
Abstract
Background
Glycoalkaloids are bioactive compounds that contribute to the defence response of plants against herbivore attack and during pathogenesis. Solanaceous plants, including cultivated and wild potato species, are sources of steroidal glycoalkaloids. Solanum plants differ in the content and composition of glycoalkaloids in organs. In wild and cultivated potato species, more than 50 steroidal glycoalkaloids were recognized. Steroidal glycoalkaloids are recognized as potential allelopathic/phytotoxic compounds that may modify the growth of target plants. There are limited data on the impact of the composition of glycoalkaloids on their phytotoxic potential.
Results
The presence of α-solasonine and α-solamargine in potato leaf extracts corresponded to the high phytotoxic potential of the extracts. Among the differentially expressed genes between potato leaf bulks with high and low phytotoxic potential, the most upregulated transcripts in sample of high phytotoxic potential were anthocyanin 5-aromatic acyltransferase-like and subtilisin-like protease SBT1.7-transcript variant X2. The most downregulated genes were carbonic anhydrase chloroplastic-like and miraculin-like. An analysis of differentially expressed proteins revealed that the most abundant group of proteins were those related to stress and defence, including glucan endo-1,3-beta-glucosidase acidic isoform, whose expression level was 47.96× higher in potato leaf extract with low phytotoxic.
Conclusions
The phytotoxic potential of potato leaf extract possessing low glycoalkaloid content is determined by the specific composition of these compounds in leaf extract, where α-solasonine and α-solamargine may play significant roles. Differentially expressed gene and protein profiles did not correspond to the glycoalkaloid biosynthesis pathway in the expression of phytotoxic potential. We cannot exclude the possibility that the phytotoxic potential is influenced by other compounds that act antagonistically or may diminish the glycoalkaloids effect.
Similar content being viewed by others

Background
Allelopathy is a broadly understood phenomenon that refers to multidirectional interactions among organisms (plants, bacteria, viruses and fungi) that involve the release of compounds called allelochemicals into the environment [1]. Plant allelopathy between donor and acceptor plants is mainly negative in nature and impairs plant growth, development and/or germination. The ability to synthetize and release allelopathic compounds, especially in plant-plant and plant-pathogen interactions, is an important aspect of allelopathy since it determines plant survival and proper development during biotic stresses [2]. To distinguish allelopathy in ecosystems from research on allelopathic interactions in laboratory, term ‘phytotoxicity’ is used, describing negative interactions between donors and acceptors [1]. A lot of laboratory research focused on recognition of phytotoxic potential use water extracts as phytotoxic factor. Water extracts most closely resemble leaching of compounds from plant organs, that occurs under natural conditions (leaching by rain, dew) [3, 4].
An integral component of allelopathic interactions are allelochemicals that are secondary metabolites derived from three biosynthetic pathways: the shikimate, isoprenoid/mevalonate and polyketide pathways [5]. Based on their origin, secondary metabolites can be divided into three groups: phenylpropanoids, terpenoids, and polyketides. These compounds are distinguished from primary metabolites by their characteristic structure (unique carbon skeleton), which makes them specialized for responses to environmental conditions and biotic stresses. Secondary metabolites are involved in allelopathic interactions but principally in plant defence against pathogens [6].
Solanaceous species have been integral parts of human civilizations as food sources and drugs for thousands of years. All of the approximately 180 tuber-bearing Solanum species are indigenous to Latin America. They occur in a wide range of environmental conditions from Mexico in the north to Chile in the south and occupy various habitats [7]. The ability of wild potatoes to adapt and acclimatize to environmental conditions makes them a rich source of variability in biotic and abiotic stress resistance [8]. Steroidal glycoalkaloids, nitrogen-containing steroidal glycosides, are secondary metabolites that occur naturally in most plant organs of Solanum species [9]. They are biosynthesized by the sterol branch of the mevalonic acid/isoprenoid pathway [10]. There are many types of glycoalkaloids (GAs) in potato germplasm. Fifty-six GAs, including α-solanine, α-solasonine, α-solamargine, α-chaconine, and leptine II, were revealed in tubers of wild species and cultivated potato [11]. The GA content in tubers is markedly lower than that in potato leaves [12].
α-Solanine and α-chaconine possess antibacterial, fungicidal and insecticidal properties [13], can be classified as phytoalexins and are synthesized in response to pathogen infection [14]. Their biological activity depends mostly on their chemical structures, and α-chaconine is more active than α-solanine on the growth of fungi such as Alternaria Brassicicola, Phytophthora. medicaginis, and Rhizoctonia solani [15]. In our previous paper, we confirmed the potato phytotoxic potential (PP) against the test plant mustard (Sinapis alba L.), specie often used as aftercorp [16]. We demonstrated a significant role of total glycoalkaloid (TGA) content in the expression of PP among wild potato species and potato hybrids, and presented that some clones with low TGA content may possess PP.
For better understanding of processes/phenomena that may be directly or indirectly implicated with PP of potato under low TGA content, we used transcriptomic and proteomic approaches. It is known that allelopathic/phytotoxic interactions are determined directly by allelopathic compounds however, their content or profile in plant’s organs is regulated not only at the genetic level but also undergo coordination of metabolic pathways. Transcriptomic and proteomic studies may provide complementary knowledge to integrative metabolite profiling and better understanding of allelopathy/phytotoxicity phenomena at plant system level [17]. In the present study, we showed that potato PP under low TGA content is directly related with GAs composition.
Contrasting F1 individuals differing in PP and GAs from a cross of an interspecific Solanum hybrid with S. chacoense were used for comprehensive analyses of transcriptomic and proteomic profiles.
Results
Evaluation of total glycoalkaloid, glycoalkaloid, total phenolic and total flavonoid contents
Two bulk samples, C and D, that had similar total glycoalkaloid (TGA) contents (2.7 and 2.6 μg ml− 1) and various PP were used (Table 1). Both samples had similar concentrations of α-solanine (0.23 and 0.26 μg ml− 1) and α-chaconine (0.14 and 0.18 μg ml− 1) (Table 1). α-Solasonine was detected in only C at a concentration of 1.69 μg ml− 1 and consisted 40.8% of GAs, while the most frequent glycoalkaloid in D was leptine II (66.7%, at a concentration of 1.22 μg ml− 1). The total flavonoids (TF) were significantly higher in D, while total phenolics (TP) were at similar levels in both samples (Table 1). The average retention time and mass of GAs found in the samples are presented in Additional File 1, and mass spectra are presented in Additional File 2.
Profiles of differentially expressed gene and protein
GAs have been recognized as compounds with protective activity against pathogens, pests and herbivores. Recently, we confirmed that GAs present in potato leaf extract (PLE) exhibit PP. PLE of wild species and hybrids inhibited mustard growth, and TGA content was negatively correlated with mustard root and seedling length [16]. Based on this finding, we addressed the question of which factors/phenomena may play a significant role in the expression of PP under low TGA (2.7 μg ml− 1) content in PLE? We analysed the PP of potato in relation to gene and protein expression in potato leaves and GA content in PLE.
Leaf RNA of bulk samples C and D was analysed using the BGISEQ-500 (BGI, China) platform. The total number of raw reads ranged from 28,723,117 to 28,804,382. A total of 23,836 differentially expressed transcripts were identified between libraries of C and D. We analysed the most differentially regulated transcripts with false discovery rates (FDR-adjusted p-value < 0.05) as a threshold, finding 3125 up- and 3479 downregulated transcripts. The results for all differentially expressed genes (DEGs) after comparison of the D vs. C data are presented in Additional File 3. The top 10 most up- and downregulated transcripts are presented in Table 2. The most upregulated transcripts in C were anthocyanin 5-aromatic acyltransferase-like and subtilisin-like protease SBT1.7, transcript variant X2, with log2 fold changes (FCs) of 9.45 and 9.19, respectively. The most downregulated genes were carbonic anhydrase chloroplastic-like and miraculin-like, with log2 FCs of − 9.94 and − 9.28, respectively.
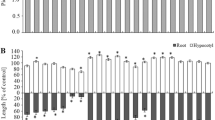
Liquid chromatography−mass spectrometry analysis of the samples C amd D resulted in 48,836 spectra that were linked to matched peptides, 2125 accessions, and annotated as 3054 proteins. All quantitatively abundant proteins are presented on Fig. 1. Thirty-three differentially expressed proteins (DEPs) were found using the established criteria (q-value< 0.05) (Table 3). The DEPs were divided into five categories according to the system of [18] and assigned based on the UniProt database: primary metabolism, amino acid metabolism, cell structure, protein turnover and stress and defence. The most abundant group of proteins were those related to stress and defence (12 proteins) with glucan endo-1,3-betaglucosidase acidic isoform, whose expression was 47.96× higher in D than in C. Among the DEPs, 3 were characteristic of C: in the category of primary metabolism, there were formamidase and non-symbiotic haemoglobin 2, and in the category of cell structure, there was tetratricopeptide repeat superfamily protein. For D, 3 DEPs were characteristic: 2 in the category cell structure (protein trichome birefringence-like, lysine histidine transporter 1) and 1 in stress and defence (basic endochitinase).

Potato leaf proteins that varied after comparison of bulks D vs. C. The volcano plot shows quantitative abundance of proteins. Only the blue and red dots possess the q-value < 0.05. Volcano plot shows quantitative abundance of proteins. Log-transformed q values (t-test) associated with individual peptides plotted against log-transformed fold change in abundance between the bulk C and D. Proteins that varied qualitatively (no. 1, 2, 13, 14, 15, 22) are not marked on volcano plot. Presented number of varied proteins correspond with protein’s numbers described in Table 2
Functional enrichment analysis
Based on the GO analysis, three functional groups were categorized: molecular function (MF), biological process (BP) and cellular component (CC). We identified 34 main GO categories, in three levels (16 GO terms in Biological process, 7 GO terms in Molecular function and 11 GO terms in Cellular component) (Fig. 2). In the MF group, the most significantly enriched GO terms were catalytic activity, binding followed by transporter activity. For the BP group, metabolic process, cellular process and response to stimulus were the GO terms most significantly enriched. With regard to CC, most enriched GO terms were cell, cell part and organelle. All unprocessed GO terms identified from the comparison of D vs. C are shown in Additional File 4.

Gene ontology (GO) analysis of potato D vs. C differentially expressed genes. The relative frequencies of GO hits for target genes assigned to the GO functional categories: cellular components, biological processes and molecular functions
Discussion
We previously demonstrate significant involvement of GAs in potato phytotoxic interactions. However, we observed that potato clones with low GAs content also possess phytotoxic abilities. We suspected that PP of clones with low GAs content is determined by various GAs composition. In this purpose we derived a diploid potato population with S. chacoense as a paternal donor of PP, to diminish effect of genetic background and selected clones of contrasting PP. S. chacoense is a potato wild relative and is known as a source of various steroidal glycoalkaloids, including leptine glycoalkaloids [19]. Depending on the S. chacoense accession a lot of glycoalkaloids can be found including leptine I and II, leptinine I and II, α-solanine, α- and β-chaconine, commersonine and demissine; other alkaloids as calystegines and clavepictine, or steroidal saponins as saponin, tuberoside, torvoside or graeculin [20,21,22,23]. In the present study, the low TGA content in C and D accompanied various GA patterns among the samples. Most frequently, the GAs α-solasonine and α-solamargine together may play a significant role in PP expression in C. Both compounds were previously recognized in ripe fruits of grey bitter-apple (Solanum incanum L.) as an inhibitors of lettuce (Lactuca sativa L.) root growth with comparable strength at a concentration of 100 mg dm− 3 [24]. Moreover, they may act synergistically when applied jointly in a 1:1 ratio, as was demonstrated on cucumber (Cucumis sativus L.) radicle growth when these two compounds were applied separately [25].
The biological activity of GAs was found to be strictly influenced by the configuration and occurrence of sugar moieties [26]. Thus, the composition of GAs may shape the expression and strength of PP. The phytotoxic effect of GAs and their derivatives on cucumber root growth increased with increasing concentration and was dependent on their type [25]. The strongest effect was observed for α-chaconine, which was followed in descending order by α-solanine, α-solamargine, α-solasonine, 6-O-sulfated chaconine and 6-O-sulfated solamargine. To our knowledge, there are no data regarding leptine II allelopathic abilities. The occurrence of foliar leptine glycoalkaloids is directly related to the degree of feeding deterrence of Colorado potato beetle (CPB) [20, 27]. Leptine II has the monoacetylated (C23) aglycone leptinidine. This chemical modification increases leptine toxicity against CPB and human tumour cell lines; however, its phytotoxicity has not yet been evaluated [20, 28].
Glycoalkaloid biosynthesis can be described as a series of actions of glycoalkyloid metabolism (GAME) genes that promote steroidal alkaloid (aglycone) yields [29]. Then, the aglycones undergo glycosylation by different UDP-glycosyltransferases to synthesize various GAs [30]. We found different expression patterns of various UDP-glycosyltransferases (see Additional File 5), especially UDP-glycosyltransferase 90A1 (log2 FC = -3.01), which confirmed different GA landscapes between the samples and may affect PP of PLE. It was recently demonstrated that the pattern of GAs in progeny derived from crosses with S. chacoense segregates and may be regulated by 38 genes that are located on chromosome VIII and coexpressed with GAME genes [31]. Comprehensive analysis of DEGs, the GO enrichment, pointing significant participation of metabolic processes (BP-30.9%) and catalytic activity (MF-62.2%) as most differentiating the bulks. This may suggest significant involvement of metabolic pathways in regulation of phytotoxic potential.
We cannot exclude the possibility that PP is influenced by other compounds that act antagonistically or may diminish the GAs effect. As was shown by [15], α-solanine applied together with gallic acid and quercetin diminished the inhibitory effect of α-solanine applied alone on mustard root growth. In the present study, D had a significantly higher TF content than C, which may influence the strength of the phytotoxic effect; however, this difference in concentration was slight.
We suggested that PP expression may be directly related to the qualitative and/or quantitative content of each GA recognized in PLE under low TGA content. Since GAs are involved in plant resistance events and the phytotoxicity of PLE, we assumed that PP of C will be accompanied by increased expression of genes and proteins involved in plant resistance to biotic stress. However, comparison of gene and protein expression profiles in D vs. C did not provide definite evidence to support this concept. Patterns of DEGs involved in the defence response, e.g., endopeptidases, were found, but they were shared between samples. Notably, a gene whose expression is increased in C encodes trans-resveratrol di-O-methyltransferase-like (pterostilbene synthase, EC 2.1.1.240), an enzyme responsible for synthesis of stilbenes, a group of phytoalexins. Most DEGs for this enzyme, which were positioned on different loci, were upregulated in C (see Additional File 3). Transgenic tobacco (Nicotiana tabacum L.) lines overexpressing stilbene synthase exhibited increased synthesis of phytoalexins, which was accompanied by resistance to Botrytis cinerea infection [32].
The protein profiles of D vs. C do not correspond to gene expression data. The most numerous group of DEPs was involved with stress and defence responses in D. An extremely high fold change was noted for glucan endo-1,3-β-glucosidase acidic isoform (47.96). Plant glucan endo-1,3-β-glucosidases are enzymes that can participate in resistance against pathogens and degradation of cell wall components (β-glucans) during plant development [33, 34]. However, their importance in pathogenesis was debated, since antisense transformants of N. sylvestris and tobacco with β-1,3-glucanase deficiency could compensate for the activity of this enzyme by producing a functionally equivalent replacement during infection by tobacco mosaic virus [35].
Protein abundance is controlled by variation at the coding gene itself and by variation mapping to other regions of the genome [36]. Protein synthesis is regulated at many levels, ranging from splicing and mRNA degradation to protein modification to ubiquitination and proteolysis in proteasomes [37]. Therefore, it is not surprising that transcriptome data did not correspond to proteomic profiles (Table 2 and Table 3). We found one DEG, threonine dehydratase biosynthetic (FC = 1.37), whose protein expression is significantly increased in D. Threonine dehydratase, also called threonine deaminase, is an enzyme for isoleucine biosynthesis [38] and acts together with protease inhibitors in the herbivore gut, decreasing dietary proteins and threonine availability. In Solanum species, the threonine dehydratase gene has a duplicated paralogue that is coexpressed with genes engaged in herbivore resistance. In lupin (Lupinus sp. L.), a high content of threonine was accompanied by an increased level of alkaloids [39]. On the other hand, a higher level of threonine induced the synthesis of alkaloid-pilocarpine in Pilocarpus vahl [40], is a good precursor of pyrrolizidine alkaloids in Senecio douglasii and participates in the direct biosynthesis of strigosine [41]. One of the proteins characterized for only C was formamidase (EC 3.5.1.49), which is engaged in nitrogen metabolism [42]. The gene encoding this enzyme was also recognized as drought-responsive and regulated by abscisic acid (ABA) [43]. Both enzymes may participate indirectly in GA synthesis due to their involvement in the metabolism of nitrogen, which is inserted into the aglycone skeleton.
Conclusion
Complex analysis of GAs, together with trancriptomic and proteomic results provided new insight in understanding the expression of phytotoxic abilities of potato. We demonstrated that PP expression under low TGA content may be connected with the GAs composition in PLE. We pointed potential involvement of metabolic pathways (GO enrichment) in direct regulation of GAs biosynthesis and accumulation (genes of GAs main biosynthesis pathway) or indirect regulation by other factors/phenomena, e.g., nitrogen metabolism, and is a result of plant reactions to biotic and abiotic stresses. Comparison of transcriptomic with proteomic revealed gene and protein of threonine dehydratase bisynthetic as common element bonding these two omics and important in expression of potato phytotoxic abilities.
Methods
Plant material
Plant material consisted of the potato diploid population 15–1 (F1 progeny, N = 166) from a cross of Solanum hybrid DG 88–89 (seed parent) and a wild species S. chacoense (pollen parent). The maternal clone was generated within the diploid potato breeding program at the Plant Breeding and Acclimatization Institute—National Research Institute, Młochów, Poland. Paternal specie was obtained from National Centre for Plant Genetic Resources, Radzików, Poland (accession POL003:333133). DG 88–89 was the multigenerational hybrid originating from crosses of diploid potato clones. In terms of the genomes, the percentage of S. tuberosum in DG 88–89 was 78.3% and that of S. chacoense was 15.7%. Maternal clone and paternal species differ with TGA concentration in the PLE and PP. DG 88–89 exhibited a low TGA concentration (5.2 μg ml− 1) and nondetectable PP, while S. chacoense had a high TGA concentration (55.6 μg ml− 1) and high PP (70%) [15]. Three replicates of each of the progeny were grown in a greenhouse from May to October 2016. In full anthesis, leaflets were collected, mixed, portioned into 0.5 and 1 g portions, frozen in liquid nitrogen and stored until use at − 80 °C.
Preparation of potato leaf extract
PLE was prepared as previously described by [16]. Briefly, 0.5 g of frozen leaves was ground in liquid nitrogen, supplemented with 50 ml of distilled water (1% w/v) and shaken for 24 h on a laboratory shaker. Freshly prepared and filtered extract was used for biochemical and mass spectrometry (MS) analysis.
Evaluation of total glycoalkaloid, total phenolic and total flavonoid contents in potato leaf extract
The TGA concentration was measured for all 166 individuals in 2016 using the colorimetric method by [44] with modification as described by [16]. Briefly, 1% PLE was concentrated fourfold in a vacuum rotary evaporator (SpeedVac Appligene Refrigerated Aspirator, Germany). TGA was extracted using 10% acetic acid and precipitated with 5 M ammonium hydroxide. Samples were suspended in 100% methanol. The colour reaction was carried out using 98% sulfuric acid and 1% paraformaldehyde.
Absorbance was measured on a Hitachi U-1900 (Japan) spectrophotometer at a wavelength of 562 nm against a blank sample. The concentration of TGA was expressed in equivalents of α-solanine (Sigma-Aldrich, S3757).
TP content was determined as in [16] with a modification of the method described by [45]. Analysis was performed on twofold-concentrated PLE using freshly prepared tenfold-diluted Folin–Ciocalteu reagent (Sigma, F9252) and 7.5% sodium carbonate (w/v), and the absorbance was measured at 760 nm against a blank sample. The concentration of TP was expressed as equivalents of gallic acid (PhytoLab, 89,198).
TF was determined as in [16] with a modified method described by [46]. Analysis was performed on twofold-concentrated PLE using 10% aluminium chloride (w/v) and 1 M potassium acetate, and the absorbance was measured at 415 nm against a blank sample. The concentration of TF was expressed as equivalents of quercetin (PhytoLab, No. 89262).
TGA, TP and TF concentrations in selected genotypes were measured in 2016–2018. All measurements were performed in three biological repetitions, and each repetition had two technical replicates.
Evaluation of phytotoxic potential of potato leaf extract
PP was measured against the test plant - mustard cv. Rota (Vera-Agra Breeding Company, Cieszków, Poland) in three biological repetitions for all 166 individuals in 2016 and for the selected plants in 2017–2018 as previously described by [16]. Briefly, 15 mustard seeds after radicle protrusion (appx. 3 mm long) were transferred into Petri dishes (square, 12 cm) filled with filter paper and moisture with distilled water or 1% PLE (PLE-treated plants). After 5 days of incubation, the lengths of the control and PLE-treated mustard seedlings were measured. PP was expressed in % as the degree of seedling length inhibition/stimulation in relation to the length of control plants (grown in water) according to the formula
Construction of bulk samples
Based on TGA concentration and PP (Additional File 6), bulk samples C and D were constructed, each with three biological replications. Bulked sample analysis allows for more effective identification of genes underlying a trait. In this approach, contrasting individuals from a segregating population are pooled and then commonly screened to identify specific markers [47]. Sample C exhibited low TGA content (2.7 μg ml− 1 in PLE) and high PP (40%); D exhibited a low TGA concentration (2.6 μg ml− 1 in PLE) and nondetectable PP (PLE-treated plants were the same length as the control). In each sample, equal amounts of frozen leaves in liquid nitrogen from three F1 individuals were ground, mixed together and stored at − 80 °C.
RNA isolation and RNA-seq analysis
RNA was isolated from samples C and D according to the protocol described in [48] using TRIzol reagent. Briefly, 0.1 g of tissue ground in liquid nitrogen was supplemented with 1 ml of TRIzol reagent. Extraction was performed twice in chloroform. The RNA was precipitated in 0.3 ml of salt solution (0.8 M sodium citrate and 1.2 M sodium chloride) and 0.3 ml of isopropanol and resuspended in sterile water. The quality and quantity of RNA were determined using a NanoDrop spectrophotometer (Thermo Scientific) at 260 nm and 280 nm and on a 2% agarose gel. Next, RNA was treated with DNase I (Thermo Scientific, EN0521) to degrade double-stranded and single-stranded DNA contaminants in RNA samples.
The mRNA was isolated using the Dynabeads® mRNA Purification Kit for mRNA enrichment (Ambion, 61,006), and a library was prepared using the MGIEasy RNA Directional Library Prep Set (MGI, 1000006386), both according to the manufacturer’s protocols.
The established cDNA libraries were sequenced on the BGISEQ-500 sequencing platform (BGI Genomics, China) to generate 100-bp paired-end reads. RNA-seq reads were generated by Genomed® (Warsaw, Poland). After filtering of adaptor sequences and low-quality reads, data were obtained for subsequent analysis. Then, the index of the reference genome (https://www.ncbi.nlm.nih.gov/assembly/GCF_000226075.1) was built using Bowtie v2.1.0, and clean reads obtained for samples C and D were aligned to the reference genome using TopHat v2.0.9 (Broad Institute, Boston, MA). Next, HTSeq v0.5.3 was used to count the number of reads mapped to each gene. The DEGs were identified by the DESeq package.
Analysis of gene ontology term enrichment
To study the biological functions of the DEGs, gene set enrichment with GO terms was performed using the topGO package. To extract the significant GO categories, Fisher’s exact test was performed with the elim algorithm. To prepare circle diagram of all significant GO terms, we used as query for finding the ontology in various functional categories on the basis of GOslim categories as:
Analysis of the glycoalkaloids profile in potato leaf extract
The GA fraction was isolated from 1% PLE of bulked samples C and D using a solid-phase extraction method (QuEChERS). First, for each PLE sample, α-solamarine (ChemFaces, CFN93102) dissolved in methanol was added as an internal standard to a final concentration of 10 ng μl− 1 to calculate the percentage of recovery of GA. In the control sample, α-solamarine was added to distilled water to the same final concentration. Both types of samples (control and PLE) were passed through sterilizing filters (0.2 μm, Nalgene™). To 750 μl of a sample, an equal amount of acetonitrile (ACN) with 1% formic acid was added, and the sample was applied onto the solid phase of QuEChERS (UTC, ECQUCHL12CT) and shaken for 30 s on a vortex mixer. Then, the supernatant obtained after GA isolation was diluted 10-fold with methanol. HPLC-MS analysis was performed on a Dionex 3000 RS-HPLC equipped with a DGP-3600 pump, a WPS-3000 TLS TRS autosampler, a TCC-3000 RS column compartment (Dionex Corporation, USA) and a Bruker micrOTOF-QII mass spectrometer (Bruker Daltonics, Germany). The chromatography column was a 50 × 3.1 (i.d)-millimetre Thermo Scientific Hyperil GOLD with 1,9-μm particles (Part No. 25002–052130, Serial No. 0110796A6, Lot No. 10922).
Chromatographic conditions: For the mobile phase, solvent A was water, and solvent B was ACN. The flow program was as follows: 0 min – 5% solvent B; 1.4 min – 5% solvent B; 22.9 min – 95% solvent B; 24.4 min – 95% solvent B; 24.5 min – 5% solvent B; 29 min – 5% solvent B. The injection sample volume was 1.5 μL. The flow rate was 0.2 ml min− 1, and the eluent was monitored by MS. The analysis was performed in negative ESI mode. Scan range: 50–1500 m/z, end plate offset: − 500 V, capillary: 4500 V, nebulizer gas (N2): 1,2 bar, dry gas (N2): 10 L/min, dry temperature: 220 °C.
Qualitative analysis of GA content was performed using the frequency of each compound in the TGA found in the sample. The qualitative GA profile was calculated using the following formula:
where
F – Frequency of compounds
An – Area of analysis compound
AAll – Area of all GAs in the sample.
Quantitative analysis was performed using the GA standards α-solanine (ChemFaces, CFN90560), α-chaconine (ChemFaces, CFN00450), α-solamargine (ChemFaces, CFN90159), and α-solasonine (PhytoLab 83,271). To quantify the compounds, calibration curves for each GA standard were generated over the concentration range of 0.1 μg ml− 1 to 10 μg ml− 1. For leptine II, a curve for α-solanine was used due to the lack of a standard. The results are expressed in μg ml− 1 and take into account the percent recovery of each compound in relation to the control sample concentration.
Protein extraction
Proteins were isolated from C and D as described in [49] with a minor modification. Then, 0.1 g of powdered tissue was suspended in 350 μl of the extraction buffer and incubated on ice for 30 min. Then, phenol solution (Roti®-Aqua-Phenol) was added in a 1:1 (v/v) ratio and incubated at room temperature for 10 min. The phenol phase was recovered twice by centrifugation at 4 °C, transferred to new tubes with extraction buffer 1:1 (v/v) and precipitated in cold methanol containing 0.1 M ammonium acetate 1:4 (v/v). The mixture was incubated overnight at − 20 °C and centrifuged at room temperature. The liquid phase was removed, and the pellet was washed once with 100% methanol pre-chilled to − 20 °C, centrifuged with 80% acetone, and centrifuged at the highest speed. The final protein pellet was air-dried and dissolved in 200 μl of 25 mM ammonium bicarbonate. The sample protein content was determined according to the method described by [50] using the bicinchoninic acid assay and bovine serum albumin as a standard. Five independent biological replicates were analysed in this study. A total of 120 μg of protein from each probe was sent to the Mass Spectrometry Laboratory at the Institute of Biochemistry and Biophysics, Polish Academy of Sciences (Warsaw, Poland), for nano-LC-MS-MS/MS (nanoliquid chromatography coupled to tandem mass spectrometry) analysis.
Comparative analysis of differentially expressed proteins
Peptide mixtures were analysed by nano-LC-MS-MS/MS using a nano-Acquity (Waters) LC system and a Q-Exactive mass spectrometer (Thermo Electron Corp., San Jose, CA) with the same equipment, buffers and parameters like in [51]. The raw data were processed by Mascot Distiller followed by Mascot Search (Matrix Science, London, UK, on-site licence) against the UniProt Solanum tuberosum database (February 2018 release). The search parameters for precursor and product ion mass tolerances were 30 ppm and 0.1 Da, respectively enzyme specificity: trypsin and missed cleavage sites. Peptides with Mascot scores exceeding the threshold value corresponding to < 5% of the expectation value as calculated by the Mascot procedure were considered positively identified. Quantitative analysis was performed as described by [52]. Shortly, the mass calibration and data filtering described above were carried out like in [51]. At the end, lists of identified peptides with corresponding abundances were exported for statistical analysis carried out with in-house developed Diffprot Software (version 1.5.19; 3.01.2013) [53]. Prior to analysis, abundances were normalized with LOWESS. Proteins with more than 90% common peptides were clustered, and only peptides unique for the cluster were used for statistical analysis. Only proteins with q-values ≤0.05 were considered differentially expressed.
Availability of data and materials
All data analysed in this study are included in this published article and its supplementary files. All raw and processed RNA-seq data have been deposited in the Gene Expression Omnibus [GEO] repository under the link [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE155583] and Sequence Read Archive [SRA] repository under the link [https://www.ncbi.nlm.nih.gov/bioproject/PRJNA650400].
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Change history
11 April 2022
A Correction to this paper has been published: https://doi.org/10.1186/s12870-022-03574-0
Abbreviations
- CPB:
-
Colorato Potato Beetle
- DEGs:
-
differentially expressed genes
- DEPs:
-
differentially expressed proteins
- GAME:
-
glycoalkaloids metabolism genes
- GO:
-
gene ontology
- PLE:
-
potato leaf extract
- PP:
-
phytotoxic potential
- GAs:
-
glycoalkaloids (measured by mass spectrometry)
- TF:
-
total flavonoids
- TGA:
-
total glycoalkaloids (measured colorimetrically)
- TP:
-
total phenolics
References
Macias FA, Marin D, Oliveros-Bastidas A, Varela RM, Simonet AM, Carrera C, et al. Allelopathy as a new strategy for sustainable ecosystems development. Biol Sci Space. 2003;17:18–23.
Trezzi MM, Vidal RA, Balbinot AA, Bittencourt H von H, Souza Filho APS. Allelopathy: driving mechanisms governing its activity in agriculture. J Plant Int 2016;11:53–60.
Gniazdowska A, Bogatek R. Allelopathic interactions between plants. Multi site action of allelochemicals. Acta Physiol Plant. 2005;27:395–407.
Findura P, Hara P, Szparaga A, Kocira S, Czerwińska E, Bartoš P, et al. Evaluation of the effects of allelopathic aqueous plant extracts, as potential preparations for seed dressing, on the modulation of cauliflower seed germination. Agriculture. 2020;10:122.
Böttger A, Vothknecht U, Bolle C, Wolf A. Plant secondary metabolites and their general function in plants. In: Lessons on caffeine, cannabis & co. Learning Materials Biosci. (eds). Cham: Springer; 2018. p. 3–17. http://doiorg-443.webvpn.fjmu.edu.cn/10.1007/978-3-319-99546-5_1.
Zaynab M, Fatimab M, Abbasc S, Sharifd Y, Umairc M, Zafare MH, et al. Role of secondary metabolites in plant defense against pathogens. Microb Pathog. 2018;124:198–202.
Gebhardt C. The historical role of species from the Solanaceae plant family in genetic research. Theor Appl Genet. 2016;129:2281–94.
Castañeda-Álvarez NP, de Haan S, Juárez H, Khoury CK, Achicanoy HA, Sosa CC, et al. Ex situ conservation priorities for the wild relatives of potato (Solanum L. section Petota). PLoS One. 2015;10:e0122599.
Friedman M, McDonald GM. Potato glycoalkaloids: chemistry, analysis, safety, and plant physiology. Crit Rev Plant Sci. 1997;16:55–132.
Morillo M, Rojas J, Lequart V, Lamarti A, Martin D. Natural and synthetic derivatives of the steroidal glycoalkaloids of Solanum genus and biological activity. Nat Prod Chem Res. 2020;8:371.
Shakya R, Navarre DA. LC-MS analysis of solanidane glycoalkaloid diversity among tubers of four wild potato species and three cultivars (Solanum tuberosum). J Agric Food Chem. 2008;56:6949–58.
Smith DB, Roddick JG, Jones JL. Potato Glycoalkaloids: some unanswered questions. Trends Food Sci Technol. 1996;7:126–31.
Siddique MAB, Brunton N. Food Glycoalkaloids: distribution, structure, cytotoxicity, extraction, and biological activity. In: Alkaloids - their importance in nature and human life; 2019. p. 13–5.
Jeandet P, Clément C, Courot E, Cordelier S. Modulation of phytoalexin biosynthesis in engineered plants for disease resistance. J Mol Sci. 2013;14:14136–70.
Fewell AM, Roddick JG. Interactive antifungal activity of the glycoalkaloids α-solanine and α-chaconine. Phytochem. 1993;33:323–8.
Sołtys-Kalina D, Murawska Z, Strzelczyk-Żyta D, Wasilewicz-Flis I, Marczewski W. Phytotoxic potential of cultivated and wild potato species (Solanum sp.): role of glycoalkaloids, phenolics and flavonoids in phytotoxicity against mustard (Sinapis alba L.). Acta Physiol Plant. 2019;41:55.
Fukushima A, Kusano M, Redestig H, Arita M, Saito K. Integrated omics approaches in plant systems biology. Curr Op Chem Biol. 2009;13:532–8.
Alexander MM, Cilia MA. Molecular tug-of-war: global plant proteome changes during viral infection. Curr Plant Biol. 2016;5:13–24.
Veilleux R, Miller AR. Hybrid breakdown in the F1 between Solanum chacoense and S. phureja and gene transfer for leptine biosynthesis. J Am Soc Hortic Sci. 1998;123:854–8.
Sinden SL, Lind LS, Deahl KL. Segregation of leptine glycoalkaloids in Solanum chacoense bitter. J Agric Food Chem. 1986;34:372–7.
Deahl KL, Sinden SL, Young RJ. Evaluation of wild tuber-bearing Solanum accessions for foliar glycoalkaloid level and composition. Am Pot J. 1993;70:61–9.
Mweetwa AM, Hunter D, Poe R, Harich KC, Ginzberg I, Veilleux RE, Tokuhisa JG. Steroidal glycoalkaloids in Solanum chacoense. Phytochemistry. 2012;75:32–40.
Joshi JR, Yao L, Charkowski AO, Heuberger AL. Metabolites from wild potato inhibit virulence factors of the soft rot and blackleg pathogen Pectobacterium brasiliense. MPMI. 2020; https://doi.org/10.1094/MPMI-08-20-0224-R.
Fukuhara K, Kubo I. Isolation of steroidal glycoalkaloids from Solanum incanum by two countercurrent chromatographic methods. Phytochem. 1991;30:485–687.
Sun F, Li S, He D, Cao G, Ni X, Tai G, et al. Effects of glycoalkaloids from Solanum plants on cucumber root growth. Phytochem. 2010;71:1534–8.
Chowański S, Adamski Z, Marciniak P, Rosiński G, Büyükgüzelet E, Büyükgüzel K, et al. A review of bioinsecticidal activity of Solanaceae alkaloids. Toxins (Basel). 2016;8:60.
Yencho GC, Kowalski SP, Kennedy GG, Sanford LL. Segregation of leptine glycoalkaloids and resistance to Colorado potato beetle (Leptinotarsa decemlineata (Say)) in F2 Solanum tuberosum (4x) ×S. chacoense (4x) potato progenies. Am J Pot Res. 2000;77:167.
Schreiber K. Steroid alkaloids, the Solanum group. In: The Alkaloids. Vol. X, Manske RHF, Holmes HL. (Eds). New York: AcademicPress; 1968. p. 1–192.
Itkin M, Heinig U, Tzfadia O, Bhide AJ, Shinde B, Cardenas PD, et al. Biosynthesis of antinutritional alkaloids in solanaceous crops is mediated by clustered genes. Science. 2013;341:175–9.
Nakayasu M, Umemoto N, Ohyama K, Fujimoto Y, Lee HJ, Watanabe B, et al. A dioxygenase catalyzes steroid 16α-hydroxylation in steroidal glycoalkaloid biosynthesis. Plant Physiol. 2017;175:120–33.
Peng Z, Wang P, Tang D, Shang Y, Li C-H, Huang S-W, et al. Inheritance of steroidal glycoalkaloids in potato tuber flesh. J Integr Agric. 2019;18:2255–63.
Hain R, Reif HJ, Krause E, Langebartels R, Kindl H, Vornam B, et al. Disease resistance results from foreign phytoalexin expression in a novel plant. Nature. 1993;361:153–6.
Bell AA. Biochemical mechanisms of disease resistance. Annu Rev Plant Physiol. 1981;32:21–81.
Morohashi Y, Matsushima H. Development of β-1,3-glucanase activity in germinated tomato seeds. J Exp Bot. 2000;51:1381–7.
Beffa RS, Neuhaus JM, Meins F Jr. Physiological compensation in antisense transformants: specific induction of an "ersatz" glucan endo-1,3-fB-glucosidase in plants infected with necrotizing viruses. Proc Natl Acad Sci U S A. 1993;19:8792–6.
Foss EJ, Radulovic D, Shaffer SA, Ruderfer DM, Bedalov A, Goodlett DR, et al. Genetic basis of proteome variation in yeast. Nat Genet. 2007;39:1369–75.
Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13:227–32.
Gonzales-Vigila E, Bianchetti CM, Phillips GN Jr, Howea GA. Adaptive evolution of threonine deaminase in plant defense against insect herbivores. PNAS. 2011;108:5897–902.
Wasilewko J, Buraczewska L. Chemical composition including content of amino acids, minerals and alkaloids in seeds of three lupin species cultivated in Poland. J Anim Feed Sci. 1999;8:1–12.
Sawaya ACHF, Abreu IN, Andreazza NL, Eberlin MN, Mazzafera P. Pilocarpine and related alkaloids in Pilocarpus Vahl (Rutaceae). In: Alkaloids: Properties, applications and pharmacological effects; 2010. p. 63–80.
Robinson T. Some miscellaneous alkaloids. In: The biochemistry of alkaloids; 1981. p. 145–7.
Fraser JA, Davis MA, Hynes MJ. The formamidase gene of Aspergillus nidulans: regulation by nitrogen metabolite repression and transcriptional interference by an overlapping upstream gene. Genetics. 2000;157:119–31.
Huang D, Weiren W, Abrams SR, Cutler AJ. The relationship of drought-related gene expression in Arabidopsis thaliana to hormonal and environmental factors. J Exp Bot. 2008;59:2991–3007.
Andreu A, Oliva C, Distel S, Daleo G. Production of phytoalexins, glycoalkaloids and phenolics in leaves and tubers of potato cultivars with different degrees of field resistance after infection with Phytophthora infestans. Am J Potato Res. 2001;44:1–9.
Maurya S, Singh D. Kr., quantitative analysis of total phenolic content in Adhatoda vasica Nees extracts. Int J pharm tech. Res. 2010;2:2403–6.
Chang CC, Yang MH, Wen HM, Chern JC. Estimation of total flavonoid content in propolis by two complementary colorimetric methods. J Food Drug Anal. 2002;10:178–82.
Zou C, Wang P, Xu Y. Bulked sample analysis in genetics, genomics and crop improvement. Plant Biotech J. 2016;14:1941–55.
Chomczyński P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–9.
Szajko K, Strzelczyk-Żyta D, Marczewski W. Comparison of leaf proteomes of potato (Solanum tuberosum L.) genotypes with ER- and HR-mediated resistance to PVY infection. Eur J Plant Pathol. 2018;150:375–85.
Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–8.
Lebecka R, Kistowski M, Debski J, Szajko K, Murawska Z, Marczewski W. Quantitative proteomic analysis of differentially expressed proteins in tubers of potato plants differing in resistance to Dickeya solani. Plant Soil. 2019;441:317–29.
Bakun M, Niemczyk M, Domanski D, Jazwiec R, Perzanowska A, Niemczyk S, et al. Urine proteome of autosomal dominant polycystic kidney disease patients. Clin Proteomics. 2012;9:13.
Malinowska A, Kistowski M, Bakun M, Rubel T, Tkaczyk M, Mierzejewska J, et al. Diffprot – software for non-parametric statistical analysis of differential proteomics data. J Proteome. 2012;75:4062–73.
Acknowledgements
The equipment used in the Mass Spectrometry Laboratory (Institute of Biochemistry and Biophysics, Polish Academy of Sciences) was sponsored in part by the Centre for Preclinical Research and Technology (CePT), a project co-sponsored by European Regional Development Fund and Innovative Economy, The National Cohesion Strategy of Poland.
We are grateful to Agata Malinowska and Bianka Świderska from Institute of Biochemistry and Biophysics, Mass Spectrometry Laboratory, Polish Academy of Sciences, for the preparation of Figure 1.
Funding
The research was financially supported by grants from The National Science Center in Poland, (UMO-2016/23/D/NZ9/02672). The funding organizations provided the financial support to the research projects, but were not involved in the design of the study, data collection, analysis of the data, or the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
KS performed most of the experiments, JC performed quantitative analysis of GA, IW-S provide and collected plant material, WM contributed useful suggestions and wrote part of the manuscript, DSK designed and conducted experiments, wrote the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: authors would like to correct the Acknowledgement section.
Supplementary Information
Additional file 1: Supplementary Table S1.
Average retention time (RT) and mass of compounds found in samples D and C.
Additional file 2: Supplementary Figure S1.
Mass spectra of GAs found in samples C and D.
Additional file 3: Supplementary Table S2.
All DEGs recognized after comparison D vs. C bulks, sorted by log2 FC.
Additional file 4: Supplementary Table S3.
GO enrichement of DEGs in molecular function, biological processes and cellular compartments after comparison D vs C.
Additional file 5: Supplementary Table S4.
Expression pattern of UDP-glucosytransferases and other genes engaged in GAs biosynthesis between the bulks D vs. C, sorted by Log2 FC.
Additional file 6: Supplementary Table S5.
TGA content and PP in individuals of 15-1 population.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Szajko, K., Ciekot, J., Wasilewicz-Flis, I. et al. Transcriptional and proteomic insights into phytotoxic activity of interspecific potato hybrids with low glycoalkaloid contents. BMC Plant Biol 21, 60 (2021). https://doi.org/10.1186/s12870-021-02825-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-021-02825-w