
Abstract
Background
The large genus Ficus comprises approximately 800 species, most of which possess high ornamental and ecological values. However, its evolutionary history remains largely unknown. Plastome (chloroplast genome) analysis had become an essential tool for species identification and for unveiling evolutionary relationships between species, genus and other rank groups. In this work we present the plastomes of ten Ficus species.
Results
The complete chloroplast (CP) genomes of eleven Ficus specimens belonging to ten species were determined and analysed. The full length of the Ficus plastome was nearly 160 kbp with a similar overall GC content, ranging from 35.88 to 36.02%. A total of 114 unique genes, distributed in 80 protein-coding genes, 30 tRNAs, and 4 rRNAs, were annotated in each of the Ficus CP genome. In addition, these CP genomes showed variation in their inverted repeat regions (IR). Tandem repeats and mononucleotide simple sequence repeat (SSR) are widely distributed across the Ficus CP genome. Comparative genome analysis showed low sequence variability. In addition, eight variable regions to be used as potential molecular markers were proposed for future Ficus species identification. According to the phylogenetic analysis, these ten Ficus species were clustered together and further divided into three clades based on different subgenera. Simultaneously, it also showed the relatedness between Ficus and Morus.
Conclusion
The chloroplast genome structure of 10 Ficus species was similar to that of other angiosperms, with a typical four-part structure. Chloroplast genome sizes vary slightly due to expansion and contraction of the IR region. And the variation of noncoding regions of the chloroplast genome is larger than that of coding regions. Phylogenetic analysis showed that these eleven sampled CP genomes were divided into three clades, clustered with species from subgenus Urostigma, Sycomorus, and Ficus, respectively. These results support the Berg classification system, in which the subgenus Ficus was further decomposed into the subgenus Sycomorus. In general, the sequencing and analysis of Ficus plastomes, especially the ones of species with no or limited sequences available yet, contribute to the study of genetic diversity and species evolution of Ficus, while providing useful information for taxonomic and phylogenetic studies of Ficus.
Similar content being viewed by others


Background
The genus Ficus, which composes one of the 50 largest genera of angiosperms with approximately 800 species, is widely distributed in the tropical and semi-tropical temperate zones [1, 2]. Plants in this genus play a vital role in the ecosystem and are considered to be key species in tropical rainforests, because they serve as an extremely important source of food for frugivores species throughout the year [3, 4]. In addition, many Ficus species are traditionally used as sources of medicines and food, as ornamental resources, religious plants, lac hosts, fodder, fuel, hedges, or enclosures by humans [5, 6]. Over the past decades, extensive investigation on pharmacological studies has elucidated the medicinal properties of Ficus species, including antioxidant [7], anti-microbial [8], anti-cancer [9], anti-inflammatory [10] and anti-diabetic [11] properties. Therefore, the superposition of dietary and medicinal values endows many Ficus species with high research value, especially to Chinese people [12].
Ficus (Moraceae) is a key group of tropical and subtropical plants with extremely important ecological significance, with the phylogenetic relationships of this group under controversy [13]. In 1965, Corner published a revised and more comprehensive classification system of Ficus, in which the Ficus genus was divided into four subgenera based on morphological characteristics including male flowers, female flowers, fruit characters, and some anatomical characters of leaves (such as the distribution of camphor), namely subgenus Urostigma, subgenus Pharmacosycea, subgenus Ficus and subgenus Sycomorus [14]. But this classification system has been questioned by Ramirez [15] and Berg [1, 2]. Hereafter, based on the morphologic study by Corner and the molecular systematics study by Weiblen [16], Berg added another two subgenera, namely Sycidium and Synoecia, in addition to the original 4 subgenera [2]. Although the classification system in those 6 subgenera has been accepted by most taxonomists, emphasis was laid on the Ficus genus, raising issues such as classification difficulties and incomplete collection of species, which renders this classification still unresolved.
With the advances of next-generation sequencing [17], the acquisition of whole genomes becomes easier than before. As an important organelle in plants, CP contains the whole enzymatic machinery, which is necessary for photosynthesis and plays a crucial role in carbon uptake [18]. Simultaneously, it possesses a small, highly conserved genome that takes the form of a circular double-stranded DNA molecule. In most angiosperms, the typical CP genome exhibits a conserved tetrad structure, formed by two IRs, one LSC region and one SSC region [19]. In general, the size of the CP genome ranges from 115 to 165 kb, owing to a contraction or expansion of the IR region. Additionally, the CP genome contains approximately 114 genes, among which there are ~ 80 protein-coding genes, 4 rRNA genes, and 30 tRNA genes [20, 21]. Even though the plant CP genome is evolutionarily conserved, it presents highly variable regions that some of them exhibit an accelerated evolution rate [22,23,24]. Based on these characteristics, the CP genome is often used for phylogenetic and evolutionary studies, and has been proved useful for screening species-specific genetic markers, i.e. DNA barcoding, SNPs, among others in recent years [25,26,27,28]. Therefore, we expect that plastome comparative genomics on more Ficus species might provide insights on Ficus taxonomic and phylogenetic concerns raised previously, and will allow the development of DNA barcodes for a reliable identification of Ficus species.
In this study, ten Ficus plastomes were obtained by Illumina NGS. Genome comparative analysis showed their quadripartite structure and their genetic diversity was assessed, including the identification of repeated regions (SSRs, large sequence repeats, among others). Barcode DNAs were developed in hypervariable regions for species molecular identification. Furthermore, the phylogenetic analysis revealed the evolutionary relationships of Ficus species, shedding light in the actual controversy among others.
Results
Features of the Ficus species chloroplast (CP) genome
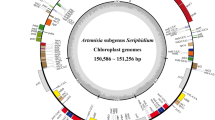
The studied Ficus CP genomes display a typical circular double-chain structure, with sizes ranging from 160,238 to 160,700 bp (Fig. 1, Table 1). The Ficus plastomes show the classic quadripartite architecture, with an LSC region (88,400–88,804 bp) and an SSC region (19,926–20,145 bp) separated by two inverted repeat (IR) regions (25,840–25,901 bp). All eleven CP genomes show similar total GC content (ranging from 35.88% to 36.02%), being significantly higher in the IR regions (Table 1).

Genome map of the average Ficus CP genome obtained in this work. The inner circle represents the quadripartite structure, with two copies of the inverted repeat (IRA and IRB), an LSC, and an SSC region in black with GC content in dark grey and AT content in light grey. External circle represents gene content, with those inside the circle transcribed clockwise, while the ones located at the outer side are counter clockwise transcribed. Genes are coloured following functional groups according to the legend show on the left bottom
While only counting one copy of those duplicated genes in the IR region, we annotated a total of 114 unique genes, consisting of 30 tRNAs, 4 rRNAs, and 80 protein-coding genes in each of the Ficus plastomes characterized. Furthermore, the overall length of the CDS region ranged from 80,334 to 80,598 bp. And the content of GC in CDS regions is slightly higher than that of the whole, varying from 37.1 to 37.2% (Table S1). In detail, in all eleven CP genomes, we identified 16 duplicated genes in the IR region, among which there are seven tRNA genes, four rRNA genes, and five protein-coding genes. A total of 63 CDS and 22 tRNA genes are present in the LSC region, while 12 CDS and one tRNA gene exist in the SSC region (Table S2). Two pseudogenes (ycf1 and rps19) are located in the boundary between IR-SSC and IR-LSC.
There were 18 genes harbouring introns, which can regulate gene expression and enhance the expression of exogenous genes at specific sites and specific times of the development of the plant [29, 30]. Among those, 12 are protein-coding genes and 6 are tRNA genes. Most genes [15] have only a single intron, whereas ycf3 and clpP genes contain two introns. The rps12 gene is so unique that it is composed of three complex exons, containing one 5’ exon and two 3’ exons. The 5’ exon is located in the LSC region, while the 3’ exons are distributed within the IR regions, which is consistent with close species such as Ficus religiosa [31], Morus celtidifolia [32], and Broussonetia kazinoki [33]. Two pseudogenes, ycf1 and rps19, are located between the IRB/ SSC and IRA /LSC, respectively. On account of the reverse repeating property of the IR region, these two genes fail to be fully duplicated and lose the ability to encode a complete protein, which leads to the presence of two pseudogenes. In addition, the trnK-UUU gene, which embodies the matK gene, has the largest intron (2,583–2,601 bp), compared to other genes (Table S3).
Identification of repeat elements
A mass of repeated sequences is widely distributed in the intergenetic spacer and intron sequences of the Ficus CP genome, which have always been the focus of genome research [34, 35]. Long repeats with a length greater than 30 bp might have functions in promoting chloroplast genome rearrangement and increasing population genetic diversity [36]. For the purpose of getting a comprehensive understanding of the long repeats within the Ficus CP genome, we classified those repeated sequences into five categories, namely tandem, forward, palindromic, reverse, and complementary repeats. These results manifested that the number of repeated sequences in the eleven Ficus CP genomes ranges from 69 (F. hispida) to 82 (F. tikoua). Among them, the number of tandem repeats were found to be the most abundant (46.4%-54.1%), varied from 32 (F. hispida) to 42 (F. sarmentosa var. lacrymans, F. microcarpa), followed by palindromic repeats (26.0%-31.7%), ranging from 20 (F. formosana, F. simplicissima) to 26 (F. tikoua), and then by forward repeats (14.9%-20.3%), with the scope of 11 (F. simplicissima) to 16 (F. sarmentosa var. impressa) (Fig. 2A). Among the ten Ficus species, the length of tandem repeats is generally distributed between 10 and 20 bp, while the size of palindromic and forward repeats is concentrated between 30 and 39 bp (Fig. 2B-D).

Repeat sequences analysis in eleven Ficus plastomes. A: Repeat types of eleven CP genomes. B: Tandem repeats in eleven CP genomes. C: Palindromic repeats in eleven CP genomes. D: Forward repeats in eleven CP genomes. Repeats with different lengths are indicated in different colours, the ordinate represents the number of repeats
Simple sequence repeats (SSRs) are composed of small repeated sequences ranging from 1 to 6 bp [37], which are extensively distributed at different locations such as intergenic region, intron region, and even protein-coding region [38]. The CP genome possesses the nature of uniparental inheritance, leaving SSRs a high level of variation within the same species [39]. Thus, chloroplast SSRs are important sources for developing molecular markers, which are widely used in phylogenetic and population genetic analysis [40, 41]. Here, a total of 299–317 SSRs were identified in these Ficus plastomes (Fig. 3), with average percentages of mononucleotide, dinucleotide, trinucleotide, tetranucleotide SSRs being 48.59%, 24.39%, 24.94%, and 3.16%, respectively. It can be found that pentanucleotide SSRs are very rare in all sequenced genomes, and we were able to detect hexanucleotide SSRs only in the plastome of Ficus simplicissima.

Analysis of number and type of SSRs in eleven Ficus plastomes. SSRs with different types are indicated in different colours
Codon usage and RNA editing sites
Codon usage patterns and nucleotide composition help to lay a theoretical foundation for genetic modifications of the CP genome [42, 43]. Here, amino acid frequency, codon usage number, and the relative synonymous codon usage (RSCU) in the eleven Ficus plastomes were analysed and summarized (Fig. 4, Table S4). A total of 64 RSCU were presented in the Ficus plastomes, and the number of codons varied from 53,412 to 53,566. Leucine and cysteine were the most and least universal amino acids, with UUU (encoding phenylalanine) and GCG (encoding alanine) as the most and least used codons in Ficus, respectively. Most of amino acid codons, except for methionine and tryptophan, had more than one synonymous codon, among which, leucine, serine, and arginine showed the maximum (six codon usage). Preferred codon is defined when its RSCU value was greater than 1.00. In the studied eleven Ficus plastomes, the number of preferred codon usage identified ranged from 28 to 32 (Fig. 4). Moreover, many of the preferred codons end with an A or T, whereas non-preferred codons ended with a C or G, supporting the reduced GC content in coding regions. This phenomenon is common in chloroplast genomes from other species [44, 45].

Codon content for the 20 amino acids and stop codons of CDS of the Ficus species CP genome. A: Codon content for CDS in the six Ficus CP genomes, each column in the bar graph represents a species. The corresponding species from left to right are F. pumila, F. tikoua, F. hispida, F. virens, F. sarmentosa var. impressa, and F. pandurata. B: Codon content for CDS in the rest five Ficus CP genomes, the corresponding species from left to right are F. microcarpa, F. formosana, F. sarmentosa var. lacrymans, F. simplicissima, and F. tinctoria
Previous studies have shown that the distribution of chloroplast RNA editing sites is uneven and more prone to protein-coding genes [46]. A total of 35 protein-coding genes were evaluated with the PREP program, to predict RNA editing sites in the Ficus plastomes. In sum, 59–65 RNA editing sites were identified (Table S5), in which amino acid conversion from S to L occurred the most frequently, while R-G occurred the least. Interestingly, it was found that all RNA editing sites appeared in the first position or second position of the corresponding codon, while no potential RNA editing sites were observed at the third position. The base conversion type is all from C to T, which is similar to those of other land plants [47, 48].
IR contraction and expansion in the Ficus CP genome
The typical quadripartite structure of the CP genome results in four boundary limits among IR, LSC, and SSC regions, namely IRB-LSC, IRB-SSC, SSC-IRA, and IRA-LSC [49, 50]. Although the inverted repeat regions (IRA and IRB) are the most conserved regions of the CP genome, shrinkage and expansion of the IR boundaries are hypothesized to help explain size differences between CP genomes beyond genus. The length of the IR region in the twelve CP genomes exhibited a modest expansion, ranging from 25,710 bp to 25,901 bp. In this work the IR-SSC and IR-LSC boundaries of Ficus species were compared to that of Morus alba var. atropurpurea (belonging to another genus within the Moraceae). Four affected protein coding genes that create some variable regions were found useful for species identification (Fig. 5).

Comparison of the borders of LSC, SSC, and IR regions among twelve CP genomes. Corresponding species from top to bottom are Morus alba var. atropurpurea, F. pumila, F. tikoua, F. hispida, F. virens, and F. sarmentosa var. impressa, F. sarmentosa var. lacrymans, F. microcarpa, F. pandurata, F. tinctoria, F.formosana, F. microcarpa, and F. simplicissima. Ψ: pseudogenes
In M. albo var. atropurpurea, the rps19 gene is entirely located within the LSC region, while it expands to the IRB region in the studied eleven Ficus plastomes, altering the boundary LSC-IRB. This fact resulted in truncated rps19 copies (ψrps19) at the junction IRA-LSC in Ficus species. Another gene crossing junction border is ycf1 that crosses the IR-SSC borders within the twelve CP genomes, creating truncated ψycf1 at the joint of IRB-SSC with a size variation from 986 to 1027 bp. It has been reported that the ycf1 gene contributes to the analyses of the CP genome variation in higher plants. Another affected gene, ndhF, covers the IRB-SSC region exhibits high similarity in ten Ficus species. Whereas the trnH gene was found to be shifted from the IRA-LSC border in all twelve species, with the longest distance (118 bp) to the border observed in F. tikoua species (Fig. 5).
Comparative genomic analysis
Interspecific comparisons employing the online software mVISTA were performed to reveal the conservation and divergence among Ficus species, as previously done with other species [39, 51]. The eleven Ficus plastomes were compared to the F. pumila plastome as the reference (Fig. S1). We found that the two IR regions were less divergent than the LSC and the SSC regions, which also occurred in almost higher plants [52]. Moreover, the non-coding region exhibited more nucleotide divergence than the coding regions. In the coding region, most genes were relatively conservative except matK, rps16, rpoC2, psbD, ndhD, and ycf1. These divergence hotspot regions identified in the eleven plastome sequences provided vast information for the development of molecular markers for phylogenetic analyses and for Ficus plant species identification.
Divergence hotspot region
Highly variable sequences can be utilized to determine the phylogenetic relationship between species and genera [53, 54]. Nucleotide diversity (Pi) values were calculated within 800-bp windows (Fig. 6) to identify sequence divergence hotspots. The result showed that the Pi value of the whole Ficus CP genome varied from 0 to 0.01543, which represents the nucleotide diversity. Eight highly variable regions (Pi > 0.009) were detected: matK-rps16, rpoB-trnC, trnT-psbD, trnL-trnF, rpl32-trnL, clpP, ndhD and ycf1. Among these, five regions (matK-rps16, rpoB-trnC, trnT-psbD, trnL-trnF, and clpP) are located in the LSC region, and the remaining three are in the SSC region (Fig. 6). This is consistent with preceding results that the IR region is generally more conserved than the LSC and the SSC regions [34, 55].

Comparative analysis of the nucleotide variability by Pi values of the eleven CP genomes presented in a sliding window (window length: 800 bp; step size: 200 bp). X-axis: position of the midpoint of a window; Y-axis nucleotide diversity in each window
Phylogenetic analysis
Phylogenetic analysis is often used to infer or evaluate evolutionary relationships [28, 56]. To examine the phylogenetic positions of the ten Ficus species and their relationships within Moraceae, ML phylogenetically analyses was performed using concatenated protein coding genes sequences from 32 CP genomes belonging to 5 genera of Moraceae and two CP genomes beyond the Moraceae family. As illustrated in Fig. 7, the phylogenetic tree has divided all species into six groups (I to VI), with almost all nodes supported with 100% bootstrap values (BP). Group I contained two species (Cannabis sativa and Rhamnus taquetii), which were set as outgroups. Those Moraceae species shaped into four paraphyletic groups. Group II and Group III consisted of Malaisia scandens from the genus Malaisia, and Artocarpus heterophyllus from the genus Artocarpus, respectively. Group IV contained six species belonging to the genus Broussonetia and Group V correspond to seven species from the genus Morus. Group VI was the most complex, and the real target of this study, which was comprised of 17 species from the genus Ficus and was further divided into three subgroups, each belonging to a different subgenus. The first subgroup contained F. microcarpa and F. virens, being clustered with F. religiosa which belongs to the subgenus Urostigma. The second subgroup contained F. tikoua and F. hispidia, being clustered with subgenus Sycomorus species (F. racemosa and F. beipeiensis). Whereas the third subgroup clustered 9 species belonging to Ficus subgenus, of which 6 species (7 sequences) were obtained in this work (Fig. 7). The Ficus clade was sistered to the Morus clade, whose common ancesteor derives from Goup II to IV, indicating a close relationship between the Ficus and Morus genera.

Phylogenetic relationships among 34 plant species based on CP genome. Phylogenetic inference was performed with concatenated protein coding genes sequences from all species shown using ML method, with branch support shown as Bootstrap values with green circles
Discussion
Ficus plastomes characterization and use for species identification
Eleven Ficus CP genomes were obtained and analysed in this study. The comparative analysis revealed highly conserved structures and genes. The plastome sizes showed slight differences, which suggested that the CP genome length in Ficus is highly conserved.
Repeat sequences, which are dispersed in CP genomes at high frequency, play a vital role in genome organization and evolution. In this work, we found resembling repeat types with similar distributions among ten Ficus species. SSRs, displaying a high level of polymorphism, are common in the CP genome as microsatellite repeats [38]. These sequences were used as a genetic marker in previous investigations [57]. The SSRs in the Ficus CP genomes were found to be particularly rich in AT, which corresponded with previous studies where proportions of polyadenine (polyA) and polythymine (polyT) were higher than polycytosine (polyC) or polyguanine (polyG) within chloroplast SSRs in many plant species [58].
RNA editing is a very common phenomenon that exists in plant CP genomes. The main functions of RNA editing include modifying mutations, correcting and regulating translation [59]. Interestingly, among the 35 protein-coding genes used to predict RNA editing sites, ndhB and ndhD have the most editing sites, and both encode subunits of the chloroplast NADH dehydrogenase complex, which is involved in electron transfer during photosynthesis [60].
The expansion and contraction of IR and SC (including LSC and SSC) boundaries are thought to be the main cause of CP genome size changes, although CP genomes in land angiosperms are highly conserved [61]. After comparing CP genomes among the ten Ficus species in our study, we found that the boundary region between the SC and two IR regions was relatively conserved, with gene distribution and specific location exhibiting high consistency. Compared with the other Morus species from the same family, the IR region of Ficus species showed expansion, mainly because the rps19 gene located at the LSC-IR boundary, expanded to the IR region by 109 bp. This indicates that the contraction and expansion of the IR regions are more common among different genera.
DNA barcoding is a method for rapid and accurate identification of species using a short and accurate DNA fragment. The concept of DNA barcoding was first proposed in 2003 by Hebert et al. [62]. Since then, an increasing number of researchers have focused on the selection of one or a few standard markers as DNA barcode(s). The earliest proposed DNA barcoding technology can identify species through ITS2, matK, psbA-trnH, rbcL and other DNA sequences [63]. However, it was found that these classical DNA barcodes were not suitable for the identification of the Ficus species of this study, due to the low nucleotide diversity in those ‘universal’ barcode fragments. Hence, finding suitable DNA markers for proper identification of these species was crucial. Here, according to nucleotide diversity analysis shown in Fig. 6, eight regions arose as putative barcoding regions, including five intergenic regions (matK-rps16, rpoB-trnC, trnT-psbD, trnL-trnF, and rpl32-trnL) and three genic regions (clpP, ndhD, and ycf1). Among these regions or genes, the ycf1 gene, as the second-largest gene in the chloroplast genome, is crucial for plant viability. Dong et al. [64] have proposed that the ycf1 is the most variable site in the chloroplast genome, showing greater variability than existing chloroplast candidate barcodes such as rbcL, matK, and trnH-psbA, and thus may have potential applications as land plant DNA barcodes. Another two genic markers clpP [65] and ndhD [66] have also been reported as a region of high variation for plant molecular identification.
Five intergenic spacer regions including matK-rps16, rpoB-trnC, trnT-psbD, trnL-trnF and rpl32-trnL, located within the SSC, are highly variable regions in the Ficus chloroplast genome, which have also been proposed as potential DNA barcodes in other species. Among them, matK-rps16 was demonstrated well utilization as DNA barcodes for Triticum plant [67] and rpoB-trnC was identified to be an effective marker for three Synstylae species [68]. Cheng et al. [69] suggested that trnT-psbD and rpl32-trnL potentially be used as molecular genetic markers for population genetics and phylogenetic studies of E. mollis. And trnL-trnF has a long history of use in plant phylogenetic studies [70], whereas this spacer often contains large A/T-rich regions that may lead to a low sequence quality [71]. Generally, although several candidate barcoding regions were identified, further research is still necessary to determine whether these highly divergent markers could be used in the identification and phylogenetic analyses of Ficus species.
Ficus phylogenetic relationships with other members of Moraceae family
The Moraceae family consists of approximately 40 genera with 1100 species, most of which are distributed in tropical and subtropical regions [72]. It mainly includes genus Ficus, Malaisia, Artocarpus, Broussonetia, Morus among others. At present, little research has been reported on the phylogeny of Moraceae, especially focusing on Ficus species. Ficus, regarded as a model system for understanding co-evolution dating back more than 75 million years, has not been able to confidently resolve phylogenomic relationships due to the lack of well-supported phylogenetic hypothesis, lack of species involved in the study or reduced dataset [73, 74]. Previously, Herre et al. (1996) performed the molecular phylogenetic studies of 15 Ficus species based on trnL-F and rbcL chloroplast markers [75]. Then, Renoult et al. (2009) revealed the potential of five non-coding chloroplast markers to address deep phylogenetic relations in Ficus, accounting for 38 species of African Ficus from the Urostigma section of Galoglychia subgenus [76]. Appearing significant conflicts when Ficus plastid phylogeny was compared with Ficus phylogeny based on ribosomal ITS and ETS [77]. These studies failed to represent what we currently know about the phylogenetic diversity within Ficus, and only sampled a maximum of 3,604 bp of plastid DNA [75, 76, 78]. More recently, Bruun-Lund et al. (2016) have examined the chloroplast genomes of 59 Ficus species and revealed that the phylogenies built from these genetic data provided both additional support to the current understanding of the evolutionary relationship of major species groups and discordance with information inferred from nuclear data [31]. In this study, eleven new CP genomes from ten Ficus species were added to solve this controversy, finding that Morus and Ficus are closely related compared with other genera. The eleven new CP genomes clustered into the same clade, with other reported Ficus species and can be distinguished from other genera of the Moraceae family (see Fig. 7). Furthermore, our study allowed unveil clustering of species within species from subgenus Urostigma, subgenus Sycomorus, and subgenus Ficus, all with high bootstrap values. These results support Berg classification system [2], in which the subgenus Ficus was further divided into the subgenus Sycomorus, and also support species differentiation based on molecular data. For example, F. tikoua, belonging to the Ficus subgenus based on morphology, helped to further differentiate a monophyletic group separating members of Ficus subgenus. Overall, these results are helpful to further understand the phylogenetic status and resolve relationships deep within Ficus.
Conclusions
In conclusion, in this work we determined the complete plastome sequence of ten Ficus species by NGS. Comparative genomics indicate that these plastomes showed the typical quadripartite structure being relatively conserved, with eight mutation hotspot regions being presented as potential molecular markers for subsequent Ficus species identification. The phylogenomic analysis performed clarified the taxonomy of the species, showing the relatedness between Ficus and Morus genera, and the split of Ficus genus into three subgenera (Ficus, Sycomorus and Urostigma). All together, these results enrich the data on the CP genome of the genus Ficus and provide additional information for future species identification and phylogenetic reconstruction of the Ficus species.
Materials and methods
Plant material, DNA extraction, and sequencing
Ten species (one of them contains two varieties), namely F. pumila, F. tikoua, F. hispida, F. virens, F. sarmentosa var. impressa, F. sarmentosa var. lacrymans, F. pandurata, F. tinctoria, F. formosana, F. microcarpa, and F. simplicissima were field-collected from the Medicinal Botanical Garden of Guangzhou University of Chinese Medicine with Longitude 113°24’ and Latitude 23°03’ (Guangzhou, Guangdong, China,). The formal identification of the plant material was undertaken by Dr. Jiaxia Su (Guangzhou University of Chinese Medicine). Permission was not necessary for collecting these species, which have not been included in the list of national key protected plants. Fresh green leaves cleaned with 75% ethanol from those collected Ficus plants were sampled. Then those leaves were dried and stored at -80 °C till DNA extraction. Total genomic DNA was extracted from 100 mg of cleaned leaves using a DNeasy Plant Mini Kit (Qiagen, German). Then, genomic DNA was examined for purity and integrity by ultraviolet spectrophotometry and gel electrophoresis (1 × TAE agarose gel), respectively.
High quality DNA was sheared to 500 bp using an ultrasonic DNA fragmentation apparatus (Covaris). Libraries were constructed with NEB Next Ultra DNA Library Prep Kit (New England Biolabs, E7370L) following the manufacturer’s protocol by the Sangon biotech High-Throughput DNA Sequencing Center. Libraries were amplified with NEB Next Q5 Hot Start HiFi PCR Master Mix kit, quantified on a Qubit 4.0 fluorometer (Thermo) and quality checked on an Agilent Technologies 2100 Bioanalyzer, prior paired-end 150 × sequencing in Illumina Hiseq 4000 sequencing platform at the Sangon biotech Sequencing Center.
Chloroplast genome assembly and annotation
After Illumina sequencing (paired-end, 150 ×), approximate 15 Gb of raw data for each sample was generated, and these raw reads were QC filtered and trimmed using the Trimmomatic (v0.39, Max Planck Institute of Molecular Plant Physiology, Potsdam, Germany) software [79] with following parameters: LEADING = 20, TRAILING = 20, SLIDINGWINDOW = 4:15, MINLEN = 36, and AVGQUAL = 20. A more detailed information related to quality control of the Illumina sequencing of the chloroplast genome of Ficus species is shown in Table S6. Taking the complete sequence of Ficus religiosa chloroplast genome (downloaded from NCBI with GenBank accession number: NC_033979) as the reference, CP-like reads were extracted from those clean reads by mapping with the bwa software (v0.7.17) [80]. Next, these CP-like reads were assembled using the SPAdes (v3.13.1) program [81], obtaining several contigs. Contigs were mapped against the F. religiosa reference genome with mummerplot (v3.5) to form a complete chloroplast genome sequence with their overlapping sequences. BLASTn (2.8.1) was conducted for self-alignment to locate the precise position of the quadripartite structure. Four regions between the IR regions and the LSC/SSC region were amplified and sequenced using specific primers (Table S7) in order to verify each CP assembly. A preliminary Ficus plastomes gene annotation was performed with the GeSeq online tool (https://chlorobox.mpimp-golm.mpg.de/geseq.html) with default parameters [82]. The annotation results were further examined and revised manually, according to reference genomes with the CLC Sequence Viewer. A detailed CP genome map for each Ficus species was drawn using the Organellar Genome DRAW (OGDRAW) v1.2 (Max Planck Institute of Molecular Plant Physiology, Potsdam, Germany) [83].
SSRs and repeat sequence analysis
Repeat sequences (including forward, reverse, complementary, palindromic) were identified by running the REPuter tools (https://bibiserv2.cebitec.uni-Bielefeld.de/reputer) [84] with a Hamming distance set at 3 and a minimum repeat size of 30 bp. Tandem repeats were analysed by the Tandem Repeats Finder (http://tandem.bu.edu/trf/trf.html), with alignment parameters set to 2, 7, and 7 for matches, mismatches, and indels. Whereas MISA was used to detect simple sequence repeats [85].
Genome structure and genome comparison
Molecular Evolutionary Genetics Analysis software MEGA v. 11 [86] (https://www.megasoftware.net/) was used to analyse codon usage distribution, GC content and phylogenomic inference as described below. Thirty-five protein-coding genes of the chloroplast genome of those eleven Ficus plastomes were used to predict potential RNA editing sites using the online program Predictive RNA Editor for Plants (PREP) suite (Mower 2009), with a cutoff value of 0.8. The mVISTA program (http://genome.lbl.gov/vista/index.shtml) in the Shuffle-LAGAN mode was used to align the obtained Ficus CP genomes with one reported CP genome (Morus atropurpurea) within the Moraceae family, whose sequence was downloaded from NCBI (GenBank accession number: KU355276) [87].
Sequence divergence and phylogenetic analysis
MAFFT (v7.419) was employed to align the CP genome sequence of ten Ficus species and then adjusted manually by Se-Al 2.024 [88]. DnaSP v5.10 software [89] was used to identify rapidly evolving molecular markers that can be applied to further phylogenetic studies, with a sliding window analysis with the step size and window length set as 200 and 800 bp.
To illustrate the phylogenetic positions and evolutionary relationships of Ficus species within the Moraceae family, the complete CP genomes of 23 species (21 from five different genera within the Moraceae, with Rhamnus taquetii and Cannabis carmagnole, that were set as out-group) were downloaded from the GenBank of NCBI (Table S8). Maximum-likelihood (ML) phylogenetic inference analysis was performed on a nucleotide alignment of 80 protein-coding genes using MEGA v.11. In detail, an ML tree inference was conducted using the general time-reversible model with a gamma distribution of substitution rate among sites (GTR + G), which was selected according to a previous model screening analysis (Model test as implemented in MEGA v11). To optimize the ML method, TBR branch switching (a fast and efficient branch switching operation), was adopted to improve the initial evolutionary tree, applying also 1,000 replicates. Bootstrap analysis to determine the support of each branch. Nucleotide and phylogeny inference models were selected after model testing in MEGA v.11.
Availability of data and materials
The sequencing datasets generated during the current study are available at China National GeneBank with project number as CNP0001337 (https://db.cngb.org/search/project/CNP0001337/). The accession numbers of eleven species are CNS0285141 (F. pumila), CNS0285142 (F. tikoua), CNS0285143 (F. hispida), CNS0285144 (F. virens), CNS0285145 (F. sarmentosa var. impressa), CNS0285146 (F. sarmentosa var. lacrymans), CNS0285147 (F. pandurata), CNS0285148 (F. tinctoria), CNS0285149 (F. formosana), CNS0285150 (F. microcarpa) and CNS0285151 (F. simplicissima).
Abbreviations
- CP:
-
Chloroplast
- SSC:
-
Small single copy
- LSC:
-
Large single copy
- IR:
-
Inverted repeat
- SSR:
-
Simple sequence repeat
- SNPs:
-
Single-nucleotide polymorphisms
- CDS:
-
Coding sequences
- RSCU:
-
Relative synonymous codon usage
- BP:
-
Bootstrap values
- polyA:
-
Polyadenine
- polyT:
-
Polythymine
- polyC:
-
Polycytosine
- polyG:
-
Polyguanine
References
Berg CC. Classification and distribution of Ficus. Experientia. 1989;45(7):605–11. https://doi.org/10.1007/BF01975677.
Berg CC, Corner EJH. Moraceae (Ficus). Flora malesiana. series I, volume 17. Nooteboom HP. eds. National herbarium Nederland, Universiteit Leiden branch, The Netherlands. 2005:1–702.
Seraia AS, Tsybulia NV, Dul’tseva GG. Role of some species of Ficus in amelioration of environment. Aviakosm Ekolog Med. 2008;42(4):66–70 (PMID: 19140478).
Harrison R. Figs and the diversity of tropical rainforests. Bioscience. 2009;55:1053–64. https://doi.org/10.1641/0006-3568(2005)055[1053:FATDOT]2.0.CO;2.
Barolo MI, Ruiz Mostacero N, López SN. Ficus carica L. (Moraceae): An ancient source of food and health. Food Chemistry. 2014;164:119–27. https://doi.org/10.1016/j.foodchem.2014.04.112.
Dangarembizi R, Erlwanger KH, Moyo D, Chivandi E. Phytochemistry, pharmacology and ethnomedicinal uses of Ficus thonningii (Blume Moraceae): a review. Afr J Tradit Complement Altern Med. 2012;10(2):203–12. https://doi.org/10.4314/ajtcam.v10i2.4.
Ayoub L, Hassan F, Hamid S, Abdelhamid Z, Souad A. Phytochemical screening, antioxidant activity and inhibitory potential of Ficus carica and Olea europaea leaves. Bioinformation. 2019;15(3):226–32. https://doi.org/10.6026/97320630015226.
Villard C, Larbat R, Munakata R, Hehn A. Defence mechanisms of Ficus: pyramiding strategies to cope with pests and pathogens. Planta. 2019;249(3):617–33. https://doi.org/10.1007/s00425-019-03098-2.
Lansky EP, Paavilainen HM, Pawlus AD, Newman RA. Ficus spp. (fig): Ethnobotany and potential as anticancer and anti-inflammatory agents. J Ethnopharmacol. 2008;119(2):195–213. https://doi.org/10.1016/j.jep.2008.06.025.
Yao J, Wang Z, Wang R, Wang Y, Xu J, He X. Anti-proliferative and anti-inflammatory prenylated isoflavones and coumaronochromones from the fruits of Ficus altissima. Bioorg Chem. 2021;113:104996–5016. https://doi.org/10.1016/j.bioorg.2021.104996.
Pandit R, Phadke A, Jagtap A. Antidiabetic effect of Ficus religiosa extract in streptozotocin-induced diabetic rats. J Ethnopharmacol. 2010;128(2):462–6. https://doi.org/10.1016/j.jep.2010.01.025.
El-Mostafa K, El Kharrassi Y, Badreddine A, Andreoletti P, Vamecq J, El Kebbaj M, et al. Nopal Cactus (Opuntia ficus-indica) as a Source of Bioactive Compounds for Nutrition, Health and Disease. Molecules. 2014;19(9):14879–901. https://doi.org/10.3390/molecules190914879.
Pothasin P, Compton SG, Wangpakapattanawong P. Riparian Ficus tree communities: the distribution and abundance of riparian fig trees in northern Thailand. PLoS ONE. 2014;9(10):489–512. https://doi.org/10.1371/journal.pone.0108945.
Corner EJH. Check-list of Ficus in Asia and Australasia with keys to identification. The Gardens’ bulletin, Singapore. 1965;21(1):1–186.
William RB. A New Classification of Ficus. Ann Mo Bot Gard. 1977;64:296–310. https://doi.org/10.2307/2395337.
Weiblen G. Phylogenetic Relationships of Functionally Dioecious Ficus (Moraceae) Based on Ribosomal DNA Sequences and Morphology. Am J Bot. 2000;87:1342–57. https://doi.org/10.2307/2656726.
Levy SE, Myers RM. Advancements in Next-Generation Sequencing. Annu Rev Genomics Hum Genet. 2016;17:95–115. https://doi.org/10.1146/annurev-genom-083115-022413.
Li W, Liu Y, Yang Y, Xie X, Lu Y, Yang Z, et al. Interspecific chloroplast genome sequence diversity and genomic resources in Diospyros. BMC Plant Biol. 2018;18(1):210–23. https://doi.org/10.1186/s12870-018-1421-3.
Feng S, Zheng K, Jiao K, Cai Y, Chen C, Mao Y, et al. Complete chloroplast genomes of four Physalis species (Solanaceae): lights into genome structure, comparative analysis, and phylogenetic relationships. BMC Plant Biol. 2020;20(1):242–68. https://doi.org/10.1186/s12870-020-02429-w.
Chen Y, Hu N, Wu H. Analyzing and Characterizing the Chloroplast Genome of Salix wilsonii. Biomed Res Int. 2019;2019:5190425. https://doi.org/10.1155/2019/5190425.
Nie L, Cui Y, Chen X, Xu Z, Sun W, Wang Y, et al. Complete chloroplast genome sequence of the medicinal plant Arctium lappa. Genome. 2020;63(1):53–60. https://doi.org/10.1139/gen-2019-0070.
Dong W, Liu J, Yu J, Wang L, Zhou S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE. 2012;7(4):e35071. https://doi.org/10.1371/journal.pone.0035071.
Wu F-H, Chan M-T, Liao D-C, Hsu C-T, Lee Y-W, Daniell H, et al. Complete chloroplast genome of Oncidium Gower Ramsey and evaluation of molecular markers for identification and breeding in Oncidiinae. BMC Plant Biol. 2010;16(10):68–86. https://doi.org/10.1186/1471-2229-10-68.
Zhang Y, Iaffaldano BJ, Zhuang X, Cardina J, Cornish K. Chloroplast genome resources and molecular markers differentiate rubber dandelion species from weedy relatives. BMC Plant Biol. 2017;17(1):34–45. https://doi.org/10.1186/s12870-016-0967-1.
Daniell H, Lin C-S, Yu M, Chang W-J. Chloroplast Genomes: Diversity, Evolution, and Applications in Genetic Engineering. Genome Biol. 2016;17(1):134–45. https://doi.org/10.1186/s13059-016-1004-2.
Kohchi T, Yamato KT, Ishizaki K, Yamaoka S, Nishihama R. Development and Molecular Genetics of Marchantia polymorpha. Annu Rev Plant Biol. 2021;72:677–702. https://doi.org/10.1146/annurev-arplant-082520-094256.
Chen H, Shao J, Zhang H, Jiang M, Huang L, Zhang Z, et al. Sequencing and Analysis of Strobilanthes cusia (Nees) Kuntze Chloroplast Genome Revealed the Rare Simultaneous Contraction and Expansion of the Inverted Repeat Region in Angiosperm. Front Plant Sci. 2018;9:324–38. https://doi.org/10.3389/fpls.2018.00324.
Huang H, Shi C, Liu Y, Mao S-Y, Gao L-Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol Biol. 2014;14:151–68. https://doi.org/10.1186/1471-2148-14-151.
Shaul O. How introns enhance gene expression. Int J Biochem Cell Biol. 2017;91:145–55. https://doi.org/10.1016/j.biocel.2017.06.016.
Qiujie Z, Ng WL, Wu W, Zhou R, Liu Y. Characterization of the complete chloroplast genome sequence of Tigridiopalma magnifica (Melastomataceae). Conserv Genet Resour. 2018;10:571–3. https://doi.org/10.1007/s12686-017-0856-4.
Bruun-Lund S, Clement WL, Kjellberg F, Rønsted N. First plastid phylogenomic study reveals potential cyto-nuclear discordance in the evolutionary history of Ficus L. (Moraceae). Mol Phylogenet Evol. 2017;109:93–104. https://doi.org/10.1016/j.ympev.2016.12.031.
Zeng Q, Chen H, Zhang C, Han M, Li T, Qi X, et al. Definition of Eight Mulberry Species in the Genus Morus by Internal Transcribed Spacer-Based Phylogeny. PLoS ONE. 2015;10(8):e0135411. https://doi.org/10.1371/journal.pone.0135411.
Payacan C, Moncada X, Rojas G, Clarke A, Chung K-F, Allaby R, et al. Phylogeography of herbarium specimens of asexually propagated paper mulberry [Broussonetia papyrifera (L.) L’Hér. ex Vent. (Moraceae)] reveals genetic diversity across the Pacific. Ann Bot. 2017;120(3):387–404. https://doi.org/10.1093/aob/mcx062.
Shen X, Guo S, Yin Y, Zhang J, Yin X, Liang C, et al. Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Aster tataricus. Molecules (Basel, Switzerland). 2018;23(10):2426–38. https://doi.org/10.3390/molecules23102426.
Zhang J, Liao M, Li X, Xu B. Characterization and phylogenetic analysis of the complete chloroplast genome sequence of xerophyta retinervis (velloziaceae). Mitochondrial DNA Part B. 2022;7:681–2. https://doi.org/10.1080/23802359.2022.2067500.
Timme RE, Kuehl JV, Boore JL, Jansen RK. A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: identification of divergent regions and categorization of shared repeats. Am J Bot. 2007;94(3):302–12. https://doi.org/10.3732/ajb.94.3.302.
Kim T-S, Booth JG, Gauch HG, Sun Q, Park J, Lee Y-H, et al. Simple sequence repeats in Neurospora crassa: distribution, polymorphism and evolutionary inference. BMC Genomics. 2008;9:31–42. https://doi.org/10.1186/1471-2164-9-31.
Qi W-H, Jiang X-M, Yan C-C, Zhang W-Q, Xiao G-S, Yue B-S, et al. Distribution patterns and variation analysis of simple sequence repeats in different genomic regions of bovid genomes. Sci Rep. 2018;8(1):14407–16. https://doi.org/10.1038/s41598-018-32286-5.
Kim K-J, Lee H-L. Complete Chloroplast Genome Sequences from Korean Ginseng ( Panax schinseng Nees) and Comparative Analysis of Sequence Evolution among 17 Vascular Plants. DNA Res. 2004;11(4):247–61. https://doi.org/10.1093/dnares/11.4.247.
Kashi Y, King DG. Simple sequence repeats as advantageous mutators in evolution. Trends Genet. 2006;22(5):253–9. https://doi.org/10.1016/j.tig.2006.03.005.
Srivastava D, Shanker A. Identification of Simple Sequence Repeats in Chloroplast Genomes of Magnoliids Through Bioinformatics Approach. Interdiscip Sci. 2015;8:327–36. https://doi.org/10.1007/s12539-015-0129-4.
Mazumdar P, Othman R, Mebus K, Ramakrishnan N, Harikrishna J. Codon usage and codon pair patterns in non-grass monocot genomes. Ann Bot. 2017;00:1–17. https://doi.org/10.1093/aob/mcx112.
Sloan D, Taylor D. Testing for Selection on Synonymous Sites in Plant Mitochondrial DNA: The Role of Codon Bias and RNA Editing. J Mol Evol. 2010;70:479–91. https://doi.org/10.1007/s00239-010-9346-y.
LaBella A, Opulente D, Steenwyk J, Hittinger C, Rokas A. Variation and selection on codon usage bias across an entire subphylum. PLoS Genet. 2019;15:e1008304. https://doi.org/10.1371/journal.pgen.1008304.
Li G, Pan Z, Gao S, He Y, Xia Q, Jin Y, et al. Analysis of synonymous codon usage of chloroplast genome in Porphyra umbilicalis. Genes Genomics. 2019;41(10):1173–81. https://doi.org/10.1007/s13258-019-00847-1.
He P, Huang S, Xiao G, Zhang Y, Yu J. Abundant RNA editing sites of chloroplast protein-coding genes in Ginkgo biloba and an evolutionary pattern analysis. BMC Plant Biol. 2016;16(1):257–65. https://doi.org/10.1186/s12870-016-0944-8.
Brenner WG, Mader M, Müller NA, Hoenicka H, Schroeder H, Zorn I, et al. High Level of Conservation of Mitochondrial RNA Editing Sites Among Four Populus Species. G3 (Bethesda). 2019;9(3):709–917. https://doi.org/10.1534/g3.118.200763.
Kawabe A, Furihata H, Tsujino Y, Kawanabe T, Fujii S, Yoshida T. Divergence of RNA editing among Arabidopsis species. Plant Sci. 2018;280:241–7. https://doi.org/10.1016/j.plantsci.2018.12.009.
Reginato M, Neubig KM, Majure LC, Michelangeli FA. The first complete plastid genomes of Melastomataceae are highly structurally conserved. PeerJ. 2016;4:e2715. https://doi.org/10.7717/peerj.2715.
Wang X, Zhou T, Bai G, Zhao Y. Complete chloroplast genome sequence of Fagopyrum dibotrys: genome features, comparative analysis and phylogenetic relationships. Sci Rep. 2018;8(1):12379–88. https://doi.org/10.1038/s41598-018-30398-6.
Asaf S, Khan A, Khan A, Waqas M, Kang S-M, Khan M, et al. Complete Chloroplast Genome of Nicotiana otophora and its Comparison with Related Species. Front Plant Sci. 2016;14(7):843–54. https://doi.org/10.3389/fpls.2016.00843.
Khakhlova O, Bock R. Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 2006;46(1):85–94. https://doi.org/10.1111/j.1365-313x.2006.02673.x.
Liu M-L, Fan W-B, Wang N, Dong P-B, Zhang T-T, Yue M, et al. Evolutionary Analysis of Plastid Genomes of Seven Lonicera L. Species: Implications for Sequence Divergence and Phylogenetic Relationships. Int J Mole Sci. 2018;19:4039–51. https://doi.org/10.3390/ijms19124039.
Wu Y, Liu F, Yang D-G, Li W, Zhou X-J, Pei X-Y, et al. Comparative Chloroplast Genomics of Gossypium Species: Insights Into Repeat Sequence Variations and Phylogeny. Front Plant Sci. 2018;21(9):376–87. https://doi.org/10.3389/fpls.2018.00376.
Yaradua S, Alzahrani D, Albokhari E, Abba A, Bello A. Complete Chloroplast Genome Sequence of Justicia flava : Genome Comparative Analysis and Phylogenetic Relationships among Acanthaceae. Biomed Res Int. 2019;2019:1–17. https://doi.org/10.1155/2019/4370258.
Jansen R, Cai Z, Raubeson L, Daniell H, dePamphilis C, Leebens-Mack J, et al. Analysis of 81 Genes From 64 Plastid Genomes Resolves Relationships in Angiosperms and Identifies Genome-Scale Evolutionary Patterns. Proc Natl Acad Sci U S A. 2008;104:19369–74. https://doi.org/10.1073/pnas.0709121104.
Tuler AC, Carrijo TT, Nóia LR, Ferreira A, Peixoto AL, da Silva Ferreira MF. SSR markers: a tool for species identification in Psidium (Myrtaceae). Mol Biol Rep. 2015;42(11):1501–13. https://doi.org/10.1007/s11033-015-3927-1.
Yang C-H, Liu X, Cui Y-X, Nie L-P, Lin Y-L, Wei X-P, et al. Molecular structure and phylogenetic analyses of the complete chloroplast genomes of three original species of Pyrrosiae Folium. Chin J Nat Med. 2020;18(8):573–81. https://doi.org/10.1016/S1875-5364(20)30069-8.
Tang W, Luo C. Molecular and Functional Diversity of RNA Editing in Plant Mitochondria. Mol Biotechnol. 2018;60(12):935–45. https://doi.org/10.1007/s12033-018-0126-z.
Sasaki T, Yukawa Y, Miyamoto T, Obokata J, Sugiura M. Identification of RNA editing sites in chloroplast transcripts from the maternal and paternal progenitors of tobacco (Nicotiana tabacum): comparative analysis shows the involvement of distinct trans-factors for ndhB editing. Mol Biol Evol. 2003;20(7):1028–35. https://doi.org/10.1093/molbev/msg098.
Zhu A, Guo W, Gupta S, Fan W, Mower J. Evolutionary dynamics of the plastid inverted repeat: The effects of expansion, contraction, and loss on substitution rates. New Phytol. 2015;209(4):1747–56. https://doi.org/10.1111/nph.13743.
Hebert P, Cywinska A, Ball SL, Dewaard J. Biological identification through DNA barcodes. Proc R Soc London B. 2003;270:313–21. https://doi.org/10.1098/rspb.2002.2218.
Cabelin VLD, Alejandro GJD. Efficiency of matK, rbcL, trnH-psbA, and trnL-F (cpDNA) to Molecularly Authenticate Philippine Ethnomedicinal Apocynaceae Through DNA Barcoding. Pharmacogn Mag. 2016;12(3):384–8. https://doi.org/10.4103/0973-1296.185780.
Dong W, Xu C, Li C, Sun J, Zuo Y, Shi S, et al. ycf1, the most promising plastid DNA barcode of land plants. Sci Rep. 2015;5:8348–52. https://doi.org/10.1038/srep08348.
Roman M, Houston R. Investigation of chloroplast regions rps16 and clpP for determination of Cannabis sativa crop type and biogeographical origin. Leg Med. 2020;47:101759–68. https://doi.org/10.1016/j.legalmed.2020.101759.
Yik M, Kong B, Siu TY, Lau D, Cao H, Shaw P-C. Differentiation of Hedyotis diffusa and Common Adulterants Based on Chloroplast Genome Sequencing and DNA Barcoding Markers. Plants. 2021;10:161–72. https://doi.org/10.3390/plants10010161.
Awad M, Fahmy RM, Mosa KA, Helmy M, El-Feky FA. Identification of effective DNA barcodes for Triticum plants through chloroplast genome-wide analysis. Comput Biol Chem. 2017;71:20–31. https://doi.org/10.1016/j.compbiolchem.2017.09.003.
Jeon J-H, Kim S-C. Comparative Analysis of the Complete Chloroplast Genome Sequences of Three Closely Related East-Asian Wild Roses (Rosa sect. Synstylae; Rosaceae). Genes. 2019;10:23–31. https://doi.org/10.3390/genes10010023.
Cheng Y, Yang Y, Fu X, Liu L, Jiang Z, Cai J. Plastid genomes of Elaeagnus mollis: comparative and phylogenetic analyses. J Genet. 2020;99:85–96. https://doi.org/10.1007/s12041-020-01243-5.
Mustapha SB, Ben Tamarzizt H, Baraket G, Abdallah D, Salhi-Hannachi A. Cytoplasmic polymorphism and evolutionary history of plum cultivars: Insights from chloroplast DNA sequence variation of trnL-trnF spacer and aggregated trnL intron & trnL-trnF spacer. Genet Mol Res. 2015;14(2):3964–79. https://doi.org/10.4238/2015.April.27.11.
Shaw J, Lickey E, Beck J, Farmer S, Liu W, Miller J, et al. The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am J Bot. 2005;92:142–66. https://doi.org/10.3732/ajb.92.1.142.
Clement W, Weiblen G. Morphological Evolution in the Mulberry Family (Moraceae). Syst Bot. 2009;34:530–52. https://doi.org/10.1600/036364409789271155.
Cheon K-S, Yoo K-O. Complete chloroplast genome sequence of Hanabusaya asiatica (Campanulaceae), an endemic genus to Korea. Mitochondrial DNA. 2014;27:1–3. https://doi.org/10.3109/19401736.2014.958702.
Raman G, Park S. The Complete Chloroplast Genome Sequence of Ampelopsis: Gene Organization, Comparative Analysis, and Phylogenetic Relationships to Other Angiosperms. Front Plant Sci. 2016;7:341–7. https://doi.org/10.3389/fpls.2016.00341.
Herre EA, Machado CA, Bermingham E, Nason JD, Windsor DM, McCafferty SS, et al. Molecular phylogenies of figs and their pollinator wasps. J Biogeogr. 1996;23(4):521–30. https://doi.org/10.1111/j.1365-2699.1996.tb00014.x.
Renoult J, Kjellberg F, Grout C, Santoni S, Khadari B. Cyto-nuclear discordance in the phylogeny of Ficus section Galoglychia and host shift in plant-pollinator associations. BMC Evol Biol. 2009;9:248–56. https://doi.org/10.1186/1471-2148-9-248.
Rønsted N, Salvo G, Savolainen V. Biogeographical and phylogenetic origins of African fig species (Ficus section Galoglychia). Mol Phylogenet Evol. 2007;43:190–201. https://doi.org/10.1016/j.ympev.2006.12.010.
Rønsted N, Yektaei E, Turk K, Clarkson J, Chase M. 9 Species-Level Phylogenetics of Large Genera: Prospects of Studying Coevolution and Polyploidy. Reconstructing the tree of life: Taxonomy and systematics of species rich taxa. 2006; 129–148. https://doi.org/10.1201/9781420009538.ch9.
Bolger A, Lohse M, Usadel B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics (Oxford, England). 2014;30(15):2114–20. https://doi.org/10.1093/bioinformatics/btu170.
Giannoulatou E, Park S-H, Humphreys DT, Ho JWK. Verification and validation of bioinformatics software without a gold standard: a case study of BWA and Bowtie. BMC Bioinformatics. 2014;15(16):15–23. https://doi.org/10.1186/1471-2105-15-s16-s15.
Bankevich A, Nurk S, Antipov D, Gurevich A, Dvorkin M, Kulikov A, et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J Comput Biol. 2012;19:455–77. https://doi.org/10.1089/cmb.2012.0021.
Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, et al. GeSeq-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017;45:6–11. https://doi.org/10.1093/nar/gkx391.
Lohse M, Drechsel O, Bock R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet. 2007;52:267–74. https://doi.org/10.1007/s00294-007-0161-y.
Kurtz S, Choudhuri J, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. REPuter: The Manifold Applications of Repeat Analysis on a Genomic Scale. Nucleic Acids Res. 2001;29:4633–42. https://doi.org/10.1093/nar/29.22.4633.
Lu X, Adedze Y, Chofong G, Mamadou G, Deng Z, Teng L, et al. Identification of high-efficiency SSR markers for assessing watermelon genetic purity. J Genet. 2018;97(5):1295–306. https://doi.org/10.1007/s12041-018-1027-4.
Tamura K, Stecher G, Kumar S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol Biol Evol. 2021;38(7):3022–7. https://doi.org/10.1093/molbev/msab120.
Mayor C, Brudno M, Schwartz J, Poliakov A, Rubin E, Frazer K, et al. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics (Oxford, England). 2000;16:1046–7. https://doi.org/10.1093/bioinformatics/16.11.1046.
Katoh K, Standley D, Katoh K, Standley DM. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–80. https://doi.org/10.1093/molbev/mst010.
Librado PJR, Rozas J. DnaSP v5: A Software for Comprehensive Analysis of DNA Polymorphism Data. Bioinformatics (Oxford, England). 2009;25:1451–2. https://doi.org/10.1093/bioinformatics/btp187.
Acknowledgements
We would like to give special thanks to Dr. Su Jia-xian, who helped us obtain and authenticate these plant samples.
Funding
This research was supported by the National Natural Science Foundation of China (Grant number: 81903741).
Author information
Authors and Affiliations
Contributions
Xiasheng Zheng conceived and designed the experiments. Yuying Huang and Jing Li, conduct the experiments, analyzed the data, and wrote the manuscript. Zerui Yang and Wenli An participated in bioinformatics analysis, Shanshan Liu and Chunzhu Xie participated in experiments and wrote the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No specific permits were required, material collection (ten Ficus species) and molecular experiments were carried on following Guangzhou University of Chinese Medicine (GZUCM) guidelines (https://zcsy.gzucm.edu.cn/index.htm) which also fulfill the current Chinese research-ethical regulations. The ten Ficus species used in this work were all sampled and collected from the Ficus plant collection of the medicinal botanical garden of GZUCM. This Botanical garden harbors ~ 10,021 (internationally-protected and unprotected) species of plants originated from China, dedicated to the protection of medicinal resources with international recognition.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Figure S1. Sequence identity plot comparison of the eleven CP genome of the ten Ficus species using mVISTA.
Additional file 2:
Table S1. Gene number and CDS nucleotide composition of the CP genomes in eleven Ficus species.
Additional file 3:
Table S2. Gene contents in the Ficus species chloroplast genome.
Additional file 4:
Table S3. Gene with introns in the Ficus species chloroplast genome and the lengths of the introns and exons.
Additional file 5:
Table S4. Codon usage of the eleven Ficus chloroplast genomes.
Additional file 6:
Table S5. Predicted RNA editing sites in eleven Ficus chloroplast genomes by the PREP program.
Additional file 7:
Table S6. Quality control of the Illumina sequencing of chloroplast genome of Ficus species.
Additional file 8:
Table S7. Universal primers for amplifying four regions between the IR regions and the LSC/SSC region.
Additional file 9:
Table S8. The 23 studied species and the corresponding chloroplast whole genome GenBank accession number.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Huang, Y., Li, J., Yang, Z. et al. Comprehensive analysis of complete chloroplast genome and phylogenetic aspects of ten Ficus species. BMC Plant Biol 22, 253 (2022). https://doi.org/10.1186/s12870-022-03643-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-022-03643-4