measured with three biological replications, and six plants were selected for each replicate.
Tissue anatomy
After 21-day waterlogging treatment, samples were obtained from the mature zone of adventitious root (approximately 6 cm from the root apex), shoot base (root node), and leaf (the first fully expanded leaf below the shoot apex). They were cut into 0.5 cm segments, and immediately immersed in 0.1 M glutaraldehyde-phosphate buffer fixative (pH 7.2) at 4 °C for at least 3 h. Subsequently, the samples were dehydrated in a graded ethanol series of 20%, 40%, 60%, 80%, 90%, 95%, and 100% (15 min each). Ethanol was replaced with propyleneoxide, and the tissues were infiltrated and embedded in SPI low-viscosity Spurr’s resin [58]. Sections of 1 μm thick were cut with a glass knife on a Leica Ultracut R (Leica Microsystems, Inc., Germany), stained with 0.5% methyl violet for 10 min, and photographed under a light microscope (Leica, Germany). Root aerenchyma area and total root cross-sectional area were measured using the Image-pro plus (IPP) software (Media Cybernetics, USA).
Physiological trait analysis
Fresh leaves and roots (0.5 g each) were rinsed thoroughly with distilled water. The crude enzymatic extracts of each line were prepared in 0.05 M phosphate buffer (pH 7.8) after grinding with a pestle and milling to powder in liquid nitrogen. The homogenate was filtered through four layers of muslin cloth and centrifuged at 12 000 g for 10 min at 4 °C. The final supernatants were used for physiological and biochemical assays. Chlorophyll content was determined by using the SPAD-502. The activities of superoxide dismutase (SOD), peroxidase (POD), catalase (CAT) and the content of malondialdehyde (MDA) and alcohol dehydrogenase (ADH) were measured using the corresponding assay kits (Institute of Jiancheng Bioengineering, Nanjing, China) according to the manufacturer’s instructions [40].
RNA-seq and Transcription analysis
The root of TX9425 and Franklin were collected after waterlogging treatment for 24 h, 72 h, and control without waterlogging. Each treatment was processed with three biological replicates. Total RNA was extracted using the Plant RNA Purification Kit (Tiangen, Beijing, China). Twelve RNA-seq libraries (two accessions × two treatment × three biological replicates) were constructed by Novogene Bioinformatics Technology (Beijing, China) and sequenced by an Illumina HiSeq 2500 platform. Detailed process of transcriptome analysis as described in previous research [59]. The sequencing data were deposited in the NCBI SRA database (Bioproject ID: PRJNA889532). DEGseq was used to identify differentially expressed genes for RNA-seq data between waterlogging treatment and control. And the DEGs were further filtered with P value ≤ 0.05 and log2 fold change (log2 FC) ≥ 1.
Quantitative real-time RT-PCR
To confirm the reality of candidate genes screened from RNA-seq. 10 candidate genes were selected to further validate by quantitative (qRT-PCR). The method of qRT-PCR was described as previous report [40]. The specific primers used for target were designed using the Primer 6. All the primers are listed in Supplementary Table S5. The Hvactin, AtACT8 genes were used as the internal control. Target genes’ relative expression levels were determined as 2−△△Ct. Three biological replicates and three technical repeats were performed in all the qRT-PCR experiments.
Cloning and bioinformatic analysis of HvADH4
Total RNA was extracted from barley leaves of TX9425, and cDNA was synthesized as a template by M-MLV reverse transcriptase (TaKaRa, Otsu, Shiga, Japan) according to the manufacturer’s instructions. Primers of HvADH4 full-length CDS were designed by Primer 6. The PCR products were detected using agarose gel electrophoresis (1.0%). The sequence accuracy of the cloned genes was confirmed by DNA sequencing. The amino acid composition was analyzed with DNAMAN 9.0 software. The molecular weight and pI were examined the online software of ExPASy ProtParam (http://web.expasy.org/protparam/). Homologs of HvADH4 in other plant species were analyzed by the BLAST tool of NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi). MEGA 7.0 program was used for phylogenetic tree mapping by the neighbor-joining method and 1000 bootstrap replicates.
Candidate gene validation by transgenic Arabidopsis
To further verify the candidate gene, transgenic Arabidopsis plants were generated by floral dipping. The detailed design and methods have been previously described [52]. The Gateway technology (Invitrogen, USA) was used to constructed transgenic lines. Through the floral dipping method, recombinant vectors were transferred into Arabidopsis (Columbia) using the Agrobacterium tumefaciens strain GV3101 (Clough and Bent,1998). The transgenic lines were selected by germinating the seeds in a MS medium containing 30 mg/L hygromycin. 40 resistant seedlings (T1 generation) were transplanted to the greenhouse after two weeks. T2 transgenic plants with a 3:1 (resistant: nonresistant) segregation ratio were selected. Seeds of 6 homozygous plants from T2 lines were screened (T3 generation) for 100% resistance. Further genetic analysis was performed using the homozygous T3 generation. Five-week-old Arabidopsis plants (T3 lines) were used for waterlogging treatment. The control plants were kept in normal conditions with regular watering. After the treatment of two weeks, the phenotypic and physiological traits were observed and recorded.
Data analysis
For phenotypic, physiological parameter, and gene expression analysis was analyzed by Student’s t-test through the SPSS software. *and** represent the significant differences at p < 0.05 and p < 0.01, respectively. All data were presented as mean ± standard deviation (SD) and were measured at least three times. And three biological replicates were set.
Availability of data and materials
The transcriptome datasets supporting the conclusions of this study are available in the NCBI (BioProject: PRJNA889532).
References
Jackson M, Colmer T. Response and adaptation by plants to flooding stress. Ann Bot. 2005;96:501–5.
Zhou M, Li H, Mendham N. Combining ability of waterlogging tolerance in barley. Crop Sci. 2007;47:278–84.
Setter T, Waters I. Review of prospects for germplasm improvement for waterlogging tolerance in wheat, barley and oats. Plant Soil. 2003;253:1–34.
Romina P, Abeledo L, Miralles D. Identifying the critical period for waterlogging on yield and its components in wheat and barley. Plant Soil. 2014;378:265–77.
Sauter M. Root responses to flooding. Curr Opin Plant Biol. 2013;16:282–6.
Lorbiecke R, Sauter M. Adventitious root growth and cell-cycle induction in deepwater rice. Plant Physiol. 1999;119:21–30.
Broughton S, Zhou G, Teakle N, Matsuda R, Zhou M, O’Leary R, Colmer T, Li C. Waterlogging tolerance is associated with root porosity in barley (Hordeum vulgare L.). Mol Breeding. 2015;35:27.
Zhang X, Shabala S, Koutoulis A, Shabala L, Johnson P, Hayes D, Nichols D, Zhou M. Waterlogging tolerance in barley is associated with faster aerenchyma formation in adventitious roots. Plant Soil. 2015;394:355–72.
Wei W, Li D, Wang L, Ding X, Zhang Y, Gao Y, Zhang X. Morpho-anatomical and physiological responses to waterlogging of sesame (Sesamum indicum L.). Plant Sci. 2013;208:102–11.
Drew M, He C, Morgan P. Programmed cell death and aerenchyma formation in roots. Trends Plant Sci. 2000;5:123–7.
Evans D. Aerenchyma formation. New Phytol. 2003;161:35–49.
Yin D, Chen S, Chen F, Guan Z, Fang W. Morphological and physiological responses of two chrysanthemum cultivars differing in their tolerance to waterlogging. Environ Exp Bot. 2009;67:87–93.
Yordanova R, Christov K, Popova L. Antioxidative enzymes in barley plants subjected to soil flooding. Environ Exp Bot. 2004;51:93–101.
Blikhina O, Virolainen E, Fagerstedt K. Antioxidants, oxidative damage and oxygen deprivation stress: a review. Ann Bot. 2003;91:179–94.
Wu F, Zhang G, Dominy P. Four barley genotypes respond differently to cadmium: lipid peroxidation and activities of antioxidant capacity. Environ Exp Bot. 2003;50:67–78.
Jaradat M, Feurtado J, Huang D, Lu Y, Cutler A. Multiple roles of the transcription factor AtMYBR1/AtMYB44 in ABA signaling, stress responses, and leaf senescence. BMC Plant Biol. 2013;13(1):192.
Rahman M, Grover A, Peacock W, Dennis E, Ellis M. Effects of manipulation of pyruvate decarboxylase and alcohol dehydrogenase levels on the submergence tolerance of rice. Funct Plant Biol. 2001;28(12):1231–41.
Bailey-Serres J, Voesenek L. Flooding stress: acclimations and genetic diversity. Annu Rev Plant Biol. 2008;59(1):313–39.
Ismond K, Dolferus R, Pauw M, Dennis E, Good A. Enhanced low oxygen survival in arabidopsis through increased metabolic flux in the fermentative pathway. Plant Physiol. 2003;132:1292–302.
Tougou M, Hashiguchi A, Yukawa K, Nanjo Y, Hiraga S, Nakamura T, Nishizawa K, Komatsu S. Responses to flooding stress in soybean seedlings with the alcohol dehydrogenase transgene. Plant Biotechnol. 2012;29:301–5.
Zhang J, Huang S, Chen Y, Wang G, Guo Z. Identification and characterization of two waterlogging responsive alcohol dehydrogenase genes (AdADH1 and AdADH2) in Actinidia deliciosa. Mol Breeding. 2017;37:52.
Borrego B, Carter A, Tucker J, Yao Z, Xu W, Badea A. Genome-wide analysis of gene expression provides new insights into waterlogging responses in barley (Hordeum vulgare L.). Plants. 2020;9:240.
Xu X, Chen M, Ji J, Xu Q, Qi X, Chen X. Comparative RNA-seq based transcriptome profiling of waterlogging response in cucumber hypocotyls reveals novel insights into the de novo adventitious root primordia initiation. BMC Plant Biol. 2017;17:129.
Sharmin R, Mashiur R, Lv W, Yu Z, Chang F, Kong J, Bhat J, Zhao T. RNA-Seq based transcriptomic analysis revealed genes associated with seed flooding tolerance in wild soybean (Glycine soja Sieb. & Zucc.). Environ Exp Bot. 2020;171:103906.
Shen C, Yuan J, Qiao H, Wang Z, Liu Y, Ren X, Wang F, Liu X, Zhang Y, Chen X, Ou X. Transcriptomic and anatomic profiling reveal the germination process of different wheat varieties in response to waterlogging stress. BMC Genet. 2020;21:93.
Huang B, Johnson J. Root respiration and carbohydrate status of two wheat genotypes in response to hypoxia. Ann Bot. 1995;75:427–32.
Liu M, Jiang Y. Genotypic ariation in growth and metabolic responses of perennial ryegrass exposed to short-term waterlogging and submergence stress. Plant Physiol Bioch. 2015;96:57–64.
Steffens B, Geske T, Sauter M. Aerenchyma formation in the rice stem and its promotion by H2O2. New Phytol. 2011;190:369–78.
Mano Y, Omori F. Relationship between constitutive root aerenchyma formation and flooding tolerance in Zea nicaraguensis. Plant Soil. 2013;370:1–14.
Xu Q, Yang L, Zhou Z, Mei F, Qu L, Zhou G. Process of aerenchyma formation and reactive oxygen species induced by waterlogging in wheat seminal roots. Planta. 2013;238:969–82.
Kaur G, Vikal Y, Kaur L, Kalia A, Mittal A, Kaur D, Yadav I. Elucidating the morpho-physiological adaptations and molecular responses under long-term waterlogging stress in maize through gene expression analysis. Plant Sci. 2021;304: 110823.
Kumutha D, Sairam RK, Ezhilmathi K, Chinnusamy V, Meena RC. Effect of waterlogging on carbohydrate metabolism in pigeon pea (Cajanus cajan L.): upregulation of sucrose synthase and alcohol dehydrogenase. Plant Sci. 2008;175:706–16.
Baud S, Vaultier MN, Rochat C. Structure and expression profile of the sucrose synthase multigene family in Arabidopsis. J Exp Bot. 2004;55:397–440.
Wang X, He Y, Zhang C, Tian Y, Lei X, Li D, Bai S, Deng X, Hong H. Physiological and transcriptional responses of Phalaris arundinacea under waterlogging conditions. J Plant Physiol. 2021;261: 153428.
Ricard B, Van T, Chourey P, Saglio P. Evidence for the critical role of sucrose synthase for anoxic tolerance of maize roots using a double mutant. Plant Physiol. 1998;116(4):1323–31.
Bieniawska Z, Paul Barratt D, Garlick A, Thole V, Kruger N, Martin C, et al. Analysis of the sucrose synthase gene family in Arabidopsis. Plant J. 2007;49:810–28.
Wang H, Sui X, Guo J, Wang Z, Cheng J, Ma S, et al. Antisense suppression of cucumber (Cucumis sativus L.) sucrose synthase 3 (CsSUS3) reduces hypoxic stress tolerance. Plant Cell Environ. 2014;37:795–10.
Luo H, Zhang J, Wang G, Jia Z, Huang S, Wang T, Guo Z. Functional characterization of waterlogging and heat stresses tolerance gene pyruvate decarboxylase 2 from Actinidia deliciosa. Int J Mol Sci. 2017;18:2377.
Steffens B, Rasmussen A. The physiology of adventitious roots. Plant Physiol. 2016;170:603–61.
Luan H, Guo B, Pan Y, Lv C, Shen H, Xu R. Morpho-anatomical and physiological responses to waterlogging stress in different barley (Hordeum vulgare L.) genotypes. Plant Growth Regul. 2018;85:399–9.
Luan H, Shen H, Pan Y, Guo B, Lv C, Xu R. Elucidating the hypoxic stress response in barley (Hordeum vulgare L.) during waterlogging: a proteomics approach. Sci Rep. 2018;8:9655.
Butsayawarapat P, Juntawong P, Khamsuk O, Somta P. Comparative transcriptome analysis of waterlogging-sensitive and tolerant Zombi Pea (Vigna vexillata) reveals energy conservation and root plasticity controlling waterlogging tolerance. Plants. 2019;8(8):264.
Khan M, Nazir F, Asgher M, Per T, Khan N. Selenium and sulfur influence ethylene formation and alleviate cadmium-induced oxidative stress by improving proline and glutathione production in wheat. J Plant Physiol. 2015;178:9–18.
Wang S, Zhou H, Feng N, Xiang H, Liu Y, Wang F, Li W, Feng S, Liu M, Zheng D. Physiological response of soybean leaves to uniconazole under waterlogging stress at R1 stage. J Plant Physiol. 2022;268: 153579.
Li Y, Shi L, Yang J, Qian Z, He Y, Li M. Physiological and transcriptional changes provide insights into the effect of root waterlogging on the aboveground part of Pterocarya stenoptera. Genomics. 2021;113:2583–90.
Tiika R, Wei J, Cui G, Ma Y, Yang H, Duan H. Transcriptome-wide characterization and functional analysis of Xyloglucan endo-transglycosylase/hydrolase (XTH) gene family of Salicornia europaea L under salinity and drought stress. BMC Plant Biol. 2021;21:491.
Moummou H, Tonfack L, Chervin C, Benichou M, Youmbi E, Ginies C, Latche A, Pech J, Der Rest BV. Functional characterization of SlscADH1, a fruit-ripening-associated short-chain alcohol dehydrogenase of tomato. J Plant Physiol. 2012;169(15):1435–44.
Kitaoka N, Wu Y, Zi J, Peters R. Investigating inducible short-chain alcohol dehydrogenases/reductases clarifies rice oryzalexin biosynthesis. Plant J. 2016;88(2):271–9.
Qin G, Qi X, Qi Y, Gao Z, Yi X, Pan H, Xu Y. Identification and expression patterns of alcohol dehydrogenase genes involving in ester volatile biosynthesis in pear fruit. J Integr Agr. 2017;16(8):1742–50.
Shen C, Yuan J, Ou X, Ren X, Li X. Genome-wide identification of alcohol dehydrogenase (ADH) gene family under waterlogging stress in wheat (Triticum aestivum). Peer J. 2021;9: e11861.
Nanjo Y, Skultety L, Ashraf Y, Komatsu S. Comparative proteomic analysis of early-stage soybean seedlings responses to flooding by using gel and gel-free techniques. J Proteome Res. 2010;9:3989–4002.
Zhou M. Accurate phenotyping reveals better QTL for waterlogging tolerance in barley. Plant Breeding. 2011;130:203–8.
Zhou M, Johnson P, Zhou G, Li C, Lance R. Quantitative trait loci for waterlogging tolerance in a barley cross of Franklin x YuYaoXiangTian and the relationship between waterlogging and salinity tolerance. Crop Sci. 2012;52:2082–8.
Zhang X, Fan Y, Shabala S, Koutoulis A, Shabala L, Johnson P, Hu H, Zhou M. A new major-effect QTL for waterlogging tolerance in wild barley (H. spontaneum). Theor Appl Genet. 2017;130:1559–68.
Zhang X, Zhou G, Shabala S, Koutoulis A, Shabala L, Johnson P, Li C, Zhou M. Identification of aerenchyma formation related QTL in barley that can be effective in breeding for waterlogging tolerance. Theor Appl Genet. 2016;2016(129):1167–77.
Li H, Vaillancourt R, Mendham N, Zhou M. Comparative mapping of quantitative trait loci associated with waterlogging tolerance in barley (Hordeum vulgare L.). BMC Genomics. 2008;9:401.
Zhang H, Xue Y, Wang Z, Yang J, Zhang J. Morphological and physiological traits of roots and their relationships with shoot growth in “super’’ rice. Field Crop Res. 2009;113:31–40.
Luan H, Shen H, Zhang Y, Zang H, Qiao H, Tao H, Chen J, Chen H. Comparative transcriptome analysis of barley (Hordeum vulgare L.) glossy mutant using RNA-Seq. Braz J Bot. 2016;40:247–56.
Luan H, Guo B, Shen H, Pan Y, Hong Y, Lv C, Xu R. Overexpression of barley transcription factor HvERF2.11 in arabidopsis enhances plant waterlogging tolerance. Int J Mol Sci. 2020;6:1982.
Acknowledgements
Not applicable.
Research involving plants
The authors confirm that all the experimental methods and plants complied with relevant institutional, national, and international guidelines and legislation.
Funding
The work was sponsored by Natural Science Foundation of Jiangsu Province (BK20201215), National Barley and Highland Barley Industrial Technology Specially Constructive Foundation of China (CARS-05). Jiangsu (Lianyungang) Modern Agricultural (Science and technology comprehensive demonstration base (JAST[2022]175).
Author information
Authors and Affiliations
Contributions
J.W. and H.L. conceived and designed the study, supervised the experiments; M.X. and S.F. compiled and finalized the article, H.Q. and C.Y. performed the experiments, Y.W. and H.T. analyzed the data, H.Y. and H.T. drafted and wrote the manuscript, Y.L. and J.Y. revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors affirm no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Table S1. The information of transcriptome libraries.
Additional file 2:
Table S2. Summary of GO results of the differentially expressed genes.
Additional file 3:
Table S3. KEGG enrichment analysis of the differentially expressed genes.
Additional file 4:
Table S4. Properties and locations of the predicted HvADH proteins in barley.
Additional file 5:
Table S5. qRT-PCR and cloning primers used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Luan, H., Li, H., Li, Y. et al. Transcriptome analysis of barley (Hordeum vulgare L.) under waterlogging stress, and overexpression of the HvADH4 gene confers waterlogging tolerance in transgenic Arabidopsis. BMC Plant Biol 23, 62 (2023). https://doi.org/10.1186/s12870-023-04081-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-023-04081-6