
Abstract
Background
The grains of foxtail millet are enriched in carotenoids, which endow this plant with a yellow color and extremely high nutritional value. However, the underlying molecular regulation mechanism and gene coexpression network remain unclear.
Methods
The carotenoid species and content were detected by HPLC for two foxtail millet varieties at three panicle development stages. Based on a homologous sequence BLAST analysis, these genes related to carotenoid metabolism were identified from the foxtail millet genome database. The conserved protein domains, chromosome locations, gene structures and phylogenetic trees were analyzed using bioinformatics tools. RNA-seq was performed for these samples to identify differentially expressed genes (DEGs). A Pearson correlation analysis was performed between the expression of genes related to carotenoid metabolism and the content of carotenoid metabolites. Furthermore, the expression levels of the key DEGs were verified by qRT-PCR. The gene coexpression network was constructed by a weighted gene coexpression network analysis (WGCNA).
Result
The major carotenoid metabolites in the panicles of DHD and JG21 were lutein and β-carotene. These carotenoid metabolite contents sharply decreased during the panicle development stage. The lutein and β-carotene contents were highest at the S1 stage of DHD, with values of 11.474 μg /100 mg and 12.524 μg /100 mg, respectively. Fifty-four genes related to carotenoid metabolism were identified in the foxtail millet genome. Cis-acting element analysis showed that these gene promoters mainly contain ‘plant hormone’, ‘drought stress resistance’, ‘MYB binding site’, ‘endosperm specific’ and ‘seed specific’ cis-acting elements and especially the ‘light-responsive’ and ‘ABA-responsive’ elements. In the carotenoid metabolic pathways, SiHDS, SiHMGS3, SiPDS and SiNCED1 were more highly expressed in the panicle of foxtail millet. The expression of SiCMT, SiAACT3, SiPSY1, SiZEP1/2, and SiCCD8c/8d was significantly correlated with the lutein content. The expression of SiCMT, SiHDR, SiIDI2, SiAACT3, SiPSY1, and SiZEP1/2 was significantly correlated with the content of β-carotene. WGCNA showed that the coral module was highly correlated with lutein and β-carotene, and 13 structural genes from the carotenoid biosynthetic pathway were identified. Network visualization revealed 25 intramodular hub genes that putatively control carotenoid metabolism.
Conclusion
Based on the integrative analysis of the transcriptomics and carotenoid metabonomics, we found that DEGs related to carotenoid metabolism had a stronger correlation with the key carotenoid metabolite content. The correlation analysis and WGCNA identified and predicted the gene regulation network related to carotenoid metabolism. These results lay the foundation for exploring the key target genes regulating carotenoid metabolism flux in the panicle of foxtail millet. We hope that these target genes could be used to genetically modify millet to enhance the carotenoid content in the future.
Similar content being viewed by others


Background
Foxtail millet [Setaria italica (L.) Beauv.] belongs to the Setaria genus of Poaceae grass family, and it is widely planted in Eurasian arid and semiarid areas as a kind of C4 crop that endures drought stress and infertile soils [1]. The ancestor of cultivated foxtail millet is green foxtail grass, which was domesticated early in China at approximately 16,000 years ago according to archaeological evidence [2]. At present, four genome sequences of foxtail millet have been published: ‘Yugu1’, ‘Zhanggu’, ‘Xiaomi’ and ‘huagu11’ [3,4,5,6]. The construction of a genome database and efficient genetic transformation system lay the foundation for investigating the functional genes, genetic evolution, plant architecture and physiology of foxtail millet, especially as a kind of C4 model and bioenergy grass system [1, 7, 8]. Foxtail millet is an important food crop in China and other Asian countries, and has small grains and higher nutritional value and contains edible fiber, protein, starch, vitamins and mineral elements [9]. Previous studies have shown that dehulled grains of foxtail millet enriched in carotenoids presents a yellow color [10, 11]. Carotenoids, as the main source of vitamin A in the human body, present various functions, such as eyesight protection, antioxidation, and anticancer effects, and they also have preventive functions in a variety of cardiovascular diseases [12]. Biofortified carotenoid food could be essential for meeting the health requirements and reduce “hidden hunger” in developing areas. According to previous reports, the carotenoid content in grains of foxtail millet ranged from 189.1 μg /100 mg to 201.3 μg /100 mg, which are 7.2-, 201.3- and 1.7- times of wheat, brown rice and sorghum [13,14,15]. Hence, the grains of foxtail millet could be an excellent carotenoid food resource.
The carotenoid metabolism pathway in plants is well known. Carotenoids and their derivatives are composed of isopentenyl pyrophosphate (IPP) and its double bond isomer and dimethylallyl diphosphate (DMAPP). In plant cells, these IPP precursors are mainly synthesized by the mevalonate (MVA) and 2-C-methyl-D-erythritol 4-phosphate (MEP) pathways and the precursors of carotenoid synthesis mainly come from the MEP metabolic pathway [16, 17]. Finally, carotenoid synthesis begins with phytoene synthase (PSY) condensation of two geranylgeranyl pyrophosphate (GGPP) molecules to form a 15 cis isomer. In the next step, all trans-lycopene is produced by phytene saturase (PDS), zeta-e carotene desaturase (ZDS), carotenoid isomerase (Crtiso), and zeta-carotene isomerase (Z-ISO). β-carotene and α-carotene are synthesized by LCYB or LCYE catalysis, and then lutein and zeaxanthin are produced by hydroxylation of nonheme carotene hydroxylase (BCH1 and BCH2) and heme hydroxylase (cytochrome P450-type monooxygenase, CYP97A3 and CYP97C1). Under the catalysis of zeaxanthin cyclooxygenase (ZEP) and pansy xanthine decyclooxygenase (VDE), luteins are produced, including anther xanthine, viola xanthine and neoxanthine [18].
Transcription factors play an important regulatory role in the carotenoid metabolism pathway. Phytochrome-interacting factor 1 (PIF1) regulated for carotenoid biosynthesis by inhibiting the expression of PSY in dark environment and also participates in the formation of chloroplasts [19]. An bZIP transcription factor named long hypocotyl 5 (HY5), also take part in carotenoid biosynthesis as a negative regulator of PIF1 during photomorphogenesis [20]. Recently, a sub-clade MADS gene of FRUITFULL transcription factor named CsMADS5 has reported in tomato that it can positively regulate the carotenoid content by up-regulating the expression of PSY/PDS/LCYB [21]. So far, some regulators have reported that revolved in carotenoid biosynthesis pathway in the transcript level in plants. However, our understanding of the transcriptional regulation of carotenoid biosynthesis still requires further investigation.
Many previous studies on genes involved in carotenoid biosynthesis and regulated networks in plants have been reported. Through introducing PSY and CRTL genes in rice, a new edible rice variety named “golden rice” was successfully developed [22]. In maize, they found that the genotype with favorable alleles of crtRB1 and lcyE had a significant effect on the β-carotene content (7.9-fold and 2.1-fold higher) compared to.
the unfavorable genotype [23]. In tomato, two PSY isoforms are responsible for divergent functions of fruit-specific carotenoid accumulation [24]. For foxtail millet, although previous reports on carotenoid components and some key gene expression analyses have been reported, the global analysis of gene expression patterns involved in carotenoid metabolism during the panicle developmental stage has not yet been reported. Therefore, in this study, we proposed to explore the dynamic pattern of carotenoid accumulation and the relationship between carotenoid content and gene expression level during panicle development periods to deeply understand the underlying molecular mechanism of carotenoid metabolism and identify target genes for genetic modification in foxtail millet.
Methods
Plant materials
Foxtail millet varieties JG21 and DHD were planted in the experimental field of Shanxi Agricultural University in April 2019 (N: 37°12′, E: 112°28′). The experiments were done in three biological replicates. Samples were collected from the middle part of the panicle at S1 (Beginning of diaspore colouring, 111 days after imbibition), S2 (Colouring of half of diaspores, 129 days after imbibition) and S3 (Colouring of almost all diaspores, 143 days after imbibition) (Fig. S1) [25]. For each cultivar, the middle of panicles were collected and mixed in one tube from the 10 independent plants. All samples were frozen and stored at - 80 °C for following analysis. (Study complied with local and national regulations for using plants.)
Extraction and determination of carotenoids
The extraction of carotenoids followed Paul’s method [26]. In brief, a fine powder generated from each sample (approximately 0.5 g) was added to 40 μL 50% KOH solution and 2 mL anhydrous ethanol with 0.1% BHT (butylated hydroxytoluene). After vortex mixing, the samples were bathed in water at 85 °C for 5 min, made up volume with 1 mL cold water. Then, 1 ml n-hexane was added and centrifuged at 5000 g for 5 min after vortexing. The supernatant was removed and transferred to a new centrifuge tube. Then, 1 mL of ultrapure water was added to the final n-hexane phase, which was vortexed and centrifuged, and then the supernatant was dried with nitrogen until it became dry matter. Finally, it was dissolved in 200 μL of acetonitrile: ethanol (1:4) added with 0.1% BHT.
The chromatographic system was a DGLC dual ternary ultrahigh-performance liquid chromatography system (Thermo, USA). A YMC Carotenoid S-3 μm (150*4.6 mm) liquid chromatographic column was used. The injection volume was 2 μl, the column temperature was 40 °C, and the detection wavelength was 450 nm. The solvent systems contained mobile phase A (methanol: water (1:1)) and mobile phase B (acetonitrile (ACN): ethyl acetate (3:1)). All solvents used were HPLC grade and filtered through a 0.2-mm filter prior to use. The gradient was 30% A:70% B for 0.5 min, then increased in intervals to 0.1% A:99.9% B for 5.5 min and then to 30% A:70% B for the last 2 min. UPLC–MS/MS was performed by Sanshu Biotechnology Co., Ltd. (ShangHai, China) (Fig. S2).
Retrieval gene sequence, collinear analysis and chromosome location
The genes involved in the map00900 and map00906 metabolic pathways were analyzed and mapped to Kyoto Encyclopedia of Genes and Genomes Database (KEGG, https://www.kegg.jp/). The sequences of key genes in the carotenoid metabolism pathway of foxtail millet were obtained from homozygous genes in Arabidopsis thaliana. TBtools analysis tools were used for the gene sequence information analysis, collinearity analysis, and chromosome mapping analysis [27]. The online websites were used for protein domain prediction analysis (http://pfam.xfam.org/search/) and subcellular location prediction (https://wolfpsort.hgc.jp/).
RNA extraction and RNA-seq analysis
The developing panicle of DHD and JG21 were separated at the S1/S2/S3 filling stages, and total RNA was isolated using a Quick RNA Isolation kit (Takara Corporation, Dalian, China) [28]. RNA quality and concentration were assessed by 1% agarose electrophoresis (electrophoresis on a denaturing agarose gel) and a NanoPhotometer® spectrophotometer (IMPLEN, CA, USA). The library construction of qualified RNA samples was carried out with a target insert size of ~ 450 bp, and the quality of the RNA-seq libraries was evaluated by an Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). After the library profile analysis, the RNA-seq libraries were sequenced on an Illumina HiSeq platform following standard methods by Novogene Life Sciences Pvt. Ltd. Beijing, China, with three biological replicates, and 150 bp paired-end reads were generated. After filtering the raw data, clean reads were mapped to the foxtail millet reference genome (https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Sitalica) using Hisat2 (http://ccb.jhu.edu/software/hisat2/index.shtml) [29].
For gene expression quantification, HTSeq was used to count the read numbers mapped to each gene, and the FPKM (fragments per kilobase per million fragments) value was used to normalize the expression level of each gene. DEGs between different samples were identified using the R package DESeq with |log2FoldChange| > 1 and P value < 0.05 as the thresholds [30]. Volcano and MA plots of DEGs were drawn by the R package ggplot2. The KEGG (Kyoto Encyclopedia of Genes and Genomes, http://www.genome.jp/kegg) and GO (Gene Ontology, http://geneontology.org/) annotations of DEGs were further performed with GOseq and KOBAS software, respectively [31]. The FPKM values of key genes involved in the carotenoid synthesis pathway at different developmental stages of ‘DHD’ and ‘JG21’ spikelets were obtained, and the correlation coefficients between the FPKM values of candidate genes and carotenoid content were calculated using SPSS 19.0 software (SPSS, Inc., Chicago, IL, USA). Heat maps of gene expression and correlation coefficients were drawn by TBtools [27]. A weighted gene coexpression network analysis of all genes was performed using the R package WGCNA. The selection of interacting genes used to construct a coexpression network is based on the following principles: 1. The modules correlated with the main carotenoid components were selected according to the correlation coefficient over 0.9 between module eigengene and specific carotenoid compound. 2. Meantime, in the correlated module, searching the carotenoid metabolism pathway genes, and extracted the top 25 transcription factors with the coexpression weight over 0.25. 3. Hub genes related to carotenoid metabolites and 25 coexpressed transcription factors were further selected to construct a coexpression network. 4.Whether transcription factors can combine the binding sites in the upstream of the promoter of carotenoid metabolism genes were predicted by using the FIMO (Find Individual Motif Occurences) tools (https://meme-suite.org/meme/tools/fimo) according to default parameters.
qRT-PCR analysis
qRT-PCR was performed to verify the expression patterns revealed by the RNA-seq study. Total RNA samples of three stages of foxtail millet panicles were extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Purified RNA samples were reverse-transcribed using the PrimeScript RT Reagent Kit with gDNA Eraser (Takara, Dalian, China) according to the manufacturer’s protocol. Four transcripts were selected for the qRT-PCR assay. Gene-specific qRT-PCR primers were designed using Primer 3 software (http://primer3.ut.ee/) (Table S1). qRT-PCR was carried out using a Bio–Rad CFX96 instrument (Bio–Rad Laboratories, USA). Each reaction mix was composed of 10 μl 2 × SYBR Green Master Mix Reagent (Vazyme Biotech, China), 2.0 μl cDNA sample, and 400 nm gene-specific primers in a final volume of 20 μl PCR conditions were as follows: 2 min at 95 °C, followed by 40 cycles of denaturation at 95 °C for 10 s and annealing at 60 °C for 40 s. The relative mRNA level for each gene was calculated using the 2−∆∆CT formula [32].
Results
Dynamic changes in carotenoid metabolites during panicle development stages
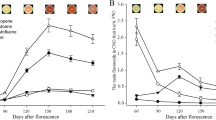
For two foxtail millet cultivars, five carotenoid metabolites, lutein, β-carotene, zeaxanthin, violaxanthin, and neoxanthin, were detected in the spikes at the three panicle development stages by HPLC. The results showed that most of carotenoid metabolite contents were higher at the S1 stage than at the other stages of the two cultivars except zeaxanthin of DHD (Fig. 1). The first major carotenoid metabolite was lutein, accounting for 43.28 to 58.18% of the total carotenoid content. The second major carotenoid metabolite was β-carotene, accounting for 27.82 to 50.36% of the total carotenoid content (Fig. S3). Moreover, we found that the contents of two major carotenoid (Lutein and β-carotene) metabolites in JG21 were lower than those in DHD at the S1&S2 but became higher at S3. The highest lutein and β-carotene contents of 11.474 μg /100 mg and 12.524 μg /100 mg, respectively, were observed at the S1 stage of DHD.

DHD and JG21 carotenoid content at the three developmental stages of panicle. (A) Lutein. (B) β-carotene. (C) Zeaxanthin. (D) Violaxanthin. (E) Neoxanthin
Sequence characteristics of genes related to the carotenoid metabolism pathway
Based on the homologous protein blast method, fifty-four genes related to carotenoid metabolism were identified from the genome database (Table 1). Of them, fifteen and fourteen genes encoded six and nine key isozymes involved in the MVA and MEP metabolism pathways (Fig. 2), respectively. The remaining genes encoded fifteen enzymes involved in the carotenoid biosynthesis and degradation pathways. The protein lengths of these genes ranged from 233 ~ 751 amino acid residues. The protein molecular weights of these genes ranged from 26,535 ~ 82,255 Da. Moreover, 34 genes could be targeted to chloroplasts according to the protein prediction of subcellular localization. The remaining 2, 6, 4, 4 and 2 genes could be targeted to the cytoskeleton, endoplasmic reticulum, mitochondrion, nucleus and plasma membrane, respectively. The SiGGPPS7b, SiLCYB and SiNNCED4 gene structures did not have any introns. The shortest gene genomic sequence length was SiGGPPS7b at 1098 bp. The longest gene genomic sequence length was SiCYP97C1 at 18373 bp. These homologous genes for each gene family contained the same specific conserved protein domain (Fig. 3).

Foxtail millet carotenoid metabolism pathway
Fifty-four genes related to the carotenoid metabolism pathway were distributed on nine chromosomes of foxtail millet. Eight genes were mapped on chromosome 1, which were SiLYCB/SiHDS/SiGGPPS7c/SiMDS/SiNCED1a/SiHMGR1/SiMDC1/SiCYP97A3. Ten genes were mapped on chromosome 2, which were SiNCED4/SiDXS2/SiZDS/SiHMGR3/SiHMGS3/SiPSY2/SiIDI1/SiIDI2/SiMVK-like/SiGGPPS7a. Seven genes were mapped on chromosome 3, which were SilDI3/SiDXS1/SiMVK/SiZ-ISO/SiNCED5/SiMDC2/SiPSY3. Two genes were mapped on chromosome 4, which were SiDXS3 and SiPSY1. Ten genes were mapped on chromosome 5, which were SiGGPPS7b/SiDXR/SiAACT1/SiLYCE/SiAACT3/SiCCD8c/SiCCD8b/SiCMK/SiMCT. Two genes were mapped on chromosome 6, which were SiHMGR2 and SiHMGS2. Six genes were mapped on chromosome 7, which were SiVDE/SiZEP2/SiZEP1/SiCCD7/SiBCH2/SiAACT2. Two genes were mapped on chromosome 8, which were SiCCD8a and SiCRTISO. Finally, seven genes were mapped on chromosome 9, which were SiHDR/SiNCED1b/SiCYP97C1/SipMVK/SiPDS/SiBCH1/SiHMGS1. A genomic collinearity analysis showed 53, 46, 33, and 37 homologous genes in the S. viridis, S. bicolor, Z. mays and O. sativa genomes, respectively, compared with the S. italica genome (Fig. 4).

Gene structure and protein structure of the carotenoid metabolism pathway infoxtail millet
Phylogenetic trees of homologous proteins related to carotenoid metabolism were constructed using the neighbor-joining method from S. viridis, Z. mays, O. sativa and A. thaliana (Fig. S4). For the MEP pathway, SiDXS1/2/3, SiIDI1/2 and SiGGPPS7a/b/c were clustered together into one group with orthologous proteins in Z. mays and O. sativa. For the MVA pathway, SiAACT1/2/3 were more highly homologous proteins with ZmAACT1/2. SiMVD1/2 were higher homologous with OsMVD. SiHMGS1/2/3 were clustered into one group with ZmHMGS and OsHMGS. Moreover, SiHMGR1/2/3 were more highly homologous with ZmHMGR1 OsHMGR3-like and OsHMGR3. For the carotenoid biosynthesis and degradation pathway, SiPSY1/2/3, SiBCH1/2, SiZEP1/2, SiNCED1a/1b/4/5, and SiCCD7/8a/8b/8c were clustered into one group with orthologous proteins of Z. mays and O. sativa.
The promoters of these genes indicated that they were mainly involved with ‘light responsiveness’, ‘plant hormone’, ‘drought stress resistance’, ‘MYB binding site’, ‘endosperm specific’ and ‘seed specific’ cis-acting elements. Except for SiMDC1 and SiNCED1a, the promoters of the remaining 52 genes contained a few ABA (abscisic acid) hormone cis-acting elements. Moreover, the promoters of all 54 genes had many MYB binding sites. Of them, the promoters of 51 and 14 genes had some ‘drought stress resistance’ and ‘light responsiveness’ cis-acting elements, respectively (Fig. S5).
Differentially expressed genes during the panicle development stage
Eighteen samples, including two cultivars at three panicle development stages, were sequenced and analyzed by RNA-seq. These samples showed higher repeatability and dependency for the same panicle development stage. In total, 125.69 Gb raw read data were produced from 18 samples by RNA-seq, with Q30 ≥ 92.31%. The range of raw read numbers mapped to the reference genome among these samples was from 83.09 to 94.63%. By comparing the gene expression levels of DHD_S1 to that of DHD_S2 and DHD_S3, 2218, 3173 and 526 differentially expressed genes were identified, respectively. DHD had a total of 2218 genes with expression differences between S1 and S2 (D1), 526 genes with expression differences between S2 and S3 (D2), and 3173 genes with expression differences between S1 and S3 (D3). Similarly, JG21 had 2209 genes with expression differences between S1 and S2 (J1), 1544 genes between S2 and S3 (J2), and 3412 genes between S1 and S3 (J3). Among them, the number of DEGs between D1 and D2 was at least 24, and the number of DEGs between D1 and D3 was at most 1784. D2 vs. D3, J1 vs. J2, J2 vs. J3, and J1 vs. J3 had 244, 187, 848 and 1184 differentially expressed genes, respectively. The number of differentially expressed genes shared by the D1, D2, and D3 stages was 209, while that of the J1, J2, and J3 stages was 175.
DEGs between DHD and JG21 at different stages of ear development were analyzed (Fig. S6). There were 2690 DEGs between DHD and JG21 (C1) in the S1 period, 2149 DEGs between DHD and JG21 (C2) in the S2 period, and 604 DEGs between DHD and JG21 (C3) in the S3 period. In C1 vs. C2, C2 vs. C3, C2 vs. C3, there were 835, 66, and 61 genes were differentially expressed simultaneously and 227 genes were differentially expressed in all three periods.
The GO enrichment analysis showed that these DEGs mapped to “cellular processes”, “environmental information processing”, “genetic information processing”, “metabolism” and “organismal system”. The KEGG enrichment analysis showed that these DEGs mainly mapped to the ‘phenylpropanoid biosynthesis’, ‘flavone and flavanol biosynthesis’ and ‘flavonoid biosynthesis’ pathways between DHD and JG21 at the three panicle development stages (Fig. S7). However, in the S1 and S3 periods, the DEGs were mainly enriched in the ‘carotenoid biosynthesis’ pathway (Fig. S8).
Gene expression characteristics related to carotenoid metabolism
To investigate the differential expression levels of genes related to the carotenoid metabolism pathway, we analyzed 54 gene expression values from the RNA-seq of JG21 and DHD at three panicle development stages (Fig. S9). In the MEP pathway, the SiHDS gene was more highly expressed at the S1 stage of DHD and JG21 than the other genes, with TPM values of 60.3 and 60.4, respectively. However, the SiIDI3 gene had nearly no expression at the three panicle development stages of DHD and JG21. In the MVA pathway, the highest expression level was observed for the SiHMGS3 gene during panicle development of DHD and JG21, with TPM values ranging from 41.0 to 70.5. However, SiMVK had the lowest expression level compared to other genes. In the carotenoid biosynthesis pathway, only SiPDS had a higher expression level, with TPM values ranging from 54.3 to 70.7. The remaining genes had both lower expression levels, with TPM values ranging from 0.0 to 25.9. Moreover, only SiNCED1a had a higher expression level than the other genes during the panicle development stage of the two cultivars. The others had lower expression levels for the two cultivars.
Relationship between carotenoid metabolites and gene expression levels
Based on Pearson’s correlation coefficient analysis, the relationship between the expression levels of genes related to carotenoid metabolism and major carotenoid metabolite contents during the panicle development stage of the two cultivars was investigated (Fig. S10). SiDXS3, SiMCT, SiHDR, SiIDI2, SiAACT1/3, SiMVK-like, SipMVK, SiPSY1, SiZDS and SiZEP1/2 were significantly positively correlated with the neoxanthin content at P < 0.05. However, SiCCD8b was significantly negatively correlated with the neoxanthin content at P < 0.05. Moreover, SiDXS3, SiMCT, SiAACT3, SiZEP1/2, and SiCCD8a/8c/8d were significantly positively correlated with the violaxanthin content at P < 0.05. SiMCT, SiAACT3, SiPSY1, SiZEP2, and SiCc8a/8d were significantly positively correlated with the lutein content at P < 0.05. Remarkably, SiZEP1/2 was significantly positively correlated with four carotenoid metabolites except zeaxanthin (r = 0.882, 0.903, 0.974, 0.894, 0.899, 0.969, 0.914 and 0.909, respectively). However, SiPSY2 and SiNCED1b/4 were significantly positively correlated with the zeaxanthin content. SiCMT, SiHDR, SiIDI2, SiAACT3, SiPSY1 and SiZEP12 were significantly positively correlated with the β-carotene content. Moreover, SiBCH1 was significantly negatively correlated with the β-carotene content (r = − 0.818, at P < 0.05). Furthermore, SiPSY1 was significantly positively correlated with the contents of neoxanthin, lutein, and β-carotene at P < 0.05 (r = 0.921, 0.818 and 0.833, respectively). SiCCD8b was negatively correlated with all carotenoids except zeaxanthin (P < 0.05, r = − 0.903 and − 0.811; P > 0.05, r = − 0.792 and − 0.804).
Carotenoid metabolism related to the gene coexpression network
The total gene expression value obtained from the RNA-seq data was used to construct a coexpression network. All genes were divided into 22 coexpression modules based on the WGCNA method (Fig. S11). The major carotenoid metabolites correlated with the coexpression network module were investigated and analyzed. Of the 22 modules, the MEcoral module was more highly correlated with lutein and β-carotene. Interestingly, the module contained 13 key genes involved in the carotenoid metabolism pathway, which were composed of SiMCT, SiIDI2, SiGGPPS7a, SipMVK-like, SipMVK, SiAACT1/3, SiPDS, SiPSY1, SiBCH1, SiZEP1/2 and SiCD8b (Fig. 5 and Table S2). Based on the previous analysis of the correlation between carotenoid metabolism genes and carotenoid content (Fig. S10), we selected 5 genes which were respectively correlated with Lutein and β-carotene to construct a coexpression network. In this coexpression network, we found that 25 transcription factors had higher weight values related to SiIDI2/SiMCT/SiAACT3/SiPSY1/SiZEP2 (Tables S3, S4 and Fig. 6). These transcription factors showed down-regulated expression patterns at different stages of DHD and JG21 panicle developments. Of all transcription factors, SiMADS8 had the highest expression level was at the S1 stage of DHD. The SiWUSCHEL9/SibHLH51/SiNAC28 had lower expression levels at all developmental stages of the two foxtail millet varieties (Fig. S12). Finally, based on analysis result of FIMO, the known binding motifs of SiMADS8/SiSPL18/SiSPT were CACATTTTTGT, GGTACGGT and ACCACGTGT located on promoters of SiMCT/SiPSY1/SiZEP2 respectively (Table S5). Specifically, the regulated SPL gene could be a conserved repressor to regulate PSY and carotenoid metabolism flux according to a previously reported reference. These results suggested that the gene coexpression network related to the carotenoid metabolism pathway was available and receivable.

(A) Subcellular location of carotenoid metabolism genes. (B) Collinearity analysis of millet carotenoid metabolism genes in close-source species. (C) Collinearity analysis of millet carotenoid metabolism genes inthemain monocotcrops

Correlation analysis of the carotenoid content in the gene coexpression network module
qRT-PCR verified the expression of key genes related to carotenoid metabolism
The relative expression levels of key genes related to carotenoid metabolism during the panicle development stage of DHD were determined by qRT-PCR. We found that SiDXS3, SiPSY1/2 and SiGGPP7a /c, as major rate-limiting enzymes during the panicle developmental stages, showed a consistent downregulation trend. But only the expression of SiPY2 and SiGGPP7c were significantly decreased Among them, the expression levels of SiDXS1, SiPSY1 and SiGGPP7a/c were 1.6-, 2.04-, 2.35- and 4.91-times higher at the S1 stage compared with the S3 stage, respectively. The expression levels of SiDXS1, SiPSY2/3 and SiGGPP7b in JG21 cells were higher than those in DHD cells at the S1 stage. The SiDXS1 expression level in JG21 was 9.58-times higher than that in DHD at the S1 stage (Fig. S13–1).
For the above coexpression gene pairs, the expression levels of SiIDI2, SiZEP2, SiMCT, SiSPL18 and SiMADS8 sharply declined during the panicle development stage. The expression level of SiSPL18 was 3.55-times higher at the S1 stage than at the S3 stage in DHD. The expression level of SiMADS8 in DHD was 1.97-times higher than that of JG21 at the S1 stage. The expression levels of SiZEP2, SiSPL18 and SiMADS8 at the S1 stage in DHD were 1.12-, 1.15- and 1.97-times higher than those in JG21. Moreover, SiIDI2 expression was 1.23-fold higher in JG21 cells than in DHD cells. Based on the correlation analysis, there was a stronger correlation between the relative expression value and TPM value, with R2 = 0.9982 (Fig. S13–2). This finding suggests that these analysis results were reliable.
Discussion
Characteristics of carotenoid content variations in plants
With an increasing number of people on our planet, food security issues and the energy crisis are increasingly outstanding and currently face challenges for developing areas [33]. Specifically, many people have presented worsening health statuses because of a lack of dietary vitamin intake for the human body. Carotenoids, as essential vitamins, cannot be synthesized by humans and animals, which play an important role in antioxidation of reactive oxygen and reducing the risk of modern civilization diseases, i.e., cancer, cardiovascular or photosensitivity disorders [12]. To date, more than 750 different carotenoid metabolites have been detected and annotated from the natural world from bacteria, algae and higher plants [34,35,36]. Although golden rice can help effectively alleviate vitamin A deficiency via the food supply for people living in developing areas, natural food enriched in vitamin A is more easily accepted than GMF (genetically modified food). Previously, reports showed that carotenoid metabolites were enriched in the grains of foxtail millet, although the dynamic changes in the carotenoid content and species are not well understood at present. For Arabidopsis leaves, the carotenoid metabolites are mainly composed of lutein, β-carotene, neoxanthin and violaxanthin. Lutein is a major carotenoid metabolite that accounts for 46.55% of the total carotenoid content [36]. In corn, lutein and zeaxanthin are the most abundant carotenoid metabolites in all immature and mature grains [37]. Among 201 corn inbred lines with different grain colors, significant differences in carotenoid content occurred, with the total carotenoid content ranging from 95.5 to 629.6 μg /100 mg. Moreover, the most abundant carotenoids in corn kernels were zeaxanthin (14.43 μg /100 mg) and lutein (12.32 μg /100 mg) [38]. In wheat and wheat bread, lutein is the main carotenoid component (0.72–3.07 μg /100 mg), followed by zeaxanthin (0.88–1.84 μg /100 mg) and β-carotene (0.07–0.33 μg /100 mg) [31, 32]. The main carotenoid metabolites in brown rice are β-carotene and lutein, up to 1.50 μg /100 mg and 1.09 μg /100 mg, respectively, while the content of zeaxanthin was as low as 0.37 μg /100 mg [14]. The total carotenoid content in mature grains of foxtail millet (192.3 μg /100 mg) was 10- to 100-times higher than that in the above cereal crops [11]. In our study, the major carotenoid metabolites were lutein (47.35%) and β-carotene (44.62%) (Fig. S3). Moreover, we found a significant difference in the total carotenoid content in the grains of foxtail millet between the two varieties at P < 0.05. A comparison between DHD and JG21, which have white color grains and yellow color grains, respectively, at the early stage of grain showed that the content of lutein and β-carotene in DHD (11.51 μg /100 mg and 12.56 μg /100 mg, respectively) was significantly higher than that in JG21 (8.94 μg /100 mg and 9.86 μg /100 mg, respectively). However, at the end of development, the contents of the two types of carotene in the white-grained variety DHD were 5.43 μg /100 mg and 2.59 μg /100 mg, respectively, which were lower than that of the yellow-grained variety JG21 (5.61 μg /100 mg and 4.21 μg /100 mg, respectively) during this period (Fig. 1).

Coexpressionnetwork of important carotenoid metabolism genes andrelated transcriptionfactors. (The black dashed line represents the coexpressionof the two genes, and the brown dashed line represents the potential combination of the two genes predicted by FIMO)
Key genes related to carotenoid metabolism controlled carotenoid metabolism flux
A total of 54 genes related to carotenoid metabolism were identified and analyzed in foxtail millet. Furthermore, we found that these genes had stronger genomic collinearity relationships among cereal crops. We also found that the genome distribution characteristics of these genes were similar to the results of Brassica napus [39], indicating that they were essential for maintaining plant development, physiology and biochemical processes in plants.
Carotenoid precursor metabolites are formed by the condensation of the 5-carbon precursors IPP and DMAPP, which are produced through the MEP pathway in plastids [40]. DXS and DXR, as important rate-limiting enzymes in the MEP pathway, play an important role in the regulation of carotenoid metabolism flux [41]. For foxtail millet, the results of the phylogenetic tree showed that the three SiDXS genes were divided into three independent branches, suggesting that each SiDXS gene could play a different role in the biosynthesis of terpenoids. Similar results existed in other plants [42,43,44]. Moreover, our results showed that SiDXS3 was specifically expressed in the panicle of foxtail millet, which indicated that this gene could be the key gene for the carotenoid accumulation at the early panicle development stage. The IDI gene was responsible for regulating the ratio of IPP and DMAPP products in the MEP and MVA pathways [45]. Overexpression of different exogenous IDI genes in E. coli could promote the biosynthesis of β-carotene, lycopene, astaxanthin and zeaxanthin in vivo [46,47,48]. Our results showed that the IDI2 gene was more highly expressed at the panicle development stage among the three IDI gene members correlated with the content of major carotenoid metabolites.
The cytoplasmic MVA pathway also contributes to the synthesis of IPP and DMAPP, which provide precursors for the biosynthesis of sesquiterpenes, polyterpenes, sterols, and glycols and the formation of ubiquinone in mitochondria. In Arabidopsis, the expression level of AtAACT2 was six times that of AtAACT1 [49]. In our study, the expression of SiAACT1 was also significantly higher than that of SiAACT2/3, which indicated that SiAACT1 plays a more important role in cytoplasmic isoprenoid biosynthesis during the panicle development stage of foxtail millet. PSY, as a rate-limiting enzyme in the carotenoid biosynthesis pathway, is easily regarded as the main bottleneck of carotenoid circulation. Ectopic expression of the PSY gene and PaCRTI gene in rice created the first- and second-generation ‘golden rice’, which had higher carotenoid contents of 16 μg /100 mg and 367 μg /100 mg, respectively [22, 50]. Moreover, overexpression of PSY1 in tomato plants significantly increased the carotenoid content in tomato fruit [51, 52]. Our results showed that the three PSY genes have differentially expressed patterns during the panicle development stage, suggesting that they have differential functions in regulating carotenoid metabolism flux [53]. Additionally, many studies have demonstrated that ZEP is an important node for fine-tuning carotenoid metabolism in Arabidopsis [54, 55]. The SNP variants of ZEP in sorghum and Arabidopsis were significantly correlated with the zeaxanthin content and lutein/zeaxanthin ratio [56, 57]. Meanwhile, we found that two members of the ZEP gene in foxtail millet were highly correlated with the content of carotenoid metabolites. However, the expression of SiZEP2 was 72-times higher than that of SiZEP1, suggesting that SiZEP2 could be a major gene for regulating carotenoid metabolism.
The function of CCDs as nonhaem iron-dependent enzymes was to cleave carotenoids by catalyzing the oxidative cleavage of a double bond to form either a ketone or an aldehyde, which reduced the carotenoid content in Arabidopsis thaliana, Chrysanthemum morifolium, Fragaria × ananassa and Solanum tuberosum [58,59,60,61,62]. In foxtail millet, SiCCD8a, SiCCD8b and SiCCD8d were tandem repeats distributed on chromosome 5. However, their expression patterns were significantly different. Moreover, we found that SiCCD8a and SiCCD8d were positively correlated with carotenoid metabolites while SiCCD8b was negatively correlated with them. Taken together, these results indicate that the molecular mechanism underlying carotenoid metabolism that maintains the balance of carotenoid synthesis and degradation during the panicle development stage in foxtail millet is relatively complex and challenging.
Transcription regulation network related to carotenoid metabolism
In recent years, a number of studies have shown that some transcription factors directly or indirectly regulate the expression of carotenoid metabolism and metabolism genes and further control carotenoid metabolism flux in plants. To date, research reports on the interactive relationship between some transcription factors and genes related to carotenoid metabolism have been identified and proven in Arabidopsis. Nevertheless, the regulatory network related to the carotenoid metabolism pathway is still unknown and lacks in-depth investigation. In our coexpression network, we found that 25 transcription factors had a stronger interaction with five genes related to carotenoid metabolism. However, only the interaction relationship between SPT and ZEP2, SPL18 and PSY1, MADS8 and MCT could be identified and predicted through the hTFtarget tool. A well-known gene named PHYTOCHROME INTERACTING FACTOR 1 (PIF1), which is a key transcription regulator of carotenoid biosynthesis, could be activated by phytochrome under red light signal treatment during the process of seedling deyellowing [63]. PIF1 is phosphorylated by phytochrome when activated by light and subsequently degraded by the proteasome by inhibiting the expression of AtPSY in the dark [19, 64]. PIF1 also initiates genes related to chlorophyll biosynthesis and chloroplast development [65]. Another important bZIP transcription factor, LONG HYPOCOTYL 5 (HY5), could antagonize PIF1 during the process of photomorphogenesis [20, 66]. Spatula (SPT) annotated a bHLH transcription factor that regulates ABA metabolism to control the gynoecium and promote vegetative growth and seed dormancy [67,68,69,70]. Hence, we speculated that SPT, as a key regulator of carotenoid metabolism to ABA, had a stronger interaction relationship with SiZEP2 in the panicle of foxtail millet [71].
In tomato, an SPL gene mutant with a colorless and immature tomato could not produce lycopene because PSY1 is not expressed [72]. Overexpression of AtmiR156b, which repressed AtSPL3 expression [73], enhanced the content of lutein and β-carotene in rape seeds [74, 75]. Hence, we suggest that the SiPSY1 gene interacts with the SPL18 gene as a potential regulator of carotenoid metabolism.
Several MADS-box regulators affect the expression of the tomato CBP gene, including tomato AGAMOUS-like 1 (TAGL1), RIPENING INHIBITOR (RIN), and FRUITFULL1/2 (FUL1/2) [76]. These MADS-box proteins directly or indirectly positively regulated the expression of SlPSY1, SlPSY2, SlZDS, SlZ-ISO, SlCRTISO and SlBCH while negatively regulating the expression of SlLCYB and SlLCYE [77,78,79,80,81,82]. Our results showed that the SiMADS8 and SiMCT genes had a stronger coexpression relationship.
Based on these results, we obtained a gene coexpression network related to carotenoid metabolism and found that three gene pairs had a stronger interaction relationship. These results would lay the foundation for exploring the underlying molecular regulation mechanism of carotenoid metabolism in the panicle of foxtail millet.
Availability of data and materials
All datasets supporting the results of this article are included within the article and its supplementary information.
References
Doust AN, et al. Foxtail millet: a sequence-driven grass model system. Plant Physiol. 2009;149(1):137–41.
Diao X, Jia G. Origin and domestication of foxtail millet. In: Doust A, Diao X, editors. Genetics and genomics of setaria. Cham: Springer International Publishing; 2017. p. 61–72.
Zhang G, et al. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat Biotechnol. 2012;30(6):549–54.
Bennetzen JL, et al. Reference genome sequence of the model plant Setaria. Nat Biotechnol. 2012;30(6):555–61.
Yang Z, et al. A mini foxtail millet with an Arabidopsis-like life cycle as a C4 model system. Nat Plants. 2020;6(9):1167–78.
Wang J, et al. De novo genome assembly of a foxtail millet cultivar Huagu11 uncovered the genetic difference to the cultivar Yugu1, and the genetic mechanism of imazethapyr tolerance. BMC Plant Biol. 2021;21(1):271.
Peng R, Zhang B. Foxtail millet: a new model for C4 plants. Trends Plant Sci. 2021;26(3):199–201.
Lata C, Gupta S, Prasad M. Foxtail millet: a model crop for genetic and genomic studies in bioenergy grasses. Crit Rev Biotechnol. 2013;33(3):328–43.
Okarter N, Liu RH. Health benefits of whole grain phytochemicals. Crit Rev Food Sci Nutr. 2010;50(3):193–208.
Shen R, et al. Identification of carotenoids in foxtail millet (Setaria italica) and the effects of cooking methods on carotenoid content. J Cereal Sci. 2015;61:86–93.
Zhang B, et al. Carotenoid composition and expression of biosynthetic genes in yellow and white foxtail millet [Setaria italica (L.) Beauv]. J Cereal Sci. 2019;85:84–90.
Fiedor J, Burda K. Potential role of carotenoids as antioxidants in human health and disease. Nutrients. 2014;6(2):466–88.
Moreau RA, et al. A comparison of the levels of oil, carotenoids, and lipolytic enzyme activities in modern lines and hybrids of grain sorghum. J Am Oil Chem Soc. 2016;93(4):569–73.
Lamberts L, Delcour JA. Carotenoids in raw and parboiled brown and milled rice. J Agric Food Chem. 2008;56(24):11914–9.
Qin X, et al. Distinct expression and function of carotenoid metabolic genes and homoeologs in developing wheat grains. BMC Plant Biol. 2016;16(1):155.
Rodriguez-Concepcion M. Supply of precursors for carotenoid biosynthesis in plants. Arch Biochem Biophys. 2010;504(1):118–22.
Vranova E, Coman D, Gruissem W. Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu Rev Plant Biol. 2013;64:665–700.
Watkins JL, Pogson BJ. Prospects for carotenoid biofortification targeting retention and catabolism. Trends Plant Sci. 2020;25(5):501–12.
Bae G, Choi G. Decoding of light signals by plant phytochromes and their interacting proteins. Annu Rev Plant Biol. 2008;59(1):281–311.
Shi H, et al. Arabidopsis DET1 degrades HFR1 but stabilizes PIF1 to precisely regulate seed germination. Proc Natl Acad Sci. 2015;112(12):3817.
Lu S, et al. A fruit ripening-associated transcription factor CsMADS5 positively regulates carotenoid biosynthesis in citrus. J Exp Bot. 2015;72(8):3028–43.
Paine JA, et al. Improving the nutritional value of Golden Rice through increased pro-vitamin a content. Nat Biotechnol. 2005;23(4):482–7.
Zunjare RU, et al. Influence of rare alleles of β-carotene hydroxylase and lycopene epsilon cyclase genes on accumulation of provitamin a carotenoids in maize kernels. Plant Breed. 2017;136(6):872–80.
Cao H, et al. A neighboring aromatic-aromatic amino acid combination governs activity divergence between tomato phytoene synthases. Plant Physiol. 2019;180(4):1988–2003.
Macrae A, et al. Phenology of the genetic model Setaria viridis (Poaceae) according to the BBCH-scale of development. Bot J Linn Soc. 2020;192(1):224–41.
Nogueira M, et al. Subchromoplast sequestration of carotenoids affects regulatory mechanisms in tomato lines expressing different carotenoid gene combinations. Plant Cell. 2013;25(11):4560–79.
Chen C, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13(8):1194–202.
Nicolaides NC, Stoeckert CJ Jr. A simple, efficient method for the separate isolation of RNA and DNA from the same cells. Biotechniques. 1990;8(2):154–6.
Pertea M, et al. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11(9):1650–67.
Wang L, et al. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26(1):136–8.
Kanehisa M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36(Database issue):D480–4.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25(4):402–8.
Brinkman H-J, et al. High food prices and the global financial crisis have reduced access to nutritious food and worsened nutritional status and health. J Nutr. 2010;140(1):153S–61S.
Digesù AM, et al. Genetic variability in yellow pigment components in cultivated and wild tetraploid wheats. J Cereal Sci. 2009;50(2):210–8.
Leenhardt F, et al. Genetic variability of carotenoid concentration, and lipoxygenase and peroxidase activities among cultivated wheat species and bread wheat varieties. Eur J Agron. 2006;25(2):170–6.
Taylor KL, et al. High-performance liquid chromatography profiling of the major carotenoids in Arabidopsis thaliana leaf tissue. J Chromatogr A. 2006;1121(1):83–91.
Song J, et al. Comparison of carotenoid composition in immature and mature grains of corn (Zea Mays L.) varieties. Int J Food Prop. 2016;19(2):351–8.
Owens BF, et al. A foundation for provitamin a biofortification of maize: genome-wide association and genomic prediction models of carotenoid levels. Genetics. 2014;198(4):1699–716.
Li P, et al. Carotenoid biosynthetic genes in Brassica rapa: comparative genomic analysis, phylogenetic analysis, and expression profiling. BMC Genomics. 2015;16(1):492.
Blanc G, Wolfe KH. Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution[W]. Plant Cell. 2004;16(7):1679–91.
Carretero-Paulet L, et al. Enhanced flux through the methylerythritol 4-phosphate pathway in Arabidopsis plants overexpressing deoxyxylulose 5-phosphate reductoisomerase. Plant Mol Biol. 2006;62(4):683–95.
Walter MH, Hans J, Strack D. Two distantly related genes encoding 1-deoxy-d-xylulose 5-phosphate synthases: differential regulation in shoots and apocarotenoid-accumulating mycorrhizal roots. Plant J. 2002;31(3):243–54.
Phillips MA, et al. Functional identification and differential expression of 1-deoxy-d-xylulose 5-phosphate synthase in induced terpenoid resin formation of Norway spruce (Picea abies). Plant Mol Biol. 2007;65(3):243–57.
Cordoba E, et al. Functional characterization of the three genes encoding 1-deoxy-D-xylulose 5-phosphate synthase in maize. J Exp Bot. 2011;62(6):2023–38.
Berthelot K, et al. Isopentenyl diphosphate isomerase: a checkpoint to isoprenoid biosynthesis. Biochimie. 2012;94(8):1621–34.
Albrecht M, Sandmann G. Light-stimulated carotenoid biosynthesis during transformation of maize etioplasts is regulated by increased activity of isopentenyl pyrophosphate isomerase. Plant Physiol. 1994;105(2):529–34.
Sun J, et al. A novel cytoplasmic isopentenyl diphosphate isomerase gene from tomato (Solanum lycopersicum): cloning, expression, and color complementation. Plant Mol Biol Report. 2010;28(3):473–80.
Gallagher C, Cervantes-Cervantes M, Wurtzel E. Surrogate biochemistry: use of Escherichia coli to identify plant cDNAs that impact metabolic engineering of carotenoid accumulation. Appl Microbiol Biotechnol. 2003;60(6):713–9.
Jin H, Song Z, Nikolau BJ. Reverse genetic characterization of two paralogous acetoacetyl CoA thiolase genes in Arabidopsis reveals their importance in plant growth and development. Plant J. 2012;70(6):1015–32.
Ye X, et al. Engineering the provitamin a (β-carotene) biosynthetic pathway into (carotenoid-free) rice endosperm. Science. 2000;287(5451):303.
Fraser PD, et al. Manipulation of phytoene levels in tomato fruit: effects on isoprenoids, plastids, and intermediary metabolism. Plant Cell. 2007;19(10):3194–211.
Tomato Genome C. The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485(7400):635–41.
Li F, Murillo C, Wurtzel ET. Maize Y9 encodes a product essential for 15-cis-zeta-carotene isomerization. Plant Physiol. 2007;144(2):1181–9.
Xi W, et al. The genes of CYP, ZEP, and CCD1/4 play an important role in controlling carotenoid and aroma volatile apocarotenoid accumulation of apricot fruit. Front Plant Sci. 2020;11:2105.
Suematsu K, et al. Comparative transcriptome analysis implied a ZEP paralog was a key gene involved in carotenoid accumulation in yellow-fleshed sweetpotato. Sci Rep. 2020;10(1):20607.
Gonzalez-Jorge S, et al. ZEAXANTHIN EPOXIDASE activity potentiates carotenoid degradation in maturing seed. Plant Physiol. 2016;171(3):1837–51.
Cruet-Burgos C, et al. Advancing provitamin a biofortification in sorghum: Genome-wide association studies of grain carotenoids in global germplasm. Plant Genome. 2020;13(1):e20013.
Schaub P, et al. Nonenzymatic β-carotene degradation in provitamin a-biofortified crop plants. J Agric Food Chem. 2017;65(31):6588–98.
Bruno M, et al. Enzymatic study on AtCCD4 and AtCCD7 and their potential to form acyclic regulatory metabolites. J Exp Bot. 2016;67(21):5993–6005.
Ohmiya A, et al. Carotenoid cleavage dioxygenase (CmCCD4a) contributes to white color formation in chrysanthemum petals. Plant Physiol. 2006;142(3):1193–201.
García-Limones C, et al. Functional characterization of FaCCD1: a carotenoid cleavage dioxygenase from strawberry involved in lutein degradation during fruit ripening. J Agric Food Chem. 2008;56(19):9277–85.
Campbell R, et al. The metabolic and developmental roles of carotenoid cleavage dioxygenase4 from potato. Plant Physiol. 2010;154(2):656–64.
Shin J, et al. Phytochromes promote seedling light responses by inhibiting four negatively-acting phytochrome-interacting factors. Proc Natl Acad Sci. 2009;106(18):7660.
Shen H, et al. Light-induced phosphorylation and degradation of the negative regulator PHYTOCHROME-INTERACTING FACTOR1 from Arabidopsis depend upon its direct physical interactions with photoactivated phytochromes. Plant Cell. 2008;20(6):1586–602.
Toledo-Ortiz G, Huq E, Rodríguez-Concepción M. Direct regulation of phytoene synthase gene expression and carotenoid biosynthesis by phytochrome-interacting factors. Proc Natl Acad Sci. 2010;107(25):11626.
Zhu L, et al. CUL4 forms an E3 ligase with COP1 and SPA to promote light-induced degradation of PIF1. Nat Commun. 2015;6(1):7245.
Alvarez J, Smyth DR. CRABS CLAW and SPATULA, two Arabidopsis genes that control carpel development in parallel with AGAMOUS. Development. 1999;126(11):2377–86.
Tsiantis M. Plant development: multiple strategies for breaking seed dormancy. Curr Biol. 2006;16(1):R25–7.
Josse E-M, et al. A DELLA in disguise: SPATULA restrains the growth of the developing arabidopsis seedling. Plant Cell. 2011;23(4):1337–51.
Penfield S, et al. Cold and light control seed germination through the bHLH transcription factor SPATULA. Curr Biol. 2005;15(22):1998–2006.
Vaistij FE, et al. MOTHER-OF-FT-AND-TFL represses seed germination under far-red light by modulating phytohormone responses in Arabidopsis thaliana. Proc Natl Acad Sci. 2018;115(33):8442.
Eriksson EM, et al. Effect of the colorless non-ripening mutation on cell wall biochemistry and gene expression during tomato fruit development and ripening. Plant Physiol. 2004;136(4):4184–97.
Wang J-W, Czech B, Weigel D. miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell. 2009;138(4):738–49.
Gandikota M, et al. The miRNA156/157 recognition element in the 3′ UTR of the Arabidopsis SBP box gene SPL3 prevents early flowering by translational inhibition in seedlings. Plant J. 2007;49(4):683–93.
Wei S, et al. Enhanced seed carotenoid levels and branching in transgenic Brassica napus expressing the Arabidopsis miR156b gene. J Agric Food Chem. 2010;58(17):9572–8.
Stanley L, Yuan Y-W. Transcriptional regulation of carotenoid biosynthesis in plants: so many regulators, so little consensus. Front Plant Sci. 2019;10:1017.
Fujisawa M, Nakano T, Ito Y. Identification of potential target genes for the tomato fruit-ripening regulator RIN by chromatin immunoprecipitation. BMC Plant Biol. 2011;11(1):26.
Fujisawa M, et al. A large-scale identification of direct targets of the tomato MADS box transcription factor RIPENING INHIBITOR reveals the regulation of fruit ripening. Plant Cell. 2013;25(2):371–86.
Giménez E, et al. Functional analysis of the Arlequin mutant corroborates the essential role of the Arlequin/TAGL1 gene during reproductive development of tomato. PLoS One. 2010;5(12):e14427.
Itkin M, et al. TOMATO AGAMOUS-LIKE 1 is a component of the fruit ripening regulatory network. Plant J. 2009;60(6):1081–95.
Martel C, et al. The tomato MADS-box transcription factor RIPENING INHIBITOR interacts with promoters involved in numerous ripening processes in a COLORLESS NONRIPENING-dependent manner. Plant Physiol. 2011;157(3):1568–79.
Shima Y, et al. Tomato FRUITFULL homologues act in fruit ripening via forming MADS-box transcription factor complexes with RIN. Plant Mol Biol. 2013;82(4):427–38.
Acknowledgments
We would like to thank Professor Donald Grierson, University of Nottingham, UK, for engaging in discussions and helping with the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (31771810, 32070366); Cultivate Scientific Research Excellence Programs of Higher Education Institutions in Shanxi (2019KJ020); Shanxi Agricultural University Science and Technology Innovation Fund Project (2020BQ12).
Author information
Authors and Affiliations
Contributions
YH and SH conceived and designed the experiments. HL and SH wrote the manuscript. GM performed the experiments and analyzed the data. QH, YH, and SH administrated the project. ZS, YH and HL revised the manuscript. All authors have read and gave final approval for publication.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Study complied with local and national regulations for using plants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of interest in this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, H., Han, S., Huo, Y. et al. Comparative metabolomic and transcriptomic analysis reveals a coexpression network of the carotenoid metabolism pathway in the panicle of Setaria italica. BMC Plant Biol 22, 105 (2022). https://doi.org/10.1186/s12870-022-03467-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-022-03467-2