
Abstract
Background
MADS-box genes play important roles in vegetative growth and reproductive development and are essential for the correct development of plants (particularly inflorescences, flowers, and fruits). However, this gene family has not been identified nor their functions analyzed in Brassica oleracea.
Results
In this study, we performed a whole-genome survey of the complete set of MADS-box genes in B. oleracea. In total, 91 MADS-box transcription factors (TFs) were identified and categorized as type I (Mα, Mβ, Mγ) and type II (MIKCC, MIKC*) groups according to the phylogeny and gene structure analysis. Among these genes, 59 were randomly distributed on 9 chromosomes, while the other 23 were assigned to 19 scaffolds and 9 genes from NCBI had no location information. Both RNA-sequencing and quantitative real-time-PCR analysis suggested that MIKC genes had more active and complex expression patterns than M type genes and most type II genes showed high flowering-related expression profiles. Additional quantitative real-time-PCR analysis of pedicel and four flower whorls revealed that the structure of the B.oleracea MIKC genes was conserved, but their homologues showed variable expression patterns compared to those in Arabidopsis thaliana.
Conclusion
This paper gives a detailed overview of the BolMADS genes and their expression patterns. The results obtained in this study provide useful information for understanding the molecular regulation of flower development and further functional characterization of MADS-box genes in B. oleracea.
Similar content being viewed by others


Background
The MADS-box family consists of genes encoding a class of transcription factors characterized by the presence of 58–60 conserved amino acids known as the MADS domain [1]. The MADS domain was named after the first four members of this family encoded by genes, MINICHROMOSOME MAINTENANCE 1 (MCM1) from yeast [2], AGAMOUS (AG) from Arabidopsis thaliana [3], DEFICIENS (DEF) from Antirrhinum majus [4], and SERUM RESPONSE FACTOR (SRF) from Homo sapiens [5]. MADS-box genes are widely distributed in eukaryotes and play important roles in an organism’s growth and development, particularly in floral organ differentiation, flowering time regulation, and fruit development and ripening in angiosperms [6, 7].
Based on phylogenetic analysis, the MADS-box genes are divided into two categories: type I and type II [8]. All type II members in yeast, animal and plant are MEF2-like genes, while type I MADS-box genes in plant are SRF-like [8]. In addition to their common MADS (M) domain, MIKC genes also include intervening (I), keratin-like (K), and C-terminal (C) domains [9, 10]. M is the most conserved domain and located in the N-terminal region, which binds specific DNA through the CArG-box (5′-CC(A/T) 6GG-3′) in the promotor region of the target gene. I is a less conserved region consisting of approximately 30 amino acids and determines the specific binding of transcription factors to DNA and dimer formation. The K domain is the second conserved region forming a coiled-coil structure, which facilitates interactions between proteins. The C-terminal domain is the least conserved region, consists mainly of hydrophobic amino acids, and plays an important role in the formation and transcriptional activation of protein complexes [6, 11, 12]. Based on sequence and structure differences of the I domain, the MIKC genes are further divided into MIKCC - and MIKC∗-types. MIKCC-type genes are the most widely studied MADS-box genes because they are essential in plant growth and development [13, 14]. According to the phylogenetic analysis of the M domain, type I genes (also known as M type genes) in plant are divided into four groups, Mа, Mß, Mγ, and Mδ. In Arabidopsis, Mδ genes are also assigned as MIKC∗-type genes based on their relatively close relationships with MIKCC-type genes [15].
Plant MIKCC genes were demonstrated to play important roles in flower organogenesis [15]. Coen and Meyerowitz first proposed the famous ABC model, and later the integral ABCDE model was developed [16,17,18]. The identity of each whorl of floral organs was shown to be determined by different combinations of A, B, C, D and E genes activities: sepals (A + E), petals (A + B + E), stamens (B + C + E), carpels (C + E), and ovules (C + D + E) [17]. In Arabidopsis, functional genes were included in this model: APETALA1 (AP1) is expressed in sepals and petals and belongs to the class A [19, 20]; PISTILLATA (PI) and AP3 are expressed in petals and stamens and belong to the class B [21]; class C gene AGAMOUS (AG) is expressed in stamens and carpels [4, 22]; class D gene AGAMOUS-LIKE 11(AGL11, another name is SEEDSTICK, STK) is expressed in ovules [23]; class E genes SEPALLATA 1 (SEP1), SEP2, SEP3 and SEP4, are expressed in all floral whorls [24, 25]. Other MIKC genes have been shown to possess different biological functions, such as genes regulating flowering time (SOC1, SVP, FLC, AGL15/18) [26,27,28,29,30]; fruit ripening genes (SHP1, SHP2, FUL) [31, 32]; and seed pigmentation genes (TT16) and root development genes (AGL12, AGL17) [33, 34].
Brassica oleracea contains multiple vegetable crops including broccoli, cauliflower, brussels sprouts, various types of cabbage, kohlrabi, and kale. Cauliflower is an economically important vegetable planted worldwide [35]. Compared with other crops of B. oleracea, cauliflower has a unique characteristic - a tight white flower curd, consisting of a large amount of shortened branches of indeterminate inflorescences [36]. Previous studies showed that the formation of cauliflower curd could be related to two MADS-box genes BoCAL and BoAP1, and in cauliflower the bocal mutant allele is present [37, 38]. However, other studies found only a weak connection between cauliflower phenotype and boap1 and bocal mutations [39, 40]. Moreover, Lan and Paterson reported that curd-related traits are affected by 86 quantitative trait loci (QTLs), indicating that the cauliflower arrest is multi-gene controlled [41]. Genome-wide studies of MADS-box genes can be conducted, as the B. oleracea genome has been sequenced [42]. In this study, a total of 91 MADS-box genes were identified in the B. oleracea genome. The phylogenetic relationships, gene structure, chromosomal locations, and conserved motifs of the encoded proteins were analyzed. The tissue-specific expression of all BolMADS genes in cauliflower were further studied. This work provides useful information regarding the molecular mechanisms underlying cauliflower curd formation and flower development.
Results
Identification of MADS-box genes in B. oleracea
A total of 95 candidate genes were identified. Based on sequence analysis, four candidate MADS-box genes composed of approximately 200 bases were confirmed to contain incomplete MADS-box domains and then were excluded from the analysis. Therefore, a total of 91 MADS-box genes with complete and functional structures were identified in the B. oleracea genome. Among them, 82 genes were found in the B. oleracea genome database (named BolMADS1- BolMADS82), and 9 genes were acquired from the NCBI database (named with NCBI access numbers).
The MADS-box genes in B. oleracea genome had coding sequence lengths of 249–1221 base pairs. The encoded proteins length ranged from 82 to 406 amino acids with predicted molecular masses of 9.52–46.31 KDa and protein isoelectric points of 4.08–10.53 (Table 1).
Phylogenetic analysis of MADS-box gene family
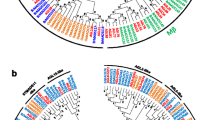
A phylogenetic tree was constructed based on the sequence of full-length MADS-box proteins from B. oleracea and A. thaliana using the neighbor joining method (Additional file 1: Figure S1). The 91 BolMADS genes were classified into two categories, type I (49) and type II (42). Independent phylogenetic trees of both types were constructed using MADS genes from both species (Fig. 1). Type I BolMADS genes were further divided into three subgroups: Mα, Mβ, and Mγ. The Mα subfamily was the largest, possessing 25 genes, while the Mβ and Mγ subgroups showed nearly the same number of genes with 11 and 13, respectively.

Phylogenetic tree of the sequence of full-length MADS-box proteins from B. oleracea and A. thaliana using neighbor-joining method. a Phylogenetic analysis of type II B. oleracea (42) and A. thaliana (45) MADS-box proteins classified into 12 MIKCC clades and one MIKC* group as marked in the Fig. b Phylogenetic analysis of type I MADS-box proteins from B. oleracea (49) and A. thaliana (59)
In type II BolMADS genes, we found 36 MIKCC-type and 6 MIKC*-type genes. A total of 12 MIKCC evolutionary branches were detected according to the known groups of A. thaliana MADS-box genes. The AP1-like branch contained the largest number (8) of BolMADS MIKCC-type genes. We found two subgroups of AGL6-like and AGL17-like genes which consisted of only one member each. In contrast with A. thaliana, B. oleracea AP1-, AGL12-, TT16/PI-, and AP3-like gene clusters are expanded by gene duplication, whereas TM3-, and AGL17-like genes do not have paralogs.
Using the neighbor joining method, another phylogenetic tree was constructed with the full-length of MADS-box protein sequences from B. oleracea and B. rapa (Additional file 2: Figure S2). In general, most MADS genes in B. rapa showed repetition and expansion compared to those from B. oleracea. Moreover, type II MADS-box genes showed higher expansion degrees than type I genes. In type II MADS genes, the TM3 subgroup showed the highest gene expansion degree, with a total of 16 TM3 genes in B. rapa but only three in B. oleracea. The AP3 and AGL12 subgroups contained nearly the same number of genes in these two Brassica species.
Gene structure and conserved motif distribution analysis
The gene structure and intron/exon arrangements of the BolMADS genes were determined by comparing their full-length cDNA and genomic DNA sequences. All type II BolMADS genes contained at least three introns except for BolMADS16 and BolMADS23. BolMADS16 showed the most conserved domain of M with no introns and BolMADS23 showed two introns. In type I genes, BolMADS34, 42, 67, 78, 80, and 82 were found to have one intron, while the others were intronless (Table 1).
The structures of proteins produced by BolMADS genes were analyzed using MEME online software. Ten conserved motifs, named as motifs 1–10, were identified (Fig. 2). Motif 1 and 5 were corresponding to the typical MADS domain. The main motif 1 was found in all BolMADS proteins. Motif 2 specified the K domain was found in most MIKCC type genes except BolMADS9, 12, 16 and 23, which contained relatively short amino acid sequences. For the six MIKC* type proteins, BolMADS29 and 33 contained motif 2, while the others have not. The K domains typical of type II proteins were missing in all type I BolMADS proteins. N-terminal motif 6 was found in all type II and in the majority of type I proteins. Motifs 4 and 8 were specific to some of the Mα proteins, motif 10 - to some of the Mα and Mβ proteins, while motifs 3 and 9 - to some of the Mγ proteins.

Distribution of conserved motifs in B. oleracea MADS-box proteins identified using MEME
Chromosome distributions of MADS-box genes
The physical locations of BolMADS genes were mapped to the 9 chromosomes of B. oleracea (Fig. 3). Of the 91 genes, 59 were randomly distributed on 9 chromosomes, 23 were assigned to 19 scaffolds and 9 from the NCBI database had no location information. For MIKC-type genes, 26 were mapped on 8 chromosomes (except Chr5). Chromosomes 3 and 9 contained the maximum number of six genes, while only one gene was found on chromosome 1. The other 7 MIKC-type genes were randomly distributed on 7 scaffolds. Type I genes showed a completely different distribution. First, of the 49 type I genes, 31 scattered nonrandomly on all 9 B. oleracea chromosomes. Chromosomes 2 and 4 had the largest number of genes and the other chromosomes contained 2–5 genes. Second, the remaining 16 genes were distributed on 12 scaffolds. Sca194_P1 had the largest number of genes, showing three genes. Sca153 and Sca192 each contained two genes. There was one gene on each of the other scaffolds.

Distribution of B. oleracea MADS-box genes on 9 chromosomes and 19 scaffolds. Chromosome numbers are written as C01–C09 above each chromosome. The genes showing segmental duplications are indicated by black vertical lines. Tandemly duplicated genes are joined by a gray shadow. The size of each chromosome was estimated as Mb on the left side of the figures
Duplicated genes including segmental and tandem duplications were detected in the MADS-box gene family of B. oleracea. There were higher frequencies of segmental duplications and a total of 10 genes contained corresponding homologous segments. In contrast, only three groups of genes were contained tandem duplications, which all related to type I genes. The first group containing BolMADS37, 38, and 34 was on chromosome 2 and the second group containing BolMADS49 and 50 was on chromosome 8. The third group contained three genes, BolMADS 72, 73, and 74, distributed on scaffold Sca194_P1.
Genome-wide expression analysis of MADS-box genes
The RNA-seq data of 82 MADS-box genes were downloaded from B. oleracea Genomics Database (http://www.ocri-genomics.org/bolbase/) and the other 9 genes from the NCBI database had no information of RNA-seq. The genes were found to be differently expressed in six tissues including the callus, root, stem, leaf, flower, and silique. Two heat maps were constructed for type I and type II genes (Fig. 4). The overall expression of type II genes was more active and diverse than that of type I genes. A total of 30 genes showed no expression or their transcription levels were too low to be detected on the heat map. Of the 30 genes with no or low expression, 22 were type I and 8 were type II. In all six tissues, only the FLC-like gene BolMADS22 was expressed.

Heat map of BolMADS genes in the callus, root, stem, leaf, flower, and silique tissues. Hierarchical clustering results are shown on the left of the heat map and relative expression values with color green (low) to red (high) are displayed at the bottom
Majority of MIKC-type genes (22 of 33), belonging to the AP1, SEP, AG, AGL12, AGL15/18, and FLC subfamilies, were expressed in the flower and /or silique. BolMADS19 and 21 (AP3-like) were consistently and specifically expressed in flowers. However, most genes within one subfamily often showed different expression patterns. Among the six AP1 genes, three (BolMADS1, 2, 3) showed high expression levels in the flower and silique and three genes (BolMADS10, 16, 20) showed no expression in the six tissues. In the AGL12-like subfamily containing three genes, BolMADS26 was highly expressed in the flower, while the other two genes showed no or very low transcription in the six tissues. Interestingly, six MIKC* genes also displayed very different expression patterns. Four genes (BolMADS29, 30, 31, 33) showed no or low expression levels. The other two genes (BolMADS28, 32) were highly expressed in flowers and BolMADS32 was detected in the leaf and callus. Compared to MIKC genes, type I genes were inert and their expression patterns were simple. A transcription of only 12 genes was detected, while the other genes were silent. These 12 genes were expressed in the silique and BolMADS75 showed high expression in the flower, leaf, stem, and callus.
Expression patterns of MADS-box genes in different tissues of cauliflower
MADS-box genes were reported to have diverse functions in plant growth and development, particularly in floral organs specification. Therefore, we examined the expression patterns of 87 B. oleracea MADS-box genes in seven different tissues of cauliflower including the root, stem, leaf, curd, bud, flower, and silique by qRT-PCR (Fig. 5). A total of 48 genes were expressed in at least one tissue, while the other 39 genes showed no or very low expression. Among the 48 expressed genes, 35 belonged to the MIKC type and the other 13 were type I.

Expression patterns of 48 BolMADS genes in cauliflower tissues of root, stem, leaf, curd, bud, flower, and silique (a) by qRT-PCR (b). A1, The tissues of root, stem and leaf from 35-day-old plant; A2, The curd of 60 days after transplanting; A3, The bud of 90 days after transplanting; A4, The fully opened flowers during the flowering period; A5, The siliques of 20 days after pollination. The standard deviations of three biological replicates are represented by the error bars
All MIKC type genes were highly expressed in reproductive organs including the curd, bud, flower, and silique except for BolMADS15 (SVP-like), 16 (AP1-like), 20 (AP1-like), and 27 (AGL12-like), which showed higher transcription levels in vegetative tissues (root, stem, and leaf). We found that not only AP1-like genes (BolMADS1, 3, 10, XP_013590290.1, XP_013589842.1), but also AP3-like BolMADS21 gene, SEP1-like BolMADS5 gene, SVP-like XP_013600280.1 gene and PI-like XP_013626307.1 gene showed relatively high expression levels in the curd, which is a specific reproductive organ in cauliflower. This result indicates that these genes play important roles in cauliflower curd formation and also confirmed the results of a previous study showing that AP1-like genes control flower primordium development. In addition, XP_013590290.1 and XP_013589842.1 were also highly expressed in the bud and flower, and XP_013590290.1 showed the highest expression level in the flower. The six MIKC* genes showed no or relatively low expression except BolMADS28, which was expressed in flower with relatively high transcription level among MIKC type genes.
For the 45 tested type I genes, 13 showed different expression patterns. BolMADS71 showed the highest expression level in the flower. BolMADS40 and 70 were specifically expressed in the root, indicating roles in root development. The other three genes (BolMADS67, 72, and 73) showed high transcription levels in the silique and may be involved in silique development.
Key MADS-box genes involved in floral organ development
A total of 21 MIKC type genes and one type I gene showing relatively high expression levels in the flower were selected to further investigate their roles in floral organ development. The expression patterns of these genes were detected in B. oleracea pedicel, sepal, petal, stamen, and pistil by qRT-PCR (Fig. 6). Three AP1/FUL-like (BolMADS1, 2, 3), two AP1/AP1-like (XP_013590290.1 and XP_013589842.1) and one AP1/CAL-like (BolMADS 10) orthologous genes showed different expression modes. BolMADS1, 2, and 3 showed relatively higher expression levels in the pistil than in other flower organs, while BolMADS10 showed low expression in all five flower organs. XP_013590290.1 and XP_013589842.1 highly expressed in sepal and petal, and XP_013590290.1 showed the highest expression level in sepal. AP3-like BolMADS19 and 21 and PI-like XP_013626307.1 and XP_013607408.1 were characterized as B class genes. These two groups of genes were specifically expressed in the petal and stamen and XP_013626307.1 showed the highest expression level in these two floral organs. BolMADS13 and 14 of the AG-like subfamily (C/D class) showed a similar expression pattern as BolMADS17 and 18 from the GGM13-like subfamily, which were characterized as B sister genes (Bs class). These two groups of genes were preferentially expressed in the pistil. In addition to expression in the pistil, another AG-like XP_013597287.1 gene also demonstrated trace expression in the stamen. BolMADS5 and 6 belonging to the SEP-like subgroup (E class) were detected in all four floral organs. The above results are consistent with the classical ABCDE flower model.

Expression profiles of 22 selected B. oleracea MADS genes in pedicel and four whorls of the flower including the sepal, petal, stamen, and pistil (a) based on qRT-PCR (b) analysis. The standard deviations of three biological replicates are represented by the error bars
XP_013630326.1 and XP_013632159.1 were SOC1 homologous gene belonging to the TM3-like subfamily. These two genes were expressed in all the five flower organs except the pistil. The overall expression level of XP_013632159.1 was much higher than that of XP_013630326.1. BolMADS28 of the MIKC* type was specifically expressed in the stamens and its expression level was the second highest among all B. oleracea MADS-box genes, indicating its involvement in stamen development. One type I gene, BolMADS71, was preferentially expressed in the petal at relatively high levels. This indicates that BolMADS71 also participates in the process of flower development.
Discussion
Cauliflower is one of the most important vegetables grown worldwide. Curd formation, floral organ development, and flowering time are important agronomic traits in cauliflower breeding and production for directly determining its adaptability and commerciality [36]. The MADS-box genes, particularly the MIKC family members, play important roles in plant flower development. Previous studies reported the characterization of MADS-box genes in several Cruciferous crops including A. thaliana and B. rapa [15, 43]. With the completion of the B. oleracea genome sequence, the BolMADS genes can be systematically identified and analyzed [42]. In this study, we identified a total of 91 BolMADS genes and comprehensively analyzed these genes to determine their phylogenetic relationships, chromosomal locations, gene structures, and expression patterns.
Causes of MADS-box genes loss in B. oleracea
Approximately 20–40 Mya, one whole genome duplication (WGD) event occurred in the Brassicaceae genome, after which the B. oleracea genome underwent another two whole genome triplication (WGT) events against Arabidopsis [44,45,46]. This caused the B. oleracea genome to become nearly 5-fold larger than the Arabidopsis genome. However, the number of MADS-box genes in B. oleracea is not directly proportional to the genome size. Only 91 BolMADS genes were identified, which is lower than the numbers in A. thaliana and B. rapa [15, 47]. This may be for two reasons. First, after genomic duplication, plants often show large areas of gene loss and chromosome rearrangements to maintain a metabolic balance [45]. Thus, most BolMADS genes may have been lost through later evolutionary processes. Second, approximately 4 million years ago, B. oleracea was differentiated as a branch [46]. Since then, this species has been subjected to strong artificial and natural selection, resulting in the loss of different BolMADS genes.
Organization of BolMADS genes
Gene organization was reported to play important roles in the evolution of multigene family [48,49,50]. In this study, the MIKC type genes contained a much larger number of introns compared to M-type genes. Among the MIKC type genes, 30 of 33 contained more than 3 introns, whereas all M-type genes had no or only one intron. The same intron distributions have been observed in other species, such as Arabidopsis, Chinese cabbage, rice, and cotton [15, 45, 51, 52]. This can be interpreted as a difference between type I and II MADS genes in their tendencies for deletion or acquisition of introns and indicate evolutionary conservation between different plant species [53]. BolMADS genes within the same group had a similar motif construction with common evolutionary ancestors. The consistency in intron number, motif structure, and phylogeny demonstrate that these genes were correctly classified.
Expression patterns of MIKC type genes in cauliflower
Most MIKC type genes were not expressed or lowly expressed in vegetative organs, including the roots, stems, and leaves, and were abundantly expressed in the reproductive organs, suggesting that MIKC type genes play important roles in the growth and development of reproductive tissues.
The classical model of flower development in plant is the ABCDE model. In A. thaliana, four A class genes have been cloned: AP1, CAL, FUL, and AGL79. AP1 acts as a floral organ identity gene to promote the development of petals and sepals and specifies floral meristem identity [16, 54]. However, CAL, which is an AP1 paralogue, has only partial functions of AP1 and promotes floral meristem formation during flower development [19, 20]. In cauliflower, two AP1 (XP_013590290.1 and XP_013589842.1) and one CAL homologous genes (BolMADS10) were detected and they were highly expressed in the curd tissue. Curd is the edible organ of cauliflower, which is composed of inflorescence meristems. This result indicates that these three genes play important roles in curd formation. FUL has a wide range of functions: it can play a role in the flower organ specification and function in carpel development [55, 56]. A total of three FUL homologous genes (BolMADS1, 2, and 3) were detected and showed similar expression patterns with relatively higher expression in the petals and ovary than in the other floral organs.
The main function of the B class genes (AP3 and PI) is to control the development of stamens and petals, particularly in the decision to form male reproductive organs [21]. Two AP3-like (BolMADS19 and 21) and two PI-like (XP_013626307.1 and XP_013607408.1) B class genes were detected in cauliflower and were highly expressed in the petals and stamens, indicating similar functions.
A total of 4 genes in Arabidopsis belong to the C/D class, including AG, SHP1 (also known as AGL1), SHP2 (also known as AGL5), and STK (also known as AGL11). These genes are mainly involved in the development of stamens, carpels, ovules, and fruit [4, 22, 23]. The BolMADS4, 13, 14 and XP_013597287.1 genes of the C/D class were preferentially expressed in the pistil, indicating their participation in pistil development.
E class genes (SEP) have obvious partially redundant functions during flower development. Moreover, these genes do not function alone in flower development, but function through interactions with ABC genes. In different species, SEP-like genes have different degrees of sub-functionalization and neo-functionalization [25, 57]. In cauliflower, three SEP-like genes were identified, with BolMADS5 showing the highest overall expression level in the flower. BolMADS5 was expressed in all five floral organs, but its expression level was highest in the petals, followed by in the ovary and stamen, indicating its roles in the development of these three floral organs.
Interestingly, BolMADS28 (MIKC* type) was specifically expressed in the stamens and its expression level was highest among all MIKC type genes. Its homologue in Arabidopsis is AGL104, which is required for pollen maturation and pollen tube growth [58]. BolMADS28 may be a new gene that plays an important role in stamen development in cauliflower.
Expression profiles of M type genes in cauliflower
Compared to MIKC MADS-box genes, the information about functions of M type genes are very limited. Several studies in Arabidopsis indicated that some M type genes participate in plant growth and reproduction, particularly in defining the female gametophyte and in the development of the embryo and endosperm [58–60]. In this study, 13 of 45 (28.89%) M type genes were detected in different tissues. Most detected M type genes were mainly expressed in reproductive tissues, while other several genes showed specific expression in the roots, such as BolMADS40 and 70. BolMADS71 showed the highest expression level in the flower tissue, mainly in the petals. This suggests the participation of BolMADS71 in petal development. BolMADS67, 72, and 73 were the top most abundantly expressed genes in the silique, indicating their roles in silique development.
Conclusions
In conclusion, 91 genes were identified in the Brassica oleracea genome. These genes were divided into 42 type II and 49 type I genes, and the type II genes were further divided into 13 subfamilies. The exon/intron structures, conserved motif distributions, and chromosomal locations of MADS-box family members in B. oleracea were also determined. Expression analysis of the BolMADS genes indicated that most MIKCC genes participated in flower development, which is consistent with the ABCDE model. Several non- MIKCC genes were also found to be highly expressed in the stamens and petals, indicating their important roles in the development of these floral organs.
Methods
Identification of MADS-box gene family in B. oleracea
The sequences of Arabidopsis MADS-box genes were obtained from the TAIR database (http://www.arabidopsis.org) and used as queries for a BLASTP algorithm-based against the B. oleracea genome database (http://www.ocri-genomics.org/bolbase/). For all candidate genes, we also examined whether they contain the MADS domain (PF00319) in the SMART (http://smart.embl-heidelberg.de) and Pfam (http://pfam.sanger.ac.uk) databases. Sequences without a MADS domain were deleted.
Conserved motifs and gene structure analysis of MADS-box proteins
Conserved motifs in MADS proteins of B. oleracea were identified using the program Multiple Em for Motif Elicitation (http://meme-suite.org/tools/meme). Default parameters were selected except that the maximum number of motifs was set to 10. The BolMADS gene structure was predicted using the program of GSDS2.0 (Gene Structure Display Server, http://gsds.cbi.pku.edu.cn/) for both genome and coding domain sequences.
Phylogenetic tree construction and protein conserved domain sequence alignment
MADS proteins of B. oleracea with A. thaliana and B. rapa [47] were aligned respectively using ClustalW with default settings. The two phylogenetic trees were constructed with MEGA5.04 software using the neighbor-joining method. The bootstrap test was executed by 1000 replications. The resulting phylogenetic tree was prepared in FigTree (v1.3.1) software.
Chromosomal location and gene duplication
The physical positions of all MADS-box genes along each chromosome were identified from the B. oleracea database. Gene duplication information was obtained by aligning all B. oleracea MADS-box genes. The criteria of both the query coverage rate and identity of the aligned region were more than 80%.
Plant materials and tissue-specific expression analysis
In this study, RNA-seq data from the B. oleracea Genomics Database (http://www.ocri-genomics.org/bolbase/) were used to analyze the expression patterns of MADS-box genes in different tissues of cultivated B. oleracea plants.
Seven types of tissues from cauliflower (a DH line from cauliflower cultivar “QingNong 65”) were used for quantitative real-time (qRT)-PCR analysis of all MADS-box genes. These tissues included the root, stem, leaf, curd, bud, flower, and silique. MADS-box genes with relatively higher transcription levels in the flower tissue were selected for qRT-PCR analysis in pedicel and four whorls of flower organs, including the sepal, petal, stamen, and pistil.
Approximately 100 mg of frozen tissue was collected for total RNA isolation using an RNA kit (RNAprep Pure Plant Kit, Tiangen, China) according to the manufacturer’s instructions. One microgram of total RNA was reverse-transcribed to the first-strand cDNA using the PrimeScript RT reagent Kit. Specific primers for all MADS-box genes were designed using Primer-Blast online software from NCBI (http://www.ncbi.nlm.nih.gov/tools/primer-blast/, Additional file 3: Table S1).
qRT-PCR assays were conducted in an ABI StepOnePlus machine using SYBR® Premix Ex Taq™ (TaKaRa, Shiga, Japan). The reaction mixture included 12.5 μl of 2 × SYBR Green PCR Master Mix (Applied Biosystems), a diluted cDNA template of 30 ng, 500 nmol of each primer in a final volume of 25 μl. The PCR conditions were as follows: 95 °C for 10 min, 40 cycles of 10 s at 95 °C, 50 °C for 15 s, and 72 °C for 30 s. The amplification efficiency of each primer pair was performed according to the method described by our previous study [61]. The 2−ΔCt value was used to measure the relative expression levels of specific genes. ACT and EF1a were used as reference genes. Each sample was analyzed in three biological and three technical replicates.
Abbreviations
- AGL :
-
AGAMOUS-LIKE
- AP1 :
-
APETALA1
- PI :
-
PISTILLATA
- SEP :
-
SEPALLATA
- WGD:
-
whole genome duplication
- WGT:
-
whole genome triplication
References
Passmore S, Maine GT, Elble R, Christ C, Tye BK. Saccharomyces cerevisiae protein involved in plasmid maintenance is necessary for mating of MATα cells. J Mol Biol. 1988;204:593–606.
Sommer H, Beltran JP, Huijser P, Pape H, Lönnig W, Saedler H, Schwarz-Sommer Z. Deficiens, a homeotic gene involved in the control of flower morphogenesis in Antirrhinum majus: the protein shows homology to transcription factors. EMBO J. 1990;9:605.
Yanofsky MF, Ma H, Bowman JL, Drews GN, Feldmann KA, Meyerowitz, E.M. The protein encoded by the Arabidopsis homeotic gene agamous resembles transcription factors. Nature. 1990;346:35–39.
Norman C, Runswick M, Pollock R, Treisman R. Isolation and properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fos serum response element. Cell. 1988;55:989–1003.
Honma T, Goto K. Complexes of MADS-BOX proteins are sufficient to convert leaves into floral organs. Nature. 2001;409:525–9.
Saedler H, Becker A, Winter KU, Kirchner C, Theissen G. MADSbox genes are involved in floral development and evolution. Acta Biochim Pol. 2001;48:351–8.
Alvarez-Buylla ER, Pelaz S, Liljegren SJ, Gold SE, Burgeff C, Ditta GS. An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc Natl Acad Sci U S A. 2000;97:5328–33.
De Bodt S, Raes J, Van de Peer Y, Theißen G. And then there were many: MADS goes genomic. Trends Plant Sci. 2003;8:475–83.
Cho S, Jang S, Chae S, Chung KM, Moon YH, Jang SK. Analysis of the C-terminal region of Arabidopsis thaliana APETALA1 as a transcription activation domain. Plant Mol Biol. 1999;40:419–29.
Kaufmann K, Melzer R, Theissen G. MIKC-type MADS-domain proteins: structural modularity, protein interactions and network evolution in land plants. Gene. 2005;347:183–98.
Riechmann JL, Krizek BA, Meyerowitz EM. Dimerization specificity of Arabidopsis MADS domain homeotic proteins APETALA1, APETALA3, PISTILLATA, and AGAMOUS. Proc Natl Acad Sci U S A. 1996;93:4793–8.
Fan HY, Hu Y, Tudor M, Ma H. Specific interactions between the K domains of AG and AGLs, members of the MADS domain family of DNA binding proteins. Plant J. 1997;12:999–1010.
Theissen G, Melzer R. Molecular mechanisms underlying origin and diversification of the angiosperm flower. Ann Bot. 2007;100:603–19.
Li C, Wang Y, Xu L, Nie S, Chen Y, Liang D. Genome-wide characterization of the MADS-box gene family in radish (Raphanus sativus L.) and assessment of its roles in flowering and floral organogenesis. Front. Plant Sci. 2016;7:1390.
Parenicová L, de Folter S, Kieffer M, Horner DS, Favalli C, Busscher J. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell Online. 2003;15:1538–51.
Coen ES, Meyerowitz EM. The war of the whorls: genetic interactions controlling flower development. Nature. 1991;353:31–7.
Zahn LM, Feng B, Ma H. Beyond the ABC-model: regulation of floral homeotic genes. Adv Bot Res. 2006;44:163–207.
Silva CS, Puranik S, Round A, Brennich M, Jourdain A, Parcy F. Evolution of the plant reproduction master regulators LFY and the MADS transcription factors: the role of protein structure in the evolutionary development of the flower. Front Plant Sci. 2015;6:1193.
Mandel MA, Gustafson-Brown C, Savidge B, Yanofsky MF. Molecular characterization of the Arabidopsis floral homeotic gene APETALA1. Nature. 1992;360:273–7.
Wang X, Cai Y, Wang H, Zeng Y, Zhuang X, Li B, Jiang L. Trans-Golgi network- located AP1 gamma adaptins mediate dileucine motif-directed vacuolar targeting in Arabidopsis. Plant Cell. 2014;26:4102–18.
Jack T, Brockman LL, Meyerowitz EM. The homeoticgene APETALA3 of Arabidopsis thaliana encodes a MADS-box and is expressed in petals and stamens. Cell. 1992;68:683–97.
Hugouvieux V, Silva CS, Jourdain A, Stigliani A, Charras Q, Conn V, Conn SJ, Carles CC, Parcy F, Zubieta C. Tetramerization of MADS family transcription factors SEPALLATA3 and AGAMOUS is required for floral meristem determinacy in Arabidopsis. Nucleic Acids Res. 2018;46:4966–77.
Pinyopich A, Ditta GS, Savidge B, Liljegren SJ, Baumann E, Wisman E, Yanofsky MF. Assessing the redundancy of MADS-box genes during carpel and ovule development. Nature. 2003;424:85–8.
Ditta G, Pinyopich A, Robles P, Pelaz S, Yanofsky MF. The SEP4 gene of Arabidopsis thaliana functions in floral organ and meristem identity. Curr Biol. 2004;14:1935–40.
Acri-Nunes-Miranda R, Mondragón-Palomino M. Expression of paralogous SEP-, FUL-, AG- and STK-like MADS-box genes in wild-type and peloric Phalaenopsis flowers. Front Plant Sci. 2014;5:76.
Hartmann U, Höhmann S, Nettesheim K, Wisman E, Saedler H, Huijser P. Molecular cloning of SVP: an egative regulator of the floral transition in Arabidopsis. Plant J. 2000;21:351–60.
Searle I, He Y, Turck F, Vincent C, Fornara F, Kröber S. The transcription factor FLC confers a flowering response to vernalization by repressing meristem competence and systemic signaling in Arabidopsis. Genes Dev. 2006;20:898–912.
Reeves PA, He Y, Schmitz RJ, Amasino RM, Panella LW, Richards CM. Evolutionary conservation of the FLOWERING LOCUS C mediated vernalization response: evidence from the sugar beet (Beta vulgaris). Genetics. 2007;176:295–307.
Lee JH, Yoo SJ, Park SH, Hwang I, Lee JS, Ahn JH. Role of SVP in the control of flowering time by ambient temperature in Arabidopsis. Genes Dev. 2007;21:397–402.
Lee J, Lee I. Regulation and function of SOC1, a flowering pathway integrator. J Exp Bot. 2010;61:2247–54.
Ferrándiz C, Liljegren SJ, Yanofsky MF. Negative regulation of the SHATTERPROOF genes by FRUITFULL during Arabidopsis fruit development. Science. 2000;289:436–8.
Liljegren SJ, Ditta GS, Eshed Y, Savidge B, Bowman JL, Yanofsky MF. SHATTERPROOF MADS-box genes control seed dispersal in Arabidopsis. Nature. 2000;404:766–70.
Nesi N, Debeaujon I, Jond C, Stewart AJ, Jenkins GI, Caboche M. The TRANSPARENT TESTA16 locus encodes the ARABIDOPSIS BSISTER MADS domain protein and is required for proper development and pigmentation of the seed coat. Plant Cell. 2002;14:2463–79.
Tapia-López R, García-Ponce B, Dubrovsky JG, Garay-Arroyo A, Pérez-Ruíz RV, Kim SH. An AGAMOUS-related MADS-box gene, XAL1 (AGL12), regulates root meristem cell proliferation and flowering transition in Arabidopsis. Plant Physiol. 2008;146:1182–92.
Picchi V, Migliori C, Scalzo RL, Campanelli G, Ferrari V, Cesare LFD. Phytochemical content in organic and conventionally grown Italian cauliflower. Food Chem. 2012;130:501–9.
Zhao ZQ, Gu HH, Sheng XG, Yu HF, Wang JS, Huang L, Wang D. Genome-wide single nucleotide poly morphisms discovery and high-density genetic map construction in cauliflower using specific locus amplified fragment sequencing. Front Plant Sci. 2016;7:334.
Duclos DV, Björkman T. Meristem identity gene expression during curd proliferation and flower initiation in Brassica oleracea. J Exp Bot. 2008;59:421–33.
Yu J, Zhao M, Wang X, Tong C, Huang S, Tehrim S, Liu Y, Hua W, Liu S. Bolbase: a comprehensive genomics database for Brassica oleracea. BMC Genomics. 2013;14:664.
Saha G, Park JI, Jung HJ, Ahmed NU, Chung MY, Hur YK, Cho YG. Genome-wide identification and characterization of MADS-box family genes related to organ development and stress resistance in Brassica rapa. BMC Genomics. 2015;16:178.
Bowers JE, Chapman BA, Rong J, Paterson AH. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature. 2003;422:433–8.
Chalhoub B, Denoeud F, Liu SY, Parkin IAP, Tang HB, Wang XY, Chiquet J, Belcram H, Tong C, Samans B, Corréa M, Da Silva C, Just J, Falentin C, Koh CS, Le Clainche I, Bernard M, Bento P, Noel B, Labadie K, Alberti A, Charles M, Arnaud D, Guo H, Daviaud C, Alamery S, Jabbari K, Zhao MX, Wincker P. Early allopolyploid evolution in the post- Neolithic Brassica napus oilseed genome. Science. 2014;345:950–3.
Liu SY, Liu YM, Yang XH, Tong CB, Edwards D, Parkin IAP, Zhao MX, Ma JX, Yu JY, Huang SM, Wang XY, Wang JY, Lu K, Fang ZY, Bancroft I, Yang TJ, Hu Q, Wang XF, Yue Z, Li HJ, Yang LF, Wu J, Zhou Q, Wang WX, King GJ, Pires JC, Lu CX, Wu ZY, Sampath P, Wang Z, Guo H, Pan SK, Yang LM, Min JM, Zhang D, Jin DC. Paterson AH the Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat Commun. 2014;5:3930.
Duan WK, Song XM, Liu TK, Huang ZN, Ren J, Hou XL, Li Y. Genome-wide analysis of the MADS-box gene family in Brassica rapa (Chinese cabbage). Mol Gen Genomics. 2015;290:239–55.
Wang PP, Li J, Gao XY, Zhang D, Li AL, Liu CG. Genome-wide screening and characterization of the Dof gene family in physic nut (Jatropha curcas L.). Int J Mol Sci 2018;19:1598.
Diao WP, Snyder JC, Wang SB, Liu JB, Pan BG, Guo GJ, Ge W. Genome-wide analyses of the NAC transcription factor gene family in pepper (Capsicum annuum L.): chromosome location, phylogeny, structure, expression patterns, cis-elements in the promoter, and interaction network. Int J Mol Sci. 2018;19:1028.
Zhao M, Chen P, Wang WY, Yuan FJ, Zhu DH, Wang Z, Ying XX. Molecular evolution and expression divergence of HMT gene family in plants. Int J Mol Sci 2018;19:1248.
Airoldi CA, Davies B. Gene duplication and the evolution of plant MADS-box transcription factors. J Genet Genomics. 2012;39:157–65.
Ren Z, Yu D, Yang Z, Li C, Qanmber G, Li Y, Li J, Liu Z, Lu L, Wang L, Zhang H, Chen Q, Li F, Yang Z. Genome-wide identification of the MIKC-type MADS-box gene family in Gossypium hirsutum L. Unravels Their Roles in Flowering. Front Plant Sci. 2017;8:384.
Nam J, Ma H, Nei M. Antiquity and evolution of the MADSbox gene family controlling flower development in plants. Mol Biol Evol. 2003;20:1435–47.
Ye L, Wang B, Zhang W, Shan H, Kong H. Gains and losses of cis-regulatory elements led to divergence of the Arabidopsis APETALA1 and CAULIFLOWER duplicate genes in the time, space, and level of expression and regulation of one paralog by the other. Plant Physiol. 2006;171:1055–69.
Bemer M, van Mourik H, Muiño JM, Ferrándiz C, Kaufmann K, Angenent GC. FRUITFULL controls SAUR10 expression and regulates Arabidopsis growth and architecture. J Exp Bot. 2017;68:3391–403.
Balanzà V, Martínez-Fernández I, Sato S, Yanofsky MF, Kaufmann K, Angenent GC, Bemer M, Ferrándiz C. Genetic control of meristem arrest and life span in Arabidopsis by a FRUITFULL-APETALA2 pathway. Nat Commun. 2018;9:565.
Soza VL, Snelson CD, Hewett Hazelton KD, Di Stilio VS. Partial redundancy and functional specialization of E-class SEPALLATA genes in an early-diverging eudicot. Dev Biol. 2016;419:143–55.
Portereiko MF, Lloyd A, Steffen JG, Punwani JA, Otsuga D, Drews GN. AGL80 is required for central cell and endosperm development in Arabidopsis. Plant Cell. 2006;18:1862–72.
Kang IH, Steffen JG, Portereiko MF, Lloyd A, Drews GN. The AGL62 MADS domain protein regulates cellularization during endosperm development in Arabidopsis. Plant Cell. 2008;20:635–47.
Bemer M, Wolters-Arts M, Grossniklaus U, Angenent GC. The MADS domain protein DIANA acts together with AGAMOUS-LIKE80 to specify the central cell in Arabidopsis ovules. Plant Cell. 2008;20:2088–101.
Sheng XG, Zhao ZQ, Yu HF, Wang JS, Zheng CF, Gu HH. In-depth analysis of internal control genes for quantitative real-time PCR in Brassica oleracea var. botrytis. Genet. Mol. Res. 2016;15(3). https://doi.org/10.4238/gmr.15038348.
Adamczyk BJ, Fernandez DE. MIKC MADS domain heterodimers are required for pollen maturation and tube growth in Arabidopsis. Plant Physiol. 2009;149:1713–23.
Acknowledgements
We thank Dr. Wan of Zhejiang Academy of Agricultural Sciences, Hangzhou, China for providing guidance on the use of relevant analytical softwares.
Funding
This work was financially supported by The National Key Research and Development Program of China (2017YFD0101805, 2017YFE0114500), Natural Science Foundation of China (31501768), Science and Technology Department of Zhejiang Province (2016C02051–5, LGN18C150010), Ningbo Science and Technology Program (2015C110008) and Zhejiang Academy of Agricultural Sciences. The funders had no role in study design, data collection, data analysis, data interpretation, the writing of the manuscript or decision to publish.
Availability of data and materials
The datasets supporting the conclusions of the present study are included within this article (and its additional files).
Author information
Authors and Affiliations
Contributions
XS was responsible for the main part of data analysis, experimental design and manuscript writing. ZZ and JW performed the qRT-PCR and participated the data analysis. HY and YS performed the primer design and plant materials preparation. XZ performed the cauliflower accessions planting and samples collection. HG was responsible for the programs and manuscript revision. All the authors have commented, read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Phylogenetic tree of MADS-box genes from B. oleracea and A. thaliana using neighbor-joining method. (DOC 414 kb)
Additional file 2:
Figure S2. Phylogenetic tree of MADS-box genes from B. oleracea and B. rapa using neighbor-joining method. (DOC 1007 kb)
Additional file 3:
Table S1. The specific primers for qRT-PCR of 87 MAD-box genes. (DOC 215 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sheng, XG., Zhao, ZQ., Wang, JS. et al. Genome wide analysis of MADS-box gene family in Brassica oleracea reveals conservation and variation in flower development. BMC Plant Biol 19, 106 (2019). https://doi.org/10.1186/s12870-019-1717-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-019-1717-y