
Abstract
Background
The WRKYs are a major family of plant transcription factors that play roles in the responses to biotic and abiotic stresses; however, a comprehensive study of the WRKY family in roses (Rosa sp.) has not previously been performed.
Results
In the present study, we performed a genome-wide analysis of the WRKY genes in the rose (Rosa chinensis), including their phylogenetic relationships, gene structure, chromosomal locations, and collinearity. Using a phylogenetic analysis, we divided the 56 RcWRKY genes into three subgroups. The RcWRKYs were unevenly distributed across all seven rose chromosomes, and a study of their collinearity suggested that genome duplication may have played a major role in RcWRKY gene duplication. A Ka/Ks analysis indicated that they mainly underwent purifying selection. Botrytis cinerea infection induced the expression of 19 RcWRKYs, most of which had undergone gene duplication during evolution. These RcWRKYs may regulate rose resistance against B. cinerea. Based on our phylogenetic and expression analyses, RcWRKY41 was identified as a candidate regulatory gene in the response to B. cinerea infection, which was confirmed using virus-induced gene silencing.
Conclusions
This study provides useful information to facilitate the further study of the function of the rose WRKY gene family.
Similar content being viewed by others


Background
Transcription factors play crucial roles in plant growth, development, metabolism, and stress responses. Transcription factors usually possess a DNA-binding domain, a transactivation domain, an oligomerization site, and a nuclear localization signal, among other domains. The WRKYs are one of the most important transcription factor families in plants. These proteins all possess at least one WRKY domain [1], a DNA-binding domain which binds the W-box (TTGACC/T) sequence of the promoter region in their target genes to regulate their expression. In addition, the C-terminal of the WRKY transcription factors usually contains a zinc finger structure. In Arabidopsis thaliana, the WRKY protein family can be divided into three different groups: Group I proteins contain two WRKY domains and Group II WRKYs contain only one WRKY domain. Group III proteins also possess a single WRKY domain, but their zinc finger structure is unique from those of the other two groups [2].
WRKY transcription factors participate in the regulation of various plant processes, including growth and development, the response to abiotic stresses, and disease resistance; for example, AtWRKY45 is involved in the regulation of plant leaf senescence through the gibberellin signaling pathway [3]. In rice (Oryza sativa), OsWRKY53 positively regulates brassinosteroid signals to influence the plant architecture [4]. The expression of AtWRKY22 in Arabidopsis is strongly induced by submergence during flooding, and its protein product binds to the promoter of TREHALASE1, which involved in stomatal function, to inhibit its expression [5]. AtWRKY8 is highly expressed in plant roots and is significantly upregulated under salt stress, with atwrky8 knockout mutants showing a greater sensitivity to salt [6]. The WRKYs are also key players in plant resistance responses against pathogens; for example, AtWRKY33 is activated by a MAPK signaling pathway and regulates the biosynthesis of phytoalexin to enhance pathogen resistance in Arabidopsis [7]. By contrast, AtWRKY38 and AtWRKY62 encode two structurally similar WRKYs that negatively regulate the defense against Pseudomonas syringae; the overexpression of these two genes decreased plant resistance to this pathogen, and the atwrky38, atwrky62, and atwrky38 atwrky62 loss-of-function mutants displayed an enhanced disease resistance [8]. These results indicate that the WRKYs play both positive and negative regulatory roles in plant basal disease resistance.
Roses (Rosa sp.) are one of the most important commercial flower crops worldwide [9]. The major rose-producing areas include the tropical plateau regions of Africa and South America (including Kenya, Ethiopia, Ecuador, and Colombia), which have suitable climatic conditions and low labor costs, while rose purchasing is largely concentrated in developed countries in Europe and North America [10]. The long-distance logistics and transportation of roses pose a challenge for their post-harvest preservation, with flowers often being affected by post-harvest diseases such as gray mold caused by the necrotrophic fungal pathogen Botrytis cinerea [11].
Some WRKYs enhance the resistance of crops and model plants such as Arabidopsis against various diseases, including B. cinerea; however, the WRKY genes involved in gray mold resistance in roses have not yet been identified. We previously explored the molecular basis of rose resistance against B. cinerea using a de novo RNA-Seq analysis, revealing that large numbers of genes, including WRKY family genes, were significantly upregulated in roses upon B. cinerea infection [10]. In the present study, we performed a genome-wide analysis of the WRKY family in roses, and used virus-induced gene silencing (VIGS) to confirm that RcWRKY41 plays an important role in rose resistance against gray mold.
Results
Identification of the RcWRKY genes in rose
To identify the rose WRKY family gene, the WRKY HMM profile (Pfam: 03106) was used as a query to search the rose genome database (Rosa chinensis Homozygous Genome v2.0; available at https://lipm-browsers.toulouse.inra.fr/pub/RchiOBHm-V2/) [12]. The HMM search led to the identification of 56 candidate RcWRKY genes in the rose genome. We examined the sequences of all candidate proteins using the Conserved Domain Database (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi), verifying that all 56 RcWRKY proteins contained the WRKY DNA-binding domain. A total of 25 of the candidate RcWRKY proteins contained two WRKY domains, while the other 31 contained one WRKY domain. All 56 RcWRKY genes could be mapped onto the rose chromosomes, and were named RcWRKY1 to RcWRKY56 according to their order on the chromosomes.
The sizes of the RcWRKY proteins varied dramatically. RcWRKY19 was the longest, containing 729 amino acids, while the shortest was RcWRKY10, comprising just 120 amino acids. The average length of the RcWRKY proteins was 359 amino acids. Details of the RcWRKY genes, including their accession numbers, chromosomal locations, number of introns and exons, protein sizes, and gene classifications, are provided in Table 1.
Phylogenetic analysis of the rose WRKY genes
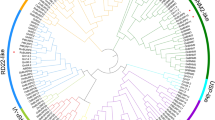
A phylogenetic analysis of the RcWRKY genes was performed using the neighbor-joining method (Fig. 1). Our subsequent analysis of the exon-intron structures of the RcWRKYs was consistent with the results of the phylogenetic analysis. The RcWRKYs contain one to six introns, and most of the RcWRKY genes in the same clade exhibited similar exon-intron structures, such as RcWRKY8, RcWRKY51, RcWRKY4, RcWRKY5, RcWRKY21, RcWRKY6, and RcWRKY22 (Table 1; Fig. 1). Some exceptions were also observed; for example, RcWRKY39 and RcWRKY40 were grouped into the same clade, but RcWRKY40 has two introns and RcWRKY39 has five. Moreover, the lengths of the RcWRKY introns are highly variable, ranging from tens to thousands of nucleotides. RcWRKY11 contains the longest intron, comprising 2369 bp, while the shortest intron (51 bp) was found in RcWRKY16. In addition, we analyzed the conserved WRKY motif sequence in the rose WRKY proteins (Fig. 2).

Phylogenetic analysis of the rose WRKY transcription factors. A complete alignment of the rose WRKYs was used to construct the phylogenetic tree

The sequence of the WRKY motif in rose WRKY proteins. These sequences were determined from the multiple alignment analysis of 56 RcWRKY transcription factors. The bit score indicates the informational content for each position in the sequence
A total of 66 AtWRKY genes were previously identified in Arabidopsis [13]. There is also increasing evidence that the WRKY transcription factors play a key role in disease resistance in various plant species (Additional file 2: Table S1). To evaluate the evolutionary relationships among the RcWRKYs, AtWRKYs, and the plant WRKYs known to be involved in the regulation of disease resistance, we generated a composite phylogenetic tree using the neighbor joining method (Fig. 3). The AtWRKYs were previously divided into three groups, according to their evolutionary relationships, with those in Group I containing two WRKY domains and those in Groups II and III containing just one WRKY domain [13]. In the present study, we found that the evolutionary relationships of the RcWRKYs were consistent with the Arabidopsis WRKY Groups; the RcWRKY genes clustered with the Group I AtWRKY genes contained two WRKY domains, while the other RcWRKY genes contained just one WRKY domain. We found that the WRKYs reported to take part in the regulation of the disease response were distributed across all three Groups.

Phylogenetic analysis of the WRKY transcription factors in rose, Arabidopsis, and other plant species. Complete alignments of the rose and Arabidopsis WRKY sequences, and the disease-resistance-related WRKY transcription factors from a variety of plant species, including cotton (Gossypium hirsutum), rice (Oryza sativa), oilseed rape (Brassica napus), grape (Vitis vinifera), tobacco (Nicotiana benthamiana), barley (Hordeum vulgare), and pepper (Capsicum annuum), were generated to construct a phylogenetic tree using the Neighbor-Joining method. The bootstrap values are indicated on the nodes of the branches. The WRKYs reported to be involved in plant disease resistance are marked in bold
Chromosomal locations, gene duplication, and Microsynteny
The RcWRKY genes are unevenly distributed across all seven rose chromosomes (Table 1; Fig. 4; Additional file 1: Figure S1). We observed a high density of RcWRKYs in several regions, including the short arm of chromosome 3 and the long arm of chromosomes 1 and 6. In contrast, RcWRKY genes were not found on the short arm of chromosomes 1 and 6. Chromosomes 2 and 5 contain the largest numbers of RcWRKY genes (10), followed by chromosome 3 (9), while the lowest numbers of RcWRKY genes (6) were found on chromosomes 4 and 6. Chromosomes 1, 3, and 7 contain RcWRKY genes from all three Groups, whereas the other chromosomes only included RcWRKY genes from Groups I and II. The imbalance of the RcWRKY locations across the rose chromosomes implied that genetic variation occurred during evolution.

Localization and synteny of the WRKY transcription factors in the rose genome. The RcWRKYs were mapped to the rose chromosomes. Those with a syntenic relationship are joined by red lines. The gray lines indicate all syntenic blocks in the rose genome
We further investigated the gene duplication events of the RcWRKYs. A total of 17 gene pairs were found in the rose genome (Table 2). Where two repeated genes are located on the same chromosome (RcWRKY2/RcWRKY7 and RcWRKY34/RhRKYY38), they are likely to be tandem repeats. Other RcWRKY gene pairs are located on different chromosomes, suggesting that segmental duplications occurred within these regions, which may have arisen during full genome duplication in the roses [12]. The collinear relationship of the RcWRKY genes across the chromosomes is shown in Fig. 4.
In order to study the selective constraints among the duplicated RcWRKY genes, the Ka/Ks nucleotide substitution ratios were calculated for the 17 gene pairs (Table 2). Generally, a Ka/Ks ratio > 1 is consistent with positive selection, while Ka/Ks < 1 indicates a purifying selection. The Ka/Ks ratios of all 17 duplicated gene pairs were < 1 (Table 2), indicating that the duplicated RcWRKYs had undergone a purifying selection with limited functional divergence during their evolutionary history.
Expression patterns of the RcWRKY genes in response to B. cinerea
There is increasing evidence to suggest that members of the WRKY family play key roles in plant defense responses against various pathogens. This involves the upregulation of WRKY expression upon pathogen infection. To study the RcWRKY responses to B. cinerea, we obtained RNA-seq transcriptomic data from rose petals exposed to this pathogen at 30 h post inoculation (hpi) and 48 hpi [10]. In rose petals, B. cinerea conidia germinate at 24 hpi, and the early response to infection is considered to occur at 30 dpi, as no visible disease lesions form by this point. The 48 hpi timepoint corresponds to the later response, when the lesions were starting to expand from the inoculation points [10].
The expression of 19 RcWRKY genes (RcWRKY2, RcWRKY4, RcWRKY7, RcWRKY8, RcWRKY13, RcWRKY18, RcWRKY21, RcWRKY23, RcWRKY28, RcWRKY29, RcWRKY30, RcWRKY33, RcWRKY34, RcWRKY35, RcWRKY38, RcWRKY41, RcWRKY46, RcWRKY51, and RcWRKY54) was significantly increased at 48 hpi with B. cinerea, suggesting they might be involved in rose resistance against this pathogen. Among these B. cinerea-induced RcWRKYs, the expression of seven RcWRKY genes was also significantly increased at 30 hpi. These results suggest these WRKYs might be specific regulators of the early stages of the defense response to B. cinerea (Table 3).
To further validate the expression profiles from RNA-Seq, transcript abundance of six RcWRKY genes were analysis using qRT-PCR. The results from the qRT-PCR analysis were generally in agreement with the expression profiles obtained using the RNA-Seq data (Fig. 5).

Validation of RNA-Seq results using qRT-PCR. RhUbi was used as an internal control. The primers used for determining transcript abundance are listed in Additional file 3: Table S2. PDB; potato dextrose broth; Values are the means of three biological replicates ± SD
RcWRKY41 is required for rose resistance against B. cinerea
Using RNA-seq data taken from rose petals infected with B. cinerea, we identified 19 B. cinerea-inducible WRKY genes. To further illustrate the potential roles of these genes in the rose resistance against B. cinerea, we knocked down the expression of RcWRKY41 using VIGS. RcWRKY41 was selected for this VIGS study because 1) its expression is induced in both the early (30 hpi) and late (48 hpi) stages of B. cinerea infection (Fig. 5; Table 3), and it is therefore considered an important candidate regulator of resistance against this pathogen; and 2) RcWRKY41 belongs to Group I of the RcWRKYs, and is closely related to many WRKYs shown to play roles in disease resistance in various plant species, such as NbWRKY7, NbWRKY8, NbWRKY9,VvWRKY33, OsWRKY53, and AtWRKY33 (Fig. 3; Additional file 2: Table S1).
To test whether RcWRKY41 is involved in providing resistance against B. cinerea, we knocked down the expression of RcWRKY41 in rose petals. To this end, we cloned a fragment of the RcWRKY41 coding sequence into pTRV2 vector [14] to generate TRV-RcWRKY41. Agrobacterium cells carrying TRV-RcWRKY41 and TRV1 [14] constructs were mixed in a 1:1 ratio, then vacuum-infiltrated into the rose petal disks to generate RcWRKY41-silenced rose petals. The silenced petals were subsequently inoculated with B. cinerea. Compared with the control petals inoculated with the empty TRV vectors (TRV-00), plants inoculated with TRV-RcWRKY41 showed more severe disease symptoms and their lesion sizes increased significantly (Fig. 6a and b). We further confirmed the silencing efficiency of VIGS by qRT-PCR (Fig. 6c). These results indicate that RcWRKY41 plays an important role in the resistance of roses against B. cinerea.

Functional analysis of rose transcription factor gene RcWRKY41. (A) Compromised B. cinerea resistance symptoms on rose petal disks upon the silencing of RcWRKY41, shown at 60 hpi (hours post inoculation). A recombinant tobacco rattle virus (TRV) targeting RcWRKY41 (TRV-RcWRKY41) was used for the gene silencing, and an empty TRV (TRV-00) was used as the control. (B) Quantification of the average diameter of the disease lesions on the control and RcWRKY41-silenced petals at 60 hpi. Error bars = standard deviation. The statistical analysis was performed using a Student’s t-test; ** P < 0.01. (C) Quantification of RcWRKY41 expression in TRV-RcWRKY41-inoculated petal discs relative to that in the control
Discussion
Transcription factors often control a cluster of functionally related genes, and are therefore appropriate targets for the genetic engineering of (broad-spectrum) resistant crops. The WRKY genes are a major family of plant transcription factors with many important functions, including in the response to pathogens. Systematic and comprehensive genome-wide analyses of the WRKY family have previously been performed in Arabidopsis [13], rice [15], tomato [16], cotton (Gossypium raimondii and G. hirsutum) [17], cucumber (Cucumis sativus) [18], poplar (Populus trichocarpa) [19], and other species; however, a comprehensive analysis of the RcWRKY gene family has not previously been reported, leaving the functions of the rose WRKYs largely unclear. The rose (R. chinensis) genome sequencing project was recently completed, providing useful tools for the genome-wide analysis of the RcWRKY gene family. In this study, we comprehensively analyzed the WRKY family in rose, including their phylogeny, gene structures, chromosomal locations, gene duplication events, and expression profiles under B. cinerea infection.
The rose RcWRKY family contains more genes (56) than were reported in cucumber (55), but fewer than the number reported in Arabidopsis (66), rice (98), tomato (81), cotton (116 in G. raimondii and 102 in G. hirsutum), and poplar (104), indicating that the WRKY family expanded to varying degrees in different plant species following various gene duplication events during their evolution. Gene duplication was found to play a very important role in the expansion of this gene family in rose; a total of 17 duplication events were identified in the 56 RcWRKYs, the majority of which (15) involved segmental duplications, while two involved tandem duplications. The Ka/Ks ratios of all 17 RcWRKY pairs were < 1, indicating that this gene family has undergone purifying selection rather than positive selection, and suggested that the RcWRKYs were highly conserved. In plants, the Resistance (R) genes encoding the immune receptors that recognize a specific pathogen are often under positive selection pressure [20]. The purifying selection detected for all RcWRKYs therefore suggests they may be involved in the basal defense of plants, rather than in race-specific resistance.
Most of the clades identified in the phylogenetic analysis contained WRKYs from both Arabidopsis and rose, implying that these two species underwent fairly conservative evolution. There are some exceptions however; for example, AtWRKY38, AtWRKY62, AtWRKY63, AtWRKY64, AtWRKY66, and AtWRKY67 belong to an evolutionary clade that does not contain any RcWRKYs. This indicated that, after diverging from their common ancestor, these WRKY genes were either lost in rose or acquired (through duplication and divergence) in Arabidopsis.
Many WRKY genes have been shown to be involved in disease resistance in plants, prompting us to search for candidate WRKY genes involved in the rose response to B. cinerea infection. The elucidation of gene expression patterns often provides clues about their functions; therefore, we examined the expression changes in the RcWRKYs when exposed to B. cinerea infection. A total of 19 RcWRKY genes were found to be significantly upregulated upon B. cinerea infection in rose petals, most of which (14 of 19) had undergone gene duplication events. We further identified the RcWRKYs that might participate in B. cinerea resistance by adding them to a phylogenetic tree of the plant WRKYs known to be involved in the disease responses. Among the 19 B. cinerea-induced RcWRKYs, RcWRKY41 was shown to be evolutionally close to a number of disease resistance WRKYs from various plant species, and its expression was found to increase from the early to late stages of the B. cinerea infection. RcWRKY41 was therefore considered to be a candidate gene participating in B. cinerea resistance, which was confirmed when VIGS was used to silence its expression in rose petals, resulting in their reduced resistance to B. cinerea. This indicates RcWRKY41 plays an important positive regulatory function in the resistance of rose petals against grey mold.
Conclusions
We performed a genome-wide analysis of the RcWRKYs, exploring their phylogenetic relationships, collinearity, and expression profiles. A total of 56 non-redundant rose RcWRKY family members were identified, which could be divided into three groups based on our analyses of their phylogeny and conserved domains; 22 of them belonged to Group I, 26 belonged to Group II, and nine belonged to Group III. Our expression analysis indicated that 19 RcWRKY family genes were induced in rose petals subjected to a B. cinerea infection. By comparing these sequences with other plant WRKYs known to be involved in disease resistance, we revealed that RcWRKY41 is involved in the regulation of gray mold resistance in rose petals, which was confirmed using VIGS. These results provide new information that may facilitate the further functional analysis of the RcWRKYs in roses.
Methods
Identification and characteristics of the WRKY genes in the rose genome
The complete rose (Rosa chinensis ‘Old Blush’) genome sequence was obtained from https://lipm-browsers.toulouse.inra.fr/pub/RchiOBHm-V2/. To identify the non-redundant WRKY genes in the rose genome, the consensus protein sequence of the WRKY Hidden Markov Model (HMM) was downloaded from Pfam (PF03106; http://pfam.xfam.org). This HMM profile was then used as a query to search the rose genome, resulting in the identification of all rose sequences containing a WRKY domain with an E-value <1e− 3. Finally, all candidate RcWRKYs were validated using the Pfam and the Conserved Domains Database (CDD; https://www.ncbi.nlm.nih.gov / Structure / cdd / wrpsb.cgi) to determine that they contained the core domains.
Phylogenetic analyses
A total of 66 Arabidopsis WRKY protein sequences were collected from The Arabidopsis Information Resource (TAIR) (http://www.arabidopsis.org/). Based on the results of previous studies, additional sequences of WRKY genes involved in plant disease resistance were collected from GenBank, including those from cotton (Gossypium hirsutum), rice (Oryza sativa), oilseed rape (Brassica napus), grape (Vitis vinifera), tobacco (Nicotiana benthamiana), barley (Hordeum vulgare), and pepper (Capsicum annuum). A phylogenetic analysis was used to determine whether orthologs of these genes are present in the rose genome. The amino acid sequences of WRKY proteins were aligned using ClustalW. The alignment of WRKY sequences was used to perform the phylogenetic analysis. Phylogenetic dendrograms were constructed using the neighbor-joining (NJ) method in MEGA 6.0 software [21]. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches. The evolutionary distances were computed using the p-distance method and are in the units of the number of amino acid differences per site. All positions with less than 50% site coverage were eliminated.
Collinearity analyses
In order to identify collinearity, a Multiple Collinearity Scan toolkit [22] was used to detect the microsyntenic relationships between the WRKY genes. The resulting microsynteny chains were then evaluated using ColinearScan (E-value <1e− 10).
Calculation of the non-synonymous (Ka) to synonymous (Ks) nucleotide substitution ratio
An analysis of the Ka/Ks ratios was used to determine the selection modes driving the evolution of the RcWRKYs. These ratios were calculated using TBtools software [23].
Expression of the RcWRKYs in response to B. cinerea
RNA-Seq data from rose petals infected with B. cinerea were obtained from the National Center for Biotechnology Information (NCBI) database (accession number PRJNA414570). Clean sequencing reads were mapped to the rose reference genome, and the number of reads per kb per million reads (RPKM) were used to determine the gene expression levels. To confirm the RNA-Seq results, the transcript abundance of 6 RcWRKY genes was analyzed using qRT-PCR. To this end, cDNA was generated from rose petals inoculated with B. cinerea, using Takara Reverse Transcriptase M-MLV (Takara). Quantitative RT-PCR was performed on a StepOnePlus Real-Time PCR System (Thermo Fisher Scientific), by using 1 μL of the first strand cDNA in the reaction with the KAPA SYBR rapid quantitative PCR kit (KAPA Biosystems). RhUbi was used as a housekeeping gene. The primers used for determining transcript abundance are listed in Additional file 3: Table S2.
VIGS
To obtain the TRV-RcWRKY41 construct, a fragment from the coding region of RcWRKY41 was amplified using the primer pairs RcWRKY41-TRV-F (5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTTTACCAAGCCACAATACCAA-3′) and RcWRKY41-TRV-R (5′-GGGGACCACTTTGTACAAGAAAGCTGGGTAACACAGCAATGATTCAAAA-3′) and cloned into the Tobacco rattle virus vector TRV2 [14]. To establish VIGS in rose petals, petals were detached from the outermost whorls of rose flowers (R. hybrida ‘Samantha’) during stage 2 of flower opening. A 15-mm disk was then punched from the center of each petal. Agrobacterium tumefaciens cultures containing constructs expressing TRV1 [14] and recombinant TRV2 were mixed in a 1:1 ratio and vacuum-infiltrated into the petal disks. At 6 days after the TRV infection, the petal disks were inoculated with B. cinerea. The VIGS was repeated at least three times using at least 48 disks. After the B. cinerea inoculation, the lesion sizes were recorded, and a Student’s t-test was conducted to identify any significant differences.
Availability of data and materials
The datasets used and/or analyzed during the current study has been included within supplemental data. The plant materials are available from the corresponding author on reasonable request.
Abbreviations
- CDD:
-
Conserved Domains Database
- HMM:
-
Hidden Markov Model
- Hpi:
-
Hours post inoculation
- NJ:
-
Neighbor-joining
- RPKM:
-
Number of reads per kb per million reads
- VIGS:
-
Virus-induced gene silencing
References
Rushton PJ, Somssich IE, Ringler P, Shen QXJ. WRKY transcription factors. Trends Plant Sci. 2010;15(5):247–58.
Zhang Y, Wang L. The WRKY transcription factor superfamily: its origin in eukaryotes and expansion in plants. BMC Evol Biol. 2005;5(1):1.
Chen L, Xiang S, Chen Y, Li D, Yu D. Arabidopsis WRKY45 interacts with the DELLA protein RGL1 to positively regulate age-triggered leaf senescence. Mol Plant. 2017;10(9):1174–89.
Tian X, Li X, Zhou W, Ren Y, Wang Z, Liu Z, Tang J, Tong H, Fang J, Bu Q. Transcription factor OsWRKY53 positively regulates Brassinosteroid signaling and plant architecture. Plant Physiol. 2017;175(3):1337–49.
Hsu FC, Chou MY, Chou SJ, Li YR, Peng HP, Shih MC. Submergence confers immunity mediated by the WRKY22 transcription factor in Arabidopsis. Plant Cell. 2013;25(7):2699–713.
Hu Y, Chen L, Wang H, Zhang L, Wang F, Yu D. Arabidopsis transcription factor WRKY8 functions antagonistically with its interacting partner VQ9 to modulate salinity stress tolerance. Plant J. 2013;74(5):730–45.
Mao G, Meng X, Liu Y, Zheng Z, Chen Z, Zhang S. Phosphorylation of a WRKY transcription factor by two pathogen-responsive MAPKs drives phytoalexin biosynthesis in Arabidopsis. Plant Cell. 2011;23(4):1639–53.
Kim KC, Lai ZB, Fan BF, Chen ZX. Arabidopsis WRKY38 and WRKY62 transcription factors interact with histone Deacetylase 19 in basal defense. Plant Cell. 2008;20(9):2357–71.
Qi W, Chen X, Fang P, Shi S, Li J, Liu X, Cao X, Zhao N, Hao H, Li Y, et al. Genomic and transcriptomic sequencing of Rosa hybrida provides microsatellite markers for breeding, flower trait improvement and taxonomy studies. BMC Plant Biol. 2018;18(1):119.
Liu X, Cao X, Shi S, Zhao N, Li D, Fang P, Chen X, Qi W, Zhang Z. Comparative RNA-Seq analysis reveals a critical role for brassinosteroids in rose (Rosa hybrida) petal defense against Botrytis cinerea infection. BMC Genet. 2018;19(1):62.
Hao Y, Cao X, Ma C, Zhang Z, Zhao N, Ali A, Hou T, Xiang Z, Zhuang J, Wu S, et al. Potential Applications and Antifungal Activities of Engineered Nanomaterials against Gray Mold Disease Agent Botrytis cinerea on Rose Petals. Front Plant Sci. 2017;8:1332.
Raymond O, Gouzy J, Just J, Badouin H, Verdenaud M, Lemainque A, Vergne P, Moja S, Choisne N, Pont C, et al. The Rosa genome provides new insights into the domestication of modern roses. Nat Genet. 2018;50(6):772.
Wang Q, Wang M, Zhang X, Hao B, Kaushik SK, Pan Y. WRKY gene family evolution in Arabidopsis thaliana. Genetica. 2011;139(8):973–83.
Liu YL, Schiff M, Dinesh-Kumar SP. Virus-induced gene silencing in tomato. Plant J. 2002;31(6):777–86.
Ross CA, Liu Y, Shen QXJ. The WRKY gene family in rice (Oryza sativa). J Integr Plant Biol. 2007;49(6):827–42.
Huang SX, Gao YF, Liu JK, Peng XL, Niu XL, Fei ZJ, Cao SQ, Liu YS. Genome-wide analysis of WRKY transcription factors in Solanum lycopersicum. Mol Gen Genomics. 2012;287(6):495–513.
Dou L, Zhang X, Pang C, Song M, Wei H, Fan S, Yu S. Genome-wide analysis of the WRKY gene family in cotton. Mol Gen Genomics. 2014;289(6):1103–21.
Ling J, Jiang W, Zhang Y, Yu H, Mao Z, Gu X, Huang S, Xie B. Genome-wide analysis of WRKY gene family in Cucumis sativus. BMC Genomics. 2011;12:471.
He H, Dong Q, Shao Y, Jiang H, Zhu S, Cheng B, Xiang Y. Genome-wide survey and characterization of the WRKY gene family in Populus trichocarpa. Plant Cell Rep. 2012;31(7):1199–217.
Mondragon-Palomino M, Meyers BC, Michelmore RW, Gaut BS. Patterns of positive selection in the complete NBS-LRR gene family of Arabidopsis thaliana. Genome Res. 2002;12(9):1305–15.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9.
Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, Lee TH, Jin H, Marler B, Guo H, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40(7):e49.
Chen C, Xia R, Chen H, He Y. TBtools, a Toolkit for Biologists integrating various HTS-data handling tools with a user-friendly interface. bioRxiv 2018. https://doi.org/10.1101/289660.
Lai Z, Vinod KM, Zheng Z, Fan B, Chen Z. Roles of ArabidopsisWRKY3 and WRKY4 transcription factors in plant responses to pathogens. BMC Plant Biol. 2008;8(1):68.
Kim K-C, Baofang F, Zhixiang C. Pathogen-induced Arabidopsis WRKY7 is a transcriptional repressor and enhances plant susceptibility to Pseudomonas syringae. Plant Physiol. 2006;142(3):1180–92.
Chen L, Zhang L, Yu D. Wounding-induced WRKY8 is involved in basal defense in Arabidopsis. Mol Plant-Microbe Interact. 2010;23(5):558–65.
Jiang CH, Huang ZY, Xie P, Gu C, Li K, Wang DC, Yu YY, Fan ZH, Wang CJ, Wang YP. Transcription factors WRKY70 and WRKY11 served as regulators in rhizobacterium Bacillus cereus AR156-induced systemic resistance to Pseudomonas syringae pv. Tomato DC3000 in Arabidopsis. Proc Prehistoric Soc. 2016;41(1):156–253.
Journot Catalino NL, Somssich IE, Dominique R, Thomas K. The transcription factors WRKY11 and WRKY17 act as negative regulators of basal resistance in Arabidopsis thaliana. Plant Cell. 2006;18(11):3289–302.
Xu X, Chen C, Fan B, Chen Z. Physical and functional interactions between pathogen-induced Arabidopsis WRKY18, WRKY40, and WRKY60 transcription factors. Plant Cell. 2006;18(5):1310–26.
Zheng Z, Mosher SL, Fan B, Klessig DF, Chen Z. Functional analysis of Arabidopsis WRKY25 transcription factor in plant defense against Pseudomonas syringae. BMC Plant Biol. 2007;7(1):2–2.
Mukhtar MS, Marco Y, Somssich IE. The Arabidopsis transcription factor WRKY27 influences wilt disease symptom development caused by Ralstonia solanacearum. Plant J Cell Mol Biol. 2010;56(6):935–47.
Chen X. Chen, Jun L, Guifang L, Airong W, Zonghua W, Guodong L: overexpression of AtWRKY28 and AtWRKY75 in Arabidopsis enhances resistance to oxalic acid and Sclerotinia sclerotiorum. Plant Cell Rep. 2013;32(10):1589–99.
Zheng Z, Qamar SA, Chen Z, Mengiste T. Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant J. 2006;48(4):592–605.
Hu Y, Dong Q, Yu D. Arabidopsis WRKY46 coordinates with WRKY70 and WRKY53 in basal resistance against pathogen Pseudomonas syringae. Plant Sci. 2012;185-186(x):288–97.
Xing D-H, Zi-Bing Z-Y. Zheng, Vinod, Bao-Fang, Zhi-Xiang, Chen: stress- and pathogen-induced Arabidopsis WRKY48 is a transcriptional activator that represses plant basal defense. Mol Plant. 2008;1(3):459–70.
Gao Q-M, Srivathsa V, Duroy N, Aardra K. Low oleic acid-derived repression of jasmonic acid-inducible defense responses requires the WRKY50 and WRKY51 proteins. Plant Physiol. 2011;155(1):464–76.
Murray SL, Ingle RA, Petersen LN, Denby KJ. Basal resistance against Pseudomonas syringae in Arabidopsis involves WRKY53 and a protein with homology to a nematode resistance protein. Mol Plant-Microbe Interact. 2007;20(11):1431–8.
Jiang Y, Yu D. The WRKY57 transcription factor affects the expression of Jasmonate ZIM-domain genes transcriptionally to compromise Botrytis cinerea resistance. Plant Physiol. 2016;171(4):2771–82.
Gao R, Liu P, Yong Y, Wong SM. Genome-wide transcriptomic analysis reveals correlation between higher WRKY61 expression and reduced symptom severity in turnip crinkle virus infectedArabidopsis thaliana. Sci Rep. 2016;6(1):24604.
Liu X, Song Y, Xing F, Wang N, Wen F, Zhu C. GhWRKY25 , a group I WRKY gene from cotton, confers differential tolerance to abiotic and biotic stresses in transgenic Nicotiana benthamiana. Protoplasma. 2015;253(5):1265–81.
Guo R, Qiao H, Zhao J, Wang X, Wang X. The Grape VlWRKY3 Gene Promotes Abiotic and Biotic Stress Tolerance in Transgenic Arabidopsis thaliana. Front Plant Sci. 2018;9:545.
Merz PR, Moser T, Höll J, Kortekamp A, Buchholz G, Zyprian E, Bogs J. The transcription factor VvWRKY33 is involved in the regulation of grapevine (Vitis vinifera) defense against the oomycete pathogen Plasmopara viticola. Physiol Plant. 2014;153(3):365.
Adachi H, Ishihama N, Nakano T, Yoshioka M, Yoshioka H. Nicotiana benthamiana MAPK-WRKY pathway confers resistance to a necrotrophic pathogen Botrytis cinerea. Plant Signal Behav. 2016;11(6):e1183085.
Han M, Ryu HS, Kim CY, Park DS, Ahn YK, Jeon JS. OsWRKY30 is a transcription activator that enhances rice resistance to the Xanthomonas oryzae pathovar oryzae. J Plant Biol. 2013;56(4):258–65.
Peng X, Hu Y, Tang X, Zhou P, Deng X, Wang H, Guo Z. Constitutive expression of rice WRKY30 gene increases the endogenous jasmonic acid accumulation, PR gene expression and resistance to fungal pathogens in rice. Planta. 2012;236(5):1485–98.
Chujo T, Koji M, Satoshi O, Yuka M, Takafumi S, Mitsuko KK, Akira T, Yoko N, Eiichi M, Hideaki N. Overexpression of phosphomimic mutated OsWRKY53 leads to enhanced blast resistance in rice. PLoS One. 2014;9(6):e98737.
Liu XQ, Bai XQ, Qian QI, Wang XJ, Chen MS, Chu CC. OsWRKY03, a rice transcriptional activator that functions in defense signaling pathway upstream of OsNPR1. Cell Res. 2005;15(8):593–603.
Wang H, Meng J, Peng X, Tang X, Zhou P, Xiang J, Deng X. Rice WRKY4 acts as a transcriptional activator mediating defense responses toward Rhizoctonia solani , the causing agent of rice sheath blight. Plant Mol Biol. 2015;89(1–2):157–71.
Abbruscato P, Nepusz T, Mizzi L, Del CM, Morandini P, Fumasoni I, Michel C, Paccanaro A, Guiderdoni E, Schaffrath U. OsWRKY22 , a monocot WRKY gene, plays a role in the resistance response to blast. Mol Plant Pathol. 2012;13(8):828–41.
Liu X, Bai X, Wang X, Chu C. OsWRKY71, a rice transcription factor, is involved in rice defense response. J Plant Physiol. 2007;164(8):969–79.
Zhang J, Zhang J, Peng Y, Guo Z. Constitutive expression of pathogen-inducible OsWRKY31 enhances disease resistance and affects root growth and auxin response in transgenic rice plants. Cell Res. 2008;18(4):508–21.
Ellis JG, Dodds PN, Lawrence GJ. The role of secreted proteins in diseases of plants caused by rust, powdery mildew and smut fungi. Curr Opin Microbiol. 2007;10(4):326–31.
Yang B, Jiang Y, Rahman MH, Deyholos MK, Kav NN. Identification and expression analysis of WRKY transcription factor genes in canola (Brassica napus L.) in response to fungal pathogens and hormone treatments. BMC Plant Biol. 2009;9(1):68.
Oh S-K, Kwang-Hyun B, Jeong Mee P, So Young Y, Seung Hun Y, Sophien K, Doil C. Capsicum annuum WRKY protein CaWRKY1 is a negative regulator of pathogen defense. New Phytol. 2010;177(4):977–89.
Acknowledgements
None.
Funding
This study was supported by the National Natural Science Foundation of China (grant number 31772344) to Zhao Zhang. The funders played no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Z.Z. and X.L. conceived and designed the experiments. X.L., D.L., and Y.X. performed the experiments. X.L. and S.Z. analyzed the data. Z.Z. and X.L. wrote the paper. All the authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable. Our research did not involve any human or animal subjects, material, or data. The plant materials used in this study were provided by the China Agricultural University and are freely available for research purposes following institutional, national and international guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Chromosomal distribution of the RcWRKY genes. The physical location of each RcWRKY gene is listed on the left side of the chromosomes.
Additional file 3: Table S2.
List of primers used in this study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liu, X., Li, D., Zhang, S. et al. Genome-wide characterization of the rose (Rosa chinensis) WRKY family and role of RcWRKY41 in gray mold resistance. BMC Plant Biol 19, 522 (2019). https://doi.org/10.1186/s12870-019-2139-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-019-2139-6