
Abstract
Background
Hybridization and introgression are vital sources of novel genetic variation driving diversification during reticulated evolution. Quercus is an important model clade, having extraordinary diverse and abundant members in the Northern hemisphere, that are used to studying the introgression of species boundaries and adaptive processes. China is the second-largest distribution center of Quercus, but there are limited studies on introgressive hybridization.
Results
Here, we screened 17 co-dominant nuclear microsatellite markers to investigate the hybridization and introgression of four oaks (Quercus acutissima, Quercus variabilis, Quercus fabri, and Quercus serrata) in 10 populations. We identified 361 alleles in the four-oak species across 17 loci, and all loci were characterized by high genetic variability (HE = 0.844–0.944) and moderate differentiation (FST = 0.037–0.156) levels. A population differentiation analysis revealed the following: allopatric homologous (FST = 0.064) < sympatric heterogeneous (FST = 0.071) < allopatric heterogeneous (FST = 0.084). A Bayesian admixture analysis determined four types of hybrids (Q. acutissima × Q. variabilis, Q. fabri × Q. serrata, Q. acutissima × Q. fabri, and Q. acutissima × Q. variabilis × Q. fabri) and their asymmetric introgression. Our results revealed that interspecific hybridization is commonly observed within the section Quercus, with members having tendency to hybridize.
Conclusions
Our study determined the basic hybridization and introgression states among the studied four oak species and extended our understanding of the evolutionary role of hybridization. The results provide useful theoretical data for formulating conservation strategies.
Similar content being viewed by others

Background
Introgressive hybridization is a common natural phenomenon among biological organisms, especially oaks [1]. Interspecific hybridization is an important source of genetic variation and drives diversification in response to changing environmental conditions [2,3,4]. Additionally, introgression may have ecological consequences, including shaping community assembly and structure [5, 6]. Interspecific introgression of adaptive genetic variation has been documented among forest tree species, including poplars [7], eucalypts [8], spruces [9], pines [10], and oaks [11]. Nuclear DNA markers, such as microsatellites (SSRs) [12], randomly amplified polymorphic DNA markers [13], and chloroplast DNA [14] have been used to detect introgression across species. With advancements in genomic studies, our understanding of the ecological, genetic, and genomic factors underlying the diverse outcomes of interspecific introgression in hybrid zones is increasing [15,16,17].
The genus Quercus L. (oaks) contains more than 400 species that are widespread in the Northern hemisphere [18]. Oak is an important model clade that offers fundamental insights into the ecological and evolutionary consequences of hybridization and introgression [19]. In Quercus, naturally occurring interspecific hybrids are common [11, 15]. Within Quercus, the American oaks and European white oaks have been well-studied [20]. For example, high quality SNPs from genic regions have been used to characterize the introgression patterns among European white oaks (Quercus petraea and Quercus robur) [21]. Eaton et al. (2015) utilized genomic RAD-seq data sampled from American live oaks (Quercus series Virentes) for phylogenetic inference and determining introgressions between lineages [22]. In China, most oak hybridization studies have used nuclear DNA markers, such as SSRs and AFLPs [23]. For conservation purposes, it is necessary to study the hybridization and introgression of oak, especially native species.
China is the second-largest distribution center of Quercus, with 62 species described in the Flora of China that are divided into five morphology-based sections: Quercus, Aegilops, Heterobalanus, Engleriana, and Echinolepides [24,25,26]. The first two sections (Quercus and Aegilops) are composed of deciduous oak species which correspond to the Quercus and Cerris proposed by Denk [27]. For our present study, Quercus acutissima Carruthers and Quercus variabilis Blume belong to Group Cerris, while Quercus fabri Hance and Quercus serrata Murray belong to Group Quercus. A new classification of Quercus L. was proposed by Denk harbouring eight sections: Cyclobalanopsis, Cerris, Ilex, Lobatae, Quercus, Ponticae, Protobalanus, and Virentes [27]. The phylogenetic positions of our study species did not change, as they remain in Cerris and Quercus Groups.
The studied four oak species (Q. acutissima, Q. variabilis, Q. fabri, and Q. serrata), having an overlapping distribution in subtropical areas, are important species in subtropical mountain and warm temperate deciduous broad-leaved forests, which occupy important positions in China’s forest ecosystem [28]. In our study, we aimed to: (1) identify the pattern of hybridization between these four oak species; (2) investigate whether, and to what extent, introgression exists among these four-oak species; and (3) compare the introgression intensity levels in sympatric and allopatric populations. This work provides an understanding of the evolutionary mechanisms of Quercus, as well as the biological processes behind their biodiversity, and has implications for forest management.
Results
The availability of microsatellite loci (SSRs)
The average values of the estimated null allele frequency at each locus across all populations were all less than 0.2 (Table 1). No evidence of significant linkage disequilibrium was observed for each pair of loci in each population at the indicative adjusted nominal level (5%) for multiple comparisons equal to 0.000368 (Table S1). The 17 polymorphic SSRs generated 361 alleles, with a mean of 21 alleles per locus. Some SSRs were highly variable, including quru-GA-0M05, Quru-GA-1H14, Quru-GA-1i15, MSQ16, ssrQpZAG36, and ssrQrZAG112, which each contained more than 21 alleles. Mean PIC, AR, and HS for all markers were 0.909 (range: 0.844 to 0.939), 10.7 (range:7.1 to 14.2), and 0.550 (range: 0.254 to 0.980), respectively (Table 1).
Comparison of DIC values across the 10 populations showed that models with null alleles had higher support than models without null alleles, and the average estimated frequency of across loci was slightly high (ranging from 0.102 to 0.188) (Table 2; Table S1). The inbreeding coefficient of the 10 populations was large (mean = 0.41) and significantly deviated from zero (P < 0.05). Two populations, LT-F and ZW-B, did not departure from Hardy–Weinberg equilibrium, and seven populations, BY-A, BY-V, LT-B, ZW-V, ZJ-A, ZJ-V, ZJ-F, and ZJ-B, did not departure from Hardy–Weinberg equilibrium after excluding the null alleles bias.
Analyses of genetic diversity among the four oaks
At the DNA level, the four oak species had high degree of genetic diversity where mean NE,AR, and HS were 6.7 (range: 5.8 to 7.9), 10.7 (range: 8.7 to 11.8), and 0.829 (range: 0.782 to 0.862), respectively (Table 2). The four species were divided into two groups belonging to Cerris and Quercus (groups AV and FB) based on the STRUCTURE analysis. The Ho of group FB (0.607) was greater than that of group AV (0.497) as determined using FSTAT (P < 0.05). The highest intraspecific genetic diversity (AR, HS, and Ho) was observed in the Q. fabri population compared with the three other investigated species and was significantly different from Q. variabilis (P < 0.05). Among the four different sites, Liangting had highest Ho, which was significantly different from that of Zhongwu. Significant differences in AR were observed between Zijin and Zhongwu while HS and FST showed no significant differences among sites.
Genetic differentiation and population structure
Null alleles have particular effect on the estimation of population differentiation, which was indicated by the almost equal values and similar 95% level CIs of global FST across all loci with (cFST = 0.085, 95% CI: 0.065—0.105) and without (FST = 0.101, 95% CI: 0.078–0.125) using the “exclusion null alleles” correction (Table 1). For each locus, cFST (range: 0.033 to 0.168) and standardized differentiation G′ST (range: 0.033 to 0.190) were relatively low at the nominal level (5%). Thus, a weak genetic differentiation occurred among the four-oak species. We also compared the genetic differentiation between pairs of populations (Fig. 1a). In this study, we identified the same species as homologous and different species as heterogeneous. Population differentiation analysis revealed the following: allopatric homologous (mean FST = 0.064) < sympatric heterogeneous (FST = 0.071) < allopatric heterogeneous (FST = 0.084). The interspecific genetic differentiation levels were more significant than genetic differentiation among regions as observed using two-sided P-values (Table 3). The PCoA confirmed a strong genetic structure existed among the populations (Fig. 1b–d).

Pairwise FST in sympatric and allopatric populations of four oak species (a: the red box represents sympatric populations and the yellow box represents the same species). and principal coordinate analysis (PCoA) based on pairwise population FST values implemented in GENALEX 6.5 (b-d)
AMOVA results indicated that 1.04% (P = 0.003) of the genetic variation occurred among regions and 9.78% (P < 0.0001) among populations within regions, with the greatest genetic differentiation of 89.17% (P < 0.0001) being harbored within populations (Table 4). The hierarchical AMOVA revealed a significant genetic differentiation among species (FCT = 0.04064, P < 0.001), with 7.2% (P < 0.0001) occurring among populations within species and the greatest genetic differentiation of 88.74% (P < 0.0001) being harbored within populations. The intraspecific gene flow of Q. acutissima, Q. variabilis, Q. fabri, and Q. serrata were estimated to be 3.31, 2.56, 4.72, and 3.8, respectively (Table 5). Simultaneously, the genetic variation among species was greater than among regions. In total, 300 samples were clustered into two branches based on Nei’s genetic distances using the NJ method (Fig. 2). Q. acutissima and Q. variabilis individuals were clustered into a large branch below the tree (group AV) and Q. fabri and Q. serrata (group FB) individuals were clustered into another large branch above the tree. The same species, along with other species, showed intraspecific differentiations and interspecies variations. Additionally, NJ graph showed considerable variation and somehow weak clustering of Q. fabri and Q. serrata. The clustering results of leaf variation were basically consistent with the molecular results. We clustered the four species and found that 14 individuals of Q. fabri and all of Q. serrata samples clustered into a single branch. These results indicated that the leaf traits were quite different from those of other Q. fabri individuals. There was a small deviation at the population-level clustering, which mainly occurred for the Q. variabilis population in Bayan, as well as for Q. fabri and Q. serrata populations in Liangting and Zijinshan (Fig. 3). This indicated that there was no great difference in morphology, but that the genetic structure had changed.

Molecular and phenotypic cluster analysis of 300 oak individuals. a NJ dendrogram showing genetic relationships implemented in MEGA 7.0 and b cluster analysis based on phenotypic traits variation

Molecular and phenotypic cluster analysis of 10 oak populations. a NJ cluster analysis implemented in MEGA 7.0 (Sudhir et al. 2016) based on the Nei’s unbiased genetic distances (Nei 1978). b Cluster analysis based on phenotypic traits variation
Admixture analysis and hybrid identification
Delta K, which is used to determine the best fit value of K, was computed using STRUCTURE HARVESTER for the given range, 1–9, and the highest value was obtained at K = 2 (Fig. 4). This was consistent with the taxonomic classifications of these four distinct species. In total, 98% of Q. acutissima and Q. variabilis individuals were assigned to cluster I (QI), with an average proportion of membership (QI = 0.98), and most Q. fabri and Q. serrata individuals (100%) were assigned to cluster II (QII = 0.99). Only three remaining individuals (two Q. acutissima and one Q. variabilis) showed different degree of admixture with assignment probabilities of < 0.90. The 10 populations were divided into two large groups (Groups AV and FB). Using K = 4, the hybridization scenario among the four oak species was more complicated. Q. fabri individuals were assigned to cluster I (QI = 0.80), Q. acutissima individuals were assigned to cluster II (QII = 0.94), Q. serrata individuals were assigned to cluster III (QIII = 0.54), and Q. variabilis individuals were assigned to cluster IV (QIV = 0.66) (Table S2).

STRUCTURE clustering results for 300 oak species individuals from 10 populations (each color represents a genetic cluster)
We observed four types of hybridization, Q. acutissima—Q. fabri, Q. acutissima—Q. variabilis, Q. fabri-Q. serrata, and Q. acutissima—Q. variabilis—Q. fabri (Table 6). A total of 188 heterozygous individuals were observed (overall Hr = 62.77%), including 67 F1 hybrids and 121 backcross individuals. Among the four sampled plots, the highest hybridization rate was found in Bayan plots (100%). Among the four-oak species, the hybridization rate was as follows: Q. acutissima (Hr = 18.0%) < Q. fabri (Hr = 50.7%) < Q. variabilis (Hr = 74.2%) < Q. serrata (Hr = 94.9%). Most hybridizations occurred in the same groups (AV or FB); however, there were also introgression individuals produced by cross breeding among different groups. Among them, individuals of Q. variabilis hybrids in Bayan were unique, having three types of ancestry (Q. acutissima—Q. variabilis—Q. fabri).
Discussion
Introgression studies usually focus on large-scale pairs of individual species [29] and sympatric related species [30], but rarely species within distinct ecological niches and evolutionarily clades. Here, we examined the extent of introgression that existed among four oaks belonging to two different groups of sympatric and allopatric populations. Oak (Quercus) are notorious for hybridization with species extinction, and this may also play a critical role in adaptive evolution [31, 32]. Hybridization and introgression in oaks has been well documented, and adaptive introgression was important in evolution as revealed by genomics [11, 32,33,34]. Our study used nSSR markers to investigate the introgression of four oaks. We identified four types of hybrids: Q. acutissima × Q. variabilis, Q. fabri × Q. serrata, Q. acutissima × Q. fabri, and Q. acutissima × Q. variabilis × Q. fabri. Introgression occurred more readily in the Quercus group than in the Cerris group, and cross-group hybridization also existed; however, it was rare. This suggested that a low frequency of interspecific hybridization occurred with incomplete reproductive isolation. Reproductive isolation mechanisms contributed to the simultaneous coexistence of the four oaks.
Genetic diversity and genetic variation of the studied four oak species
Introgression is a vital source of novel genetic variation and drives diversification during reticulate evolution [3]. Owing to the critical variation introduced by hybridization, and the hybrid constitution itself, the diversification rate shifts [2]. In this study, we found that the studied 10 oak populations, all had high levels of genetic diversity, with Q. fabri in the Liangting and Zijing sites having the highest value. Long-lived trees span broad geographical ranges through a combination of adaptive plasticity and adaptive differentiation, as well as the standing genetic variation that occurs in response to environmental changes [4]. A high genetic diversity provides adaptability that ensures the survival of the four oaks when facing extreme climatic events. In our study, the FST values of the 10 populations ranged from 0.05 to 0.15, which indicates moderate differentiation. Additionally, the order of of genetic differentiation among the 10 populations was as follows: allopatric homologous < sympatric heterogeneous < allopatric heterogeneous. Q. acutissima and Q. variabilis diverged 13.2 Ma, and the divergence time of Q. fabri and Q. acutissima was 12.33 Ma [35, 36]. During the quaternary ice period, the three species (Q. acutissima, Q. variabilis, and Q. fabri) showed different migration routes in response to climatic changes. In addition, the strong uplift of the Tibetan Plateau in China led to the intensification of the Asian monsoon climate, which may promote species differentiation [37,38,39]. The main cause of the genetic differentiation was the long-term evolutionary divergence of the four species, followed by adaptive differentiation caused by interspecific introgression [40].
By studying 17 pairs of polymorphic loci with different FST values, we speculated that gene flow occurred among these oaks. The limited gene flow prevented hybridization-induced genetic swamping, instead of providing more sources of genetic variation for heterozygotes. Additionally, we found that genetic variation among the four oaks mainly occurred within populations (88.74%), with only a small percentage occurring among populations (7.2%). Ramırez-Valiente et al. (2018) found that drought drove the evolution of genetic differences in functional traits among oaks [41]. The cluster analysis of leaf variation of the 300 individuals also showed large phenotypic variation. Allelic and associated phenotypic changes of species are strategies used to respond to the threat of global climatic changes during the adaptive evolutionary progress [42]. Although a small number of nuclear loci were used in this experiment to verify this hypothesis, we could not determine a phenotype associated with introgressed loci. A future genome-wide association analysis will further analyze the ingression regions and phenotypic associations.
Gene flow and hybridization among the four-oak species
Compared with intraspecific gene flow dynamics, interspecific introgression is more complex owing to the interactions of multiple species. Gene flow levels were distinguished as high (Nm > 1), intermediate (0.25 < Nm < 0.99), and low (Nm < 0.25) by Govindaraju [43]. In our study, the intraspecific gene flows of Q. acutissima, Q. variabilis, Q. fabri, and Q. serrata were estimated to be 3.31, 2.56, 4.72, and 3.8, respectively. Compared with species in other families, such as Picea asperata (0.75) and Liriodendron chinense (1.028), the gene flow was stronger, being similar to those of other oak species, such as Quercus aquifolioides (3.749). Gene flow is an important attribute affecting species ability to spread its genes [44]. Most oak trees have larger pollen dispersal distances. The dispersal capacity in the genus Quercus is also related to many factors, such as tree height, vegetation density, and leaf area [45]. A small amount of gene flow enables all breeding populations to maintain similar alleles and thus high heterozygosity, so the NJ clustering graph showed weak differentiation between Q.fabri and Q. serrata groups while the STRUCTURE clustering revealed similar genetic separation. However, it may also be caused by variation within population, as stated above, the proportion of variation within population is very high (88.74%). We found recurrent interspecific gene flow among the four oaks. The STRUCTURE clustering revealed that most populations contained hybrids between two species. For example, the ZJ-A population showed F1 hybrids between Q. acutissima and Q. variabilis, while BY-A population showed hybridization between Q. acutissima and Q. fabri. Interestingly, a large number of individuals in the BY-V population appeared to be trihybrids. Additionally, STRUCTURE clustering showed that ZJ-F and ZJ-B populations did not form two strong groups and their hybrid individuals are relatively uniform, probably representing a mixed group. However, the phenotypic data indicated that these are two distinct species. Additionally, these two populations, BY-V and ZW-B, have more uniform hybrids, and we are not sure whether this is due to ancestral lineage or recent gene flow, and furthermore we did not rule out the existing possibility of shared variation. Because no suitable outgroups were utilized in the present study, the observed complex admixture patterns maybe are attributable to their ancestral contributions from other Quercus spp. Additional samples of related species are needed to elucidate this case. At the same time, the Bayan site appeared to be an especially active zone of introgression among these three oaks.
Many F1 progeny and some backcrosses were hybrids. In section Cerris, the most hybrids occurred among Q. variabilis backcrosses, while in section Quercus, the most hybrids occurred among Q. serrata backcrosses. This may have resulted from our sampling of individuals displaying more than one species traits, but we cannot rule out a bias toward introgressions between parental species. Such asymmetric introgression is common [46], as in the introgression from the coastal oak species Quercus dentata to an ecotype of the inland oak species Quercus mongolica var. crispula, which has colonized the coastal environment in northern Japan [34]. The environment in which hybrid individuals are grown is an important cause of asymmetric introgression [47,48,49]. Oaks are wind-pollinated, and gene flow mainly occurs through pollen [50]. Therefore, differentiation in seed and pollen dispersal between hybrids and parental species are crucial. Future sampling should target a broader range of individuals in unique environments, like the edge of a hybrid zone, to determine whether their asymmetric introgression is occurring at the genome level. In addition, the reproductive isolation patterns between the hybrid individuals and their parents would be experimentally verified.
Implications for forest management
Natural hybridization is a common phenomenon in plants, occurring in 25% of existing species [51, 52]. Fertile hybrids backcross with their parents, leading to introgression. Introgression is an evolutionary creative force that introduces new, possibly adaptive alleles into a population [53, 54]; however, excessive gene flow may result in genetic swamping and species extinction [55]. For conservationists, whether to prevent or encourage interspecific gene flow is a difficult question. Oaks are “notorious” for their widespread hybridization and are exceptional forest trees with high levels of genetic diversity. Oaks tend to hybridize, and introgression may influence community structure and increasing genetic diversity [6]. In the face of climatic changes, interspecific gene flow transfers adaptive alleles to avoid extinction [56, 57]. The studied four oak species (Q. acutissima, Q. variabilis, Q. fabri, and Q. serrata) are all widely distributed in China. A palaeo distributive simulation showed that Q. acutissima, Q. variabilis, and Q. fabri tended toward expansion and contraction in glacial and interglacial periods, respectively [37,38,39]. Adaptive evolution in response to changing environment allows an organism to avoid extinction. Such adaptive evolution is related to the adaptive introgression of the species. In our study, hybridization and introgression existed among the studied four oak species. Although we did not confirm whether this introgression was adaptive, other experiments have confirmed this view [53, 54]. In future studies, we will use genome-wide data to correlate phenotypic data with fitness to explore the adaptive introgression of Quercus. To increase the efficacy of conservation strategies, conservation management should consider evolutionary theory. Thus, managers should consider whether hybrids co-exist with the parental species when formulating conservation strategies because natural hybrids have conservation value. Natural hybridization zones are good sites for studying species adaptive evolution using newly developed genomics techniques.
Conclusions
The long-term evolutionary divergence of the studied four oaks, and the resulting interspecific introgression, led to their high levels of genetic diversity and moderate differentiation. The genetic variation among the four oaks mainly occurred within populations, with only a small percentage occurring among populations. Four types of hybrids (Q. acutissima × Q. variabilis, Q. fabri × Q. serrata, Q. acutissima × Q. fabri, and Q. acutissima × Q. variabilis × Q. fabri) were determined, accompanied by asymmetric introgression. We concluded that interspecific hybridization is commonly observed within the section and that section Quercus has a high tendency to hybridize. In future work, we hope to use genomic data to study the dynamic nature of gene flow and adaptive introgression in these hybrid populations.
Methods
Sampling and study sites
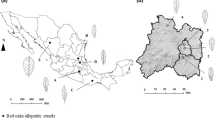
In this study, we used the new classification of Quercus proposed by Denk et al. (2017) in which Quercus is divided into eight sections: Cyclobalanopsis, Cerris, Ilex, Lobatae, Quercus, Ponticae, Protobalanus, and Virentes [27]. We selected four species in section Quercus, two wild populations of Q. acutissima, three of Q. variabilis, two of Q. fabri, and three of Q. serrata, for genetic analyses, and they were sampled randomly within four plots distributed in Jiangsu Province (Plot Zijing) and Anhui Province (Plots Bayan, Liangting, and Zhongwu) (Fig. 5). Within each locality, selected individuals were at least 30 m apart to avoid sampling the same plant. A total of 300 adult individuals were sampled, and their geographic coordinates and altitudes were recorded using a global positioning system (Table S3). From each individual plant, three to five leaves were collected, quickly dried in silica gel, and stored at room temperature for molecular experiment. At the same time, we randomly collected 10–15 leaves for morphological analysis. A single branch was collected for identification by Y.F. and then preserved in Nanjing Forestry University herbarium.

Geographic distribution of the sampled four oak plots. Pie charts with different color represent different species (Q. acutissima: blue; Q. variabilis: green; Q. fabri: yellow; Q. serrata: red). Pie area is proportional to the sample size
Measurement of leaf morphological traits
Nine leaf morphological attributes were measured using 10 mature leaves from each of the 300 trees (Table 4 S). The observed variables were scored as indicated in Table 4S. The dimensional characteristics were measured with a ruler. The epidermal hairs were observed under a microscope at 40 × magnification.
DNA extraction and microsatellite genotyping
Total genomic DNA was extracted from 30–40 mg dried leaves per individual using a Plant Genomic DNA Kit (Tiangen, China). The quality and concentration of the genomic DNA were evaluated using a One-drop spectrophotometer (OD–1000, Shanghai Cytoeasy Biotech Co., Ltd., China) and electrophoresis was conducted in 1% agarose gels. DNA samples were diluted to 20 ng/μL and stored at -20 °C.
All DNA samples were genotyped at 17 nuclear microsatellite loci (nSSRs) (QM58TGT [58]; quru-GA-0I01, quru-GA-0M05, quru-GA-0M07, Quru-GA-Oi21, Quru-GA-1H14, Quru-GA-1i15 [59, 60]; MSQ16 [61]; ssrQpZAG1/5, ssrQpZAG15, ssrQpZAG36 [62]; ssrQrZAG 7, ssrQrZAG 31, ssrQrZAG 74, ssrQrZAG 87, ssrQrZAG 96, and ssrQrZAG 112 [63]), using primers developed for oak tree species (Table S5). PCR reactions were performed in an Eppendorf thermal cycler (Eppendorf, Hamburg, Germany) using the following parameters: initial denaturation at 94 °C for 4 min, 30 cycles of denaturation at 94 °C for 45 s, 45 s of annealing at a primer specific temperature, and extension at 72 °C for 45 s, followed by a final extension of 8 min at 72 °C. The PCR mixtures had total volumes of 20 μL, containing 2 × Taq PCR MasterMix (Tiangen), 10 μM of each primer, 20–40 ng of template DNA, and ddH2O. PCR products were separated on an ABI3730xl automated Genetic Analyzer using ROX-500 as an internal standard (Applied Biosystems, USA). Allele sizes were determined manually using Genemarker version 2.2.0 (Applied Biosystems).
Statistical analyses
We used the dimensional characteristics, transformed variables, counted variables, and observed variables described in the literature to detect the intraspecific variation. The morphological characteristics data was analyzed using SPSS 19.0 (SPSS, Inc., Chicago, IL, USA). Using the Euclidean distances, nine phenotypic characteristics were clustered at the individual and population level. The obtained population clustering graphs and individual clustering diagrams were used along with the molecular results in a comparative analysis.
For the SSR data, we used INEST, version 2.2 [64] to detect null allele frequency at each locus simultaneously in each population using the individual inbreeding model, which includes three parameters, null alleles (n), inbreeding coefficients (f), and genotyping failures (b). The number of Markov Chain Monte Carlo iterations and burn-in were set at 500,000 and 50,000 cycles, respectively. Thinning maintained every 50th update. To conduct a Bayesian model comparison, we performed the analysis using the model with null alleles (nfb) and without null alleles (fb) to evaluate the significance of null alleles within each population. We set nfb significance using low deviance information criterion (DIC). For this method, the thresholds were set to a maximum of less than 0.2 and a mean of less than 0.1, which resulted in the final selection of all 17 loci for subsequent analyses. Linkage disequilibrium for all the locus pairs in each population and significant deviations from Hardy–Weinberg equilibrium were determined using Genepop version 4.6.9 [65].
Genetic diversity statistics of each population and each locus were estimated using POPGENE, version 1.32 [66]. The statistics included number of alleles (N), observed (HO) and expected (HE) heterozygosity and effective number of alleles (NE). Polymorphic information content (PIC) was estimated in Cervus 2.0 [67], while allelic richness (AR), genetic diversity within populations (HS), and inbreeding coefficient (FIS) were estimated using FSTAT version 2.9.3.2 [68]. The corrected inbreeding coefficient (FIS′) for each population was also evaluated using INEST version 2.2 [64] with the full model (nfb). The significance of FIS′ was assessed by comparing DIC values of inbreeding and without inbreeding models.
We estimated global FST and cFST [69] both with and without the “exclusion null allele” correction per loci using FreeNA [70]. The 95% level confidence intervals (CIs) of both global FST and cFST across loci were obtained through bootstrap resampling in the same program. The FST of each pair of populations was estimated in GENALEX 6.5 [71] and visualized in the form of a heatmap using Heml 1.0.33 [72]. Gene flow was indirectly estimated using Wright’s (1951) formula: Nm = 1—FST/4 FST. Additionally, we performed a principal coordinate analysis based on pairwise population FST values using GENALEX 6.5 [71]. We tested the significance of differences in AR, HO, HS, and FST among geographic regions and among species using FSTAT, version 2.9.3.2 [68]. The two-sided P-values were obtained after 10,000 permutations. To examine the genetic variances of allopatric and heterogeneous types we conducted analysis of molecular variance (AMOVA) using F-statistics using ARLEQUIN, version 3.5 [73]. Significances of fixation indices (FCT, FSC, and FST) were tested with 10,000 permutations.
STRUCTURE software [74] was used to infer population structure. To identify the number of populations (K) capturing the data major structure, a burn-in period of 500,000 Markov Chain Monte Carlo iterations was used, with a 500,000-run length. In total, 20 independent runs were performed for each simulated K value, ranging from 1 to 9. All the iterations were run with the admixture model, which assumes that individuals may have mixed ancestry, because of the likelihood of inter-population and interspecific crossing among the four oaks. The optimal K value was identified from the maximum value of ΔK [75] as implemented using STRUCTURE HARVESTER 0.6.93 [76]. Clusters of 20 runs were permuted using CLUMPP, version 1.1.2 [77], and DISTRUCT 1.1 [78] was employed to envisage the STRUCTURE results after processing with CLUMPP. On the basis of the binary matrices, Nei’s unbiased genetic distance matrix was calculated using GENALEX 6.5 [71], and was employed to construct a dendrogram using MEGA 7.0 [79] with the neighbor-joining (NJ) clustering method [80].
The admixture coefficient (q-value) generated from STRUCTURE was used to classify individuals into purebreds and hybrids, with a threshold q-value of = 0.1, where samples with q-values < 0.1 or > 0.9 were classified as purebreds, and those with 0.1 < q-values < 0.9 as hybrids, including the F1 generation and backcrosses [81, 82]. F1 hybrids have q-values = 0.5, but the coefficients of backcrosses are biased toward one of the parental species and produce q-values between 0.5 and 0.9 [81]. Taking errors into consideration, individuals with 0.6 < q-values < 0.9 were recognized as backcrosses.
Availability of data and materials
The SSRs genotyping data from this study have been deposited in GitHub (https://github.com/Lixuan221/SSR-Database).
Abbreviations
- SSRs:
-
Simple sequence repeats
- AFLPs:
-
Amplified fragment length polymorphisms
- RAD-seq:
-
Restriction-site associated DNA sequencing
- SNPs:
-
Single nucleotide polymorphism
- N A :
-
Number of alleles
- N E :
-
Effective number of alleles
- A R :
-
Allelic richness
- H O :
-
Observed heterozygosity averaged across loci
- H E :
-
Expected heterozygosity averaged across loci
- H S :
-
Genetic diversity within populations averaged across loci
- PIC :
-
Polymorphism information content
- F ST :
-
Genetic differentiation index
- cF ST :
-
Corrected genetic differentiation index using the “exclusion null alleles” (ENA) method
- G' ST :
-
Standardized genetic differentiation index
- T A :
-
Annealing temperature
- null:
-
Null allele frequency averaged across populations
- DIC:
-
Deviance information criterion value
- n :
-
Model with null alleles
- f :
-
Inbreeding coefficients
- b :
-
Genotyping failures
References
Rushton BS. Natural hybridization within the genus Quercus L. Ann For Sci. 1993;50(Supplement):73s–90s.
Cannon CH, Manuel L. Variable mating behaviors and the maintenance of tropical biodiversity. Front Genet. 2015;6:183.
Goulet BE, Roda F, Hopkins R. Hybridization in plants: old ideas, new techniques. Plant Physiol. 2016;173(1):1340–2016.
Cannon CH, Scher CL. Exploring the potential of gametic reconstruction of parental genotypes by F1 hybrids as a bridge for rapid introgression. Genome. 2017;60(9):713–9.
Gailing O, Curtu AL. Interspecific gene flow and maintenance of species integrity in oaks. Ann For Res. 2014;57(1):5–18.
Valencia-Cuevas L, Piñero D, Mussali-Galante P, et al. Effect of a red oak species gradient on genetic structure and diversity of Quercuscastanea (Fagaceae) in Mexico. Tree Genet Genomes. 2014;10(3):641–52.
Chhatre VE, Evans LM, DiFazio SP, et al. Adaptive introgression and maintenance of a trispecies hybrid complex in range-edge populations of Populus. Mol Ecol. 2018;27(3):4820–38.
Larcombe MJ, Holland B, Steane DA, et al. Patterns of reproductive isolation in Eucalyptus - a phylogenetic perspective. Mol Biol Evol. 2015;32(7):1833–46.
deLafontaine G, Prunier J, Gérardi S, et al. Tracking the progression of speciation: variable patterns of introgression across the genome provide insights on the species delimitation between progenitor-derivative spruces (Piceamariana×P.rubens). Mol Ecol. 2015;24(20):5229–47.
Vasilyeva G, Bondar A, Goroshkevich S. What does a mixed population of Pinussibirica and P.pumila from the southern Baikal region suggest about the structure of their hybrid zone? Eur J Forest Res. 2020;139:311–9.
Crowl AA, Manos PS, McVay JD, et al. Uncovering the genomic signature of ancient introgression between white oak lineages (Quercus). New Phytol. 2020;226(4):1158–70.
Heredia ULD, Sánchez H, Soto A. Molecular evidence of bidirectional introgression between Quercussuber and Quercusilex. iForest Biogeosci Forestry. 2018;11:338–43.
González-Rodríguez A, Arias DM, Oyama K. Genetic variation and differentiation of populations within the Quercusaffinis – Quercuslaurina (Fagaceae) complex analyzed with RAPD markers. Can J Bot. 2020;83(2):155–62.
Amar MH, Magdy M, Wang L, et al. Peach chloroplast genome variation architecture and phylogenomic signatures of cpDNA introgression in Prunus. Can J Plant Sci. 2019;99(6):885–96.
Ortego J, Gugger PF, Sork VL. Genomic data reveal cryptic lineage diversification and introgression in Californian golden cup oaks (section Protobalanus). New Phytol. 2018;218(2):804–18.
He S, Wang P, Zhang Y, et al. Introgression leads to genomic divergence and responsible for important traits in upland cotton. Front Plant Sci. 2020;11:929.
Li Z, Tirado SB, Kadam DC, et al. Characterizing introgression-by-environment interactions using maize near isogenic lines. Theor Appl Genet. 2020. https://doi.org/10.1007/s00122-020-03630-z.
Aldrich PR, Cavender-Bares J. Genomics and breeding of oaks and their slightly less-domesticated wild oak relatives. In: Kole C, editor. Wild crop relatives: genomic and breeding resources. New York: Springer; 2011. p. 89–129.
Cavender-Bares J. Diversification, adaptation, and community assembly of the American oaks (Quercus), a model clade for integrating ecology and evolution. New Phytol. 2018;2(221):669–92.
Pettenkofer T, Burkardt K, Ammer C, et al. Genetic diversity and differentiation of introduced red oak (Quercusrubra) in Germany in comparison with reference native North American populations. Eur J Forest Res. 2019;138:275–85.
Tiange Lang PAVL. High-quality SNPs from genic regions highlight introgression patterns among European white oaks (Quercus petraea and Q. robur). bioRxiv preprint. 2018. https://doi.org/10.1101/388447.
Eaton DAR, Hipp AL, Gonzalez-Rodriguez A, et al. Historical introgression among the American live oaks and the comparative nature of tests for introgression. Evolution. 2015;69(10):2587–601.
An M, Deng M, Zheng SS, et al. Introgression threatens the genetic diversity of Quercusaustrocochinchinensis (Fagaceae), an endangered oak: a case inferred by molecular markers. Front Plant Sci. 2017;8:229.
Zhou Z, Wilkinson H, Wu Z. Taxonomical and evolutionary implications of the leaf anatomy and architecture of Quercus L. subgenus Quercus from China. Cathaya. 1995;7:1–34.
Pu C, Zhou Z, Luo Y. A cladistic analysis of Quercus (Fagaceae) in China based on leaf epidermic and architecture. Acta Bot Yunnanica. 2002;24(6):689–98.
Peng YS, Chen L, Li JQ. Study on numerical taxonomy of Quercus L. (Fagaceae) in China. J Wuhan Bot Res. 2007;25:149–57.
Denk T, Grimm GW, Manos PS, et al. An updated infrageneric classification of the oaks: review of previous taxonomic schemes and synthesis of evolutionary patterns. 2017. https://doi.org/10.1007/978-3-319-69099-5_2.
Huang Z. The fundamental features of zonal vegetation in Jiangsu province and its distribution patter. Acta Phytoecologica Sinica. 1982;6(3):237–46.
Leroy T, Roux C, Villate L, et al. Extensive recent secondary contacts between four European white oak species. New Phytol. 2017;214(2):865–78.
Beatty GE, Montgomery WI, Spaans F, et al. Pure species in a continuum of genetic and morphological variation: sympatric oaks at the edge of their range. Ann Bot-London. 2016;117(4):541–9.
Burger WC. The Species Concept in Quercus. Taxon. 1975;24(1):45–50.
Platt A, Gugger PF, Pellegrini M, et al. Genome-wide signature of local adaptation linked to variable CpG methylation in oak populations. Mol Ecol. 2015;24(15):3823–30.
Kim BY, Wei X, Fitz-Gibbon S, et al. RADseq data reveal ancient, but not pervasive, introgression between Californian tree and scrub oak species (Quercus sect. Quercus: Fagaceae). Mol Ecol. 2018;27(22):4556–71.
Nagamitsu T, Uchiyama K, Izuno A, et al. Environment-dependent introgression from Quercusdentata to a coastal ecotype of Quercusmongolica var. crispula in northern Japan. New Phytol. 2020;226(4):1018–28.
Chen X. Population genetics structure and phylogeography of Quercusfabri. Northwestern University. 2018.
Zhang H. Study on population genetics and demographic history of the three closely related species of Section Aegilops occurred in China. Northwestern University. 2018.
Li Yao, Zhang XW, Fang YM. Predicting the impact of global warming on the geographical distribution pattern of Quercusvariabilis in China. Chin J Plant Ecol. 2014;12:3381–9.
Zhang XW, Li Yao, Fang YM. Geographical distribution and prediction of potential ranges of Quercusacutissima in China. Acta Botanica Sinica. 2014;34(8):1685–92.
Li X, Li Y, Fang YM. Prediction of potential suitable distribution areas of Quercusfabri in China based on an optimized maxent model. Scientia Silvae Sinicae. 2018;54(8):153–64.
Yang YC. Plastid genomes of Quercus in China and the key genera in Fagaceae: comparative and phylogenetic analysis. Northwestern University. 2018.
Ramirez-Valiente JA, Deacon NJ, Etterson J, et al. Natural selection and neutral evolutionary processes contribute to genetic divergence in leaf traits across a precipitation gradient in the tropical oak Quercusoleoides. Mol Ecol. 2018;27(9):2176–92.
Hamilton JA, Miller JM. Adaptive introgression as a resource for management and genetic conservation in a changing climate. Conserv Biol. 2016;30(1):33–41.
Govindaraju DR. Variation in gene flow levels among predominantly self-pollinated plants. J Evolution Biol. 2010;2(3):173–81.
Gömöry D, Schmidtová J. Extent of nuclear genome sharing among white oak species (Quercus L. subgen. Lepidobalanus (Endl.) Oerst.) in Slovakia estimated by allozymes. Plant Syst Evol. 2007;266(3–4):253–64.
Tauber H. Investigations of aerial pollen transport in a forested area. Rev Palaeobotan Palynol. 1967;3(1-4):277–86.
Petrova EA, Zhuk EA, Popov AG, et al. Asymmetric introgression between Pinussibirica and Pinuspumila in the Aldan plateau (Eastern Siberia). Silvae Genet. 2018;67(1):66–71.
Dodd RS, Afzalrafii Z. Selection and dispersal in a multispecies oak hybrid zone. Evolution. 2004;58:261–9.
Peñalozaramírez JM, Gonzálezrodríguez A, Mendozacuenca L, et al. Interspecific gene flow in a multispecies oak hybrid zone in the Sierra Tarahumara of Mexico. Ann Bot-London. 2010;105(3):389–99.
Curtu AL, Gailing O, Leinemann L, et al. Genetic variation and differentiation within a natural community of five oak species (Quercus spp.). Plant Biol. 2007;9(1):116–26.
Schueler S, Schlünzen KH, Scholz F. Viability and sunlight sensitivity of oak pollen and its implications for pollen-mediated gene flow. Trees. 2005;19(2):154–61.
Mallet J. Hybridization as an invasion of the genome. Trends Ecol Evol. 2005;20(5):229–37.
Whitney KD, Ahern JR, Campbell LG, et al. Patterns of hybridization in plants. Perspect Plant Ecol. 2010;12(3):175–82.
Tigano A, Friesen VL. Genomics of local adaptation with gene flow. Mol Ecol. 2016;25(10):2144–64.
Suarez-Gonzalez A, Lexer C, Cronk QCB. Adaptive introgression: a plant perspective. Biol Letters. 2018;14(3):20170688.
Petit RJ. Biological invasions at the gene level. Divers Distrib. 2004;10(3):159–65.
Khodwekar S, Gailing O. Evidence for environment-dependent introgression of adaptive genes between two red oak species with different drought adaptations. Am J Bot. 2017;104(7):1088–98.
Stejskal J, Horák J, Typta J. Effect of hybridization in the firs: artificial hybridization may lead to higher survival rate. Eur J Forest Res. 2016;135:1097–105.
Isagi Y, Suhandono S. PCR primers amplifying microsatellite loci of Quercusmyrsinifolia Blume and their conservation between oak species. Mol Ecol. 1997;6(9):897–9.
Aldrich PR, Michler CH, Sun W, et al. Microsatellite markers for northern red oak (Fagaceae: Quercusrubra). Mol Ecol Notes. 2002;2(4):472–4.
Aldrich PR, Jagtap M, Michler CH, et al. Amplification of North American red oak microsatellite markers in European white oaks and Chinese chestnut. Silvae Genet. 2003;52(3–4):176–9.
Xiaolin Xu. Genetic diversity of microsatellites (SSRs) of natural populations of Quercusvariabilis. Hereditas. 2004;26(5):683–8.
Steinkellner H, Fluch S, Turetschek E, et al. Identification and characterization of (GA/CT)n-microsatellite loci from Quercuspetraea. Plant Mol Biol. 1997;33(6):1093–6.
Kampfer S, Lexer C, Glössl J, et al. Characterization of (GA)n microsatellite loci from Quercusrobur. Hereditas. 1998;129(2):183–6.
Chybicki IJ. Burczyk J Simultaneous estimation of null alleles and inbreeding coefficients. J Hered. 2009;100(1):106–13.
Rousset F. GENEPOP (version 4.0) GENEPOP’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour. 2008;8(1):103–6.
Yeh F, Yang R, Boyle T. POPGENE Version 1.32 Microsoft Windows-based freeware for populations genetic analysis. Edmonton: University of Alberta; 1999.
Marshall TC, Slate J, Kruuk LEB, et al. Statistical confidence for likelihood-based paternity inference in natural populations. Mol Ecol. 2010;7(5):639–55.
Goudet J. FSTAT (version 2.9.3) : A program to estimate and test gene diversities and fixation indices. 2001. www.unil.ch/izea/softwares/fstat.html
Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38(6):1358–70.
Chapuis MP, Estoup A. Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol. 2007;24(3):621–31.
Peakall R, Smouse PE. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research--an update. Bionformatics. 2012;28(19):2537–9.
Deng W, Wang Y, Liu Z, et al. HemI: a toolkit for illustrating heatmaps. PLoS ONE. 2014;9:e111988.
Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–7.
Pritchard JK, Stephens M, Donnelly P, et al. Inferences of population structure using multilocus genotype data. 2000.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters in individuals using the software STRUCTURE: a simlation study. Mol Ecol. 2005;14(8):2611–20.
Earl DA, VonHoldt BM. HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2012;4(2):359–61.
Jakobsson M, Rosenberg N. CLUMPP1: CLUster Matching and Permutation Program Version 1.1 Bioinformatics. 2007.
Rosenberg NA. Distruct: a program for the graphical display of population structure. Mol Ecol Notes. 2010;4(1):137–8.
Sudhir K, Glen S, Koichiro T. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Bio Evol. 2016;33(7):1870–4.
Nei M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics. 1978;89:583–90.
Lepais O, Petit RJ, Guichoux E, et al. Species relative abundance and direction of introgression in oaks. Mol Ecol. 2010;18(10):2228–42.
Vähä JP, Primmer CR. Efficiency of model-based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Mol Ecol. 2010;15(1):63–72.
Acknowledgements
We thank all the colleagues in our laboratory for help, advice, and discussion as well as the editor and reviewers for their constructive comments.
Funding
This research was supported by the National Natural Science Foundation of China (31770699), the State Scholarship Fund, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), Nanjing Forestry University Excellent Doctoral Thesis Fund (2171700124), and the Jiangsu Innovation Engineering Fund (KYCX18_0989). The funding bodies were not involved in the design of the study, collection, analysis, and interpretation of data, and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
X.L performed the writing of the manuscript and participated in the data analysis; G.W. performed most of the experiments, Y. F. and Y. A. E., supervised the project and provided suggestions for the manuscript. The authors have read and approved the manuscript. The authors declare that there is no conflict of interest.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare absence of competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
12870_2021_3007_MOESM1_ESM.docx
Additional file 1:Table S1. Deviance information criterion values for nfb, nb, and fb models in 10 populations obtained by INEST. Table S2. Probability of membership to each genetic cluster in each population of four oaks when K = 2 and 4. Table S3. Locations of 10 populations of four oak species. Table 4S.. Leaf macromorphological features examined. Table S5. SSR primer sets, number and size of alleles amplified in four oak species study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li , X., Wei, G., El-Kassaby, Y.A. et al. Hybridization and introgression in sympatric and allopatric populations of four oak species. BMC Plant Biol 21, 266 (2021). https://doi.org/10.1186/s12870-021-03007-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-021-03007-4