
Abstract
Background
Zygophyllum xanthoxylum is a succulent xerophyte with remarkable tolerance to diverse abiotic stresses. Previous studies have revealed important physiological mechanisms and identified functional genes associated with stress tolerance. However, knowledge of the regulatory genes conferring stress tolerance in this species is poorly understood.
Results
Here, we present a comprehensive analysis of regulatory genes based on the transcriptome of Z. xanthoxylum roots exposed to osmotic stress and salt treatments. Significant changes were observed in transcripts related to known and obscure stress-related hormone signaling pathways, in particular abscisic acid and auxin. Significant changes were also found among key classes of early response regulatory genes encoding protein kinases, transcription factors, and ubiquitin-mediated proteolysis machinery. Network analysis shows a highly integrated matrix formed by these conserved and novel gene products associated with osmotic stress and salt in Z. xanthoxylum. Among them, two previously uncharacterized NAC (NAM/ATAF/CUC) transcription factor genes, ZxNAC083 (Unigene16368_All) and ZxNAC035 (CL6534.Contig1_All), conferred tolerance to salt and drought stress when constitutively overexpressed in Arabidopsis plants.
Conclusions
This study provides a unique framework for understanding osmotic stress and salt adaptation in Z. xanthoxylum including novel gene targets for engineering stress tolerance in susceptible crop species.
Similar content being viewed by others


Background
Drought and salinity are two major environmental stressors that impact crop yields worldwide [1]. Salinity threatens approximately one-fifth of cultivated lands globally. Meanwhile, up to one third of land surfaces are exposed to drought [2, 3]. Many experts agree that using molecular genetics to breed crops with higher yields and improved tolerance to abiotic stresses is an effective strategy in safeguarding food supplies. Yet, this task remains one of the greatest challenges faced by modern agriculture [4]. Deciphering key genes and regulatory mechanisms in salt and drought tolerance and adaptation is a key step in engineering stress-tolerant crop plants [5, 6].
Mechanisms of abiotic stress tolerance have mainly been studied in model plants. High throughput sequencing and functional genomics tools in model plants have yielded numerous abiotic stress tolerance genes grouped into two major classes: functional genes or regulatory genes [7]. Functional genes include important enzymes and metabolic proteins including detoxification enzymes, water channels, ion transporters, heat shock proteins and late embryogenesis abundant proteins, which directly function to protect cells from stress. Regulatory genes include important signaling and regulatory proteins that modulate protein activity during stress exposure including hormone and stress signaling pathway components, transcription factors, and ubiquitin-mediated proteolysis machinery. A large number of studies show that scope for enhancing stress tolerance in plants by altering single functional genes is limited, due to the complexity of stress responses [8, 9]. Thus, alteration of key regulatory genes is desirable because this can mimic or enhance stress signals by regulating a large spectrum of downstream stress-responsive genes in conferring tolerance [10, 11]. Knowledge of regulatory genes is therefore important from an genetic engineering perspective in plants [11, 12].
Despite vast knowledge derived from Arabidopsis thaliana (Arabidopsis) and Oryza sativa (rice) model plants, a low capacity for stress tolerance limits their usefulness as discovery tools. By contrast, xerophyte and halophyte species, widely distributed in arid and saline regions, have evolved multiple protective mechanisms that allow them to grow successfully under hostile conditions [6, 13, 14]. A detailed understanding of salt and drought protective mechanisms in naturally tolerant species, and the identification of key regulatory genes, is a promising new strategy for breeding salt and drought tolerant crops [13, 15].
Zygophyllum xanthoxylum is a succulent xerophyte with a highly developed root system and strong stress tolerance. The natural range of Z. xanthoxylum includes arid and semiarid lands in northwestern China and Mongolia [16]. This shrub is widely planted in China for protecting fragile desert ecosystems and improving vegetation coverage [17]. Previous investigations in Z. xanthoxylum have focused on growth properties, nutritive characteristics, and transpiration resulting in the characterization of several drought and salt stress response functional genes [17,18,19,20,21,22]. Previously, we ge nerated transcriptome datasets of roots and leaves of Z. xanthoxylum to identify differentially expressed genes (DEGs) under osmotic stress and salt treatments [20]. Attention was focused on several important classes of functional genes traditionally associated with drought and salt stress responses, including ion transporters, reactive oxygen species (ROS) scavenging systems, and photosynthesis [20].
Here, we further analyzed the transcriptome and digital gene expression profiling data of Z. xanthoxylum roots under osmotic stress and salt treatments to identify potential upstream regulators of these functional genes (Fig. 1). Our transcriptome analysis focused on signaling pathways important for stress tolerance, transcription factors important for signal output, and ubiquitin proteasome system enzymes important for protein turnover (Fig. 1). To further analyze this transcriptome, DEGs in Z. xanthoxylum roots were matched to putative orthologs in Arabidopsis, which allowed us to perform in silico functional inference, including gene network analysis for protein function, protein subcellular localization, and gene co-expression. Finally, candidate genes were selected and functionally characterized. Among these, Z. xanthoxylum unigenes orthologous to Arabidopsis NAC transcription factor genes NAC083 and NAC035 conferred drought and salt tolerance when constitutively overexpressed in Arabidopsis plants, confirming relevance of the dataset for crop engineering.

An overview of relationships between the regulatory gene groups studied in this work and their downstream functional genes. The 6 h and 24 h DEG transcriptomes of Z. xanthoxylum roots under osmotic stress and salt treatment were analyzed. Regulatory gene categories, blue boxes. Functional gene categories, gray boxes
Results and discussion
Transcriptome profile of roots during osmotic stress and salt treatments
To learn more about how Z. xanthoxylum adapts to drought and saline environments, we further analyzed the transcriptome data of seedling roots exposed to osmotic stress and salt treatments for 6 h and 24 h [20]. Roots were studied because they are the first organs to be exposed to osmotic and salt stress conditions [23]. 6063 and 6258 DEGs were identified in Z. xanthoxylum roots exposed to osmotic stress or salt treatments for 6 h, respectively (Additional file 1: Figure S1a). In osmotic-stressed seedling roots, 4000 DEGs were up-regulated and 2063 DEGs were down-regulated. In salt-treated seedling roots, 4140 DEGs were up-regulated and 2118 DEGs were down-regulated (Additional file 1: Figure S1a). Venn-diagram analysis shows that a large number of 6 h DEGs overlap in expression, including 2780 up-regulated genes and 1402 down-regulated genes that are present in both osmotic stress and salt treatments, suggesting that signaling pathways controlling these responses in Z. xanthoxylum are interacting (Additional file 1: Figure S1a). By comparison, only 2708 and 1307 DEGs were identified in roots exposed to osmotic stress or salt treatments for 24 h, respectively (Additional file 1: Figure S1b). In osmotic-stressed seedling roots, 1723 DEGs were up-regulated and 985 DEGs were down-regulated. In salt-treated seedling roots, 657 DEGs were up-regulated and 750 DEGs were downregulated (Additional file 1: Figure S1b). Similarly, Venn-diagram analysis revealed that 329 up-regulated genes and 309 down-regulated genes overlapped in osmotic stress- and salt-treated seedling roots. Overall, the 6 h dataset was more complex than treatment for 24 h, reflecting that regulatory genes are generally early responders to environmental signals [7, 20]. We therefore selected the 6 h dataset as the focus of further analysis.
Functional assignment of DEGs under osmotic stress and salt treatment
To evaluate the biological pathways and molecular function of genes participating in osmotic stress and salt response, DEGs in 6 h-treated Z. xanthoxylum roots were functionally annotated by aligning their sequences to proteins in the KEGG database (see Methods). 3028 DEGs and 3167 DEGs were functionally annotated under osmotic stress and salt treatment, respectively. Eighteen functional pathways were identified as significantly enriched (p ≤ 0.05) during both treatments (Fig. 2a). In osmotic-stressed Z. xanthoxylum, the dominant pathways were “metabolic pathways” (737 DEGs), “biosynthesis of secondary metabolites” (357 DEGs), “plant hormone signal transduction” (197 DEGs) “plant-pathogen interaction” (179 DEGs), “ribosome” (161 DEGs), “RNA transport” (135 DEGs) and “spliceosome” (116 DEGs). In salt-treated Z. xanthoxylum, the dominant pathways were “metabolic pathways” (784 DEGs), “biosynthesis of secondary metabolites” (400 DEGs), “ribosome” (230 DEGs), “plant-pathogen interaction” (180 DEGs), “plant hormone signal transduction” (179 DEGs), “RNA transport” (139 DEGs) and “spliceosome” (118 DEGs) (Fig. 2a).

KEGG analysis of DEGs in Z. xanthoxylum roots. a Top 18 enriched functional categories and distribution of DEGs in (b, c) ABA and (d, e) auxin signaling pathways under (b, d) osmotic stress and (c, e) salt treatment. a X-axis indicates the number of DEGs in KEGG pathways, and Y-axis represents the specific category of DEGs in the pathway. Black bars, number of up-regulated genes. White bars, number of down-regulated genes. Black arrows, total number of up-regulated genes. White arrows, total number of down-regulated genes
Among hormone signaling pathways, abscisic acid (ABA) signaling components were significantly enriched followed by auxin, ethylene, and cytokinin signaling pathway terms. ABA plays a well-established role in plant stress response signaling including salinity, drought, osmotic, and cold stresses [24]. During stress responses, cellular ABA levels increase via ABA biosynthesis. PYL/PYR/RCAR (PYRABACTIN RESISTANCE1/PYR1-LIKE/REGULATORY COMPONENTS OF ABA RECEPTORS) receptors bind to ABA and interact with PP2C (protein phosphatase 2C) negative regulators thereby releasing SnRKs (sucrose non-fermenting like kinases). Activated SnRKs phosphorylate downstream proteins, including bZIP (basic leucine zipper) transcription factors, thereby activating stress tolerance genes [24, 25]. DEGs encoding all of these core positive components of ABA signaling were upregulated in Z. xanthoxylum roots under osmotic stress and salt treatments (Fig. 2b and c, Additional file 2: Table S1). PP2C components showed the most change, with 8 up-regulated and 3 down-regulated genes under osmotic stress conditions and 8 up-regulated and 1 down-regulated gene under salt conditions (Fig. 2b and c, Additional file 2: Table S1). These data indicate that ABA signaling is an important factor in Z. xanthoxylum stress tolerance, similar to other plants. ABA signaling directly activates stress tolerance genes as well as initiates secondary mechanisms for stress tolerance [26]. Auxin is an important regulator of plant growth and development but its role in abiotic stress responses is poorly studied [27]. Interestingly, numerous auxin signaling components were differentially expressed in osmotic-stressed and salt-treated Z. xanthoxylum roots. Under osmotic stress, 28 auxin signaling pathway genes were upregulated, including 2 TIR1/AFB (Transport Inhibitor Response1/Auxin Signaling F-box) receptor genes, 12 AUX/IAA (Auxin/Indole-3-Acetic Acid) repressor genes, 3 ARF (Auxin Response Factor) transcription factor genes, 3 GH3 (Gretchen Hagen 3) and 8 SAUR (Small Auxin-Up RNA) auxin early-response genes (Fig. 2d and Additional file 2: Table S2). Under salt stress, 24 auxin signaling pathway genes were upregulated including 2 TIR/AFB genes, 8 AUX/IAA genes, 4 ARF genes, 4 GH3 genes, and 6 SAUR genes (Fig. 2e and Additional file 2: Table S2). This enrichment suggests that auxin signaling is an important element in Z. xanthoxylum stress tolerance. Unlike Arabidopsis, Z. xanthoxylum can effectively maintain growth and regulate its root architecture under osmotic stress and salinity [18,19,20]. In turn, auxin plays a well-characterized role in stimulating root growth [27]. Collectively, steep changes in ABA and auxin signaling genes at 6 h under osmotic-stress and salt treatments suggests an important interconnecting role for these hormone pathways in the stress response.
Osmotic stress- and salt-responsive regulatory genes in Z. xanthoxylum roots
Perception of stress signals leads to signal transduction and the activation of protective physiological and metabolic responses [12, 28]. To further analyze signal transduction genes in osmotic or salt-treated Z. xanthoxylum DEG libraries, we used Gene Ontology (GO) enrichment analysis. Over-represented categories were found to include protein kinases (253 and 204 kinase genes in osmotic-stressed and salt-treated DEGs libraries, respectively) (Fig. 3a and b), transcription factors (126 and 143 transcription factor genes in osmotic-stressed and salt-treated DEGs libraries, respectively) (Fig. 3c and d) and ubiquitin proteasome system (UPS) genes (80 and 85 UPS genes in osmotic-stressed and salt-treated DEGs libraries, respectively) (Fig. 3e and f).

Distribution of osmotic stress- and salt-responsive DEGs encoding (a, b) protein kinases, (c, d) transcription factors, and (e, f) ubiquitin proteasome system-related enzymes in Z. xanthoxylum roots. Black bars, number of up-regulated genes. White bars, number of down-regulated genes. Black arrows, total number of up-regulated genes. White arrows, total number of down-regulated genes
Protein kinases
Protein kinases play key roles in the perception of stress-related signals. Major classes involved in signal perception are leucine-rich repeat receptor-like kinases (LRR-RLKs), cell-wall associated kinases, lectin-domain-containing receptor kinases, and receptor-like cytoplasmic kinases [29, 30]. A large number of these genes are rapidly induced in osmotic or salt stress treated Arabidopsis plants [30, 31]. Several have confirmed roles in drought and salt tolerance, including FERRONIA [32] and RECEPTOR-LIKE PROTEIN KINASE 1 [33]. Numerous DEGs encoding RLKs are upregulated in our dataset (Fig. 3a and b and Additional file 3: Figure S2a). For example, the Z. xanthoxylum FERRONIA-like Unigene61098_All is up-regulated 4.6- and 7.5-fold during osmotic stress and salt treatment, respectively (Additional file 2: Table S3). Many RLK genes not previously linked to stress are also differentially upregulated in Z. xanthoxylum roots including LRR-RLK-like Unigene9800_All, Unigene176_All, and Unigene12756_All with highly significant blast hits to Hevea brasiliensis BAK1 (BRASSINOSTEROID INSENSITIVE 1-ASSOCIATED RECEPTOR KINASE 1) [34], Ricinus communis ERECTA [35], and Prunus avium BAM3 (BARELY ANY MERISTEM 3) [36], respectively (Additional file 2: Table S3). The Arabidopsis counterparts of these genes are well studied but not in the context of abiotic stress responses [34, 36,37,38,39]. Future work to evaluate the role of these genes in stress-related functions using Arabidopsis model plants is a valuable future direction.
Protein kinases are also important for signal transmission. Major classes include CDPKs (calcium-dependent protein kinases) and MAPKs (mitogen-activated protein kinases) [40]. In Arabidopsis, several CDPKs and MAPKs are involved in abiotic stress signaling pathways [41,42,43]. Arabidopsis plants overexpressing CPK6 have enhanced tolerance to salt and drought stresses [42]. Similarly, transgenic barley (Hordeum vulgare) plants overexpressing HvMPK1 showed greater tolerance to salt and other abiotic stresses [44]. Our DEG libraries include 31 and 28 predicted calcium response protein kinase genes, and 28 and 24 predicted MAPK genes in salt- and osmotic stress-treated Z. xanthoxylum roots, respectively (Fig. 3a and b, Additional file 3: Figure S2b and Additional file 2: Table S3). These data are consistent with known roles for Arabidopsis CDPKs and MAPKs in abiotic stress signaling.
Transcription factors
Transcription factors are important outputs of signaling pathways, directly responsible for the activation or repression of stress-responsive genes [1, 12, 45]. Major classes include NAC (NAM/ATAF/CUC), AP2/ERF (APETALA2 and ethylene-responsive element binding proteins), bHLH (basic helix-loop-helix), MYB (myeloblastosis), WRKY (WRKY-domain) and bZIP transcription factors whose members confer drought and salt stress tolerance in various plants [1]. For example, many rice WRKY genes exhibit obviously different functions under drought or salt stress [46] and soybean WRKY54 over-expressed in transgenic Arabidopsis confers tolerance to drought and salt stress [47]. bZIP24 was identified by screening salt-inducible transcripts in Arabidopsis and a closely-related halophyte species with functional analysis showing that repression increased salt tolerance and the transcript abundance of numerous stress response genes [48]. Multiple bHLH transcription factors also have stress-related roles such as MYC2 (Myelocytomatosis protein 2) involved in jasmonic acid signaling and AIB (ABA-inducible bHLH-type transcription factor) whose overexpression enhances the drought tolerance of transgenic plants [49, 50]. Consistent with these studies, 126 and 144 transcription factors DEGs were identified in Z. xanthoxylum under osmotic stress and salt treatment, respectively. NAC, AP2/ERF, bHLH, MYB, WRKY, and bZIP families were the most abundant (Fig. 3c and d). Many of these DEGs matched to putative orthologs in other plants with a characterized role in stress biology. For example, CL8432.Contig3_All, CL6477.Contig2_All, Unigene2817_All, Unigene2222_All, Unigene2285_All were up-regulated in osmotic-stressed and salt-treated Z. xanthoxylum (Additional file 3: Figure S2c and Additional file 2: Table S4). These genes encode predicted orthologs of Arabidopsis ERF96 [51], bHLH106 [52], MYB3 [53], WRKY22 [54], and bZIP53 [55], which are previously identified as salt or drought responsive genes with important roles in abiotic stress signaling pathways.
Numerous other transcription factor genes with less-defined roles in abiotic stress were up- or down-regulated in osmotic-stressed and salt-treated Z. xanthoxylum roots (Fig. 3c and d and Additional file 2: Table S4). Examples include Unigene57160_All, Unigene16368_All, CL6534.Contig1_All and Unigene9789_All, which are orthologous to Arabidopsis MYB40, NAC083, NAC035, and WRKY69, respectively (Additional file 2: Table S4). Several transcription factor families not previously associated with abiotic stress mechanisms were also identified. Examples include GATA, GRAS (GAI/RGA/SCR) and ARF transcription factors. In rice, abiotic stress signaling by ABA and salt treatment causes the induction and differential splicing of 28 GATA transcription factor genes revealing tight regulation of transcript abundance and splice variants and possible diverse roles in abiotic stress signaling [56]. Within these families, the expression level of some genes changed significantly. For example, CL11183.Contig1_All predicted to encode a GRAS transcription factor and was up-regulated more than 10-fold in response to osmotic stress and salt treatment in Z. xanthoxylum (Additional file 2: Table S4). Some of these previously uncharacterized transcription factors might represent novel regulators of osmotic stress or salt tolerance.
UPS enzymes
Ubiquitination is an important mechanism for regulating protein turnover in response to stimuli and UPS enzymes play a central role [57, 58]. Accordingly, numerous Z. xanthoxylum DEGs induced under osmotic-stress and salt treatment encode UPS-related components such as ubiquitins, proteasome subunits, E1 ubiquitin-activating enzymes, E2 ubiquitin-conjugating enzymes, and E3 ubiquitin ligases (Fig. 3e and f and Additional file 3: Figure S2d). Among these, 22 F-box E3 genes were induced both in osmotic stress- and salt-treated Z. xanthoxylum (Fig. 3e and f). Four of these genes match to Arabidopsis orthologs with known roles in stress signaling including CL7586.Contig2_All (Additional file 2: Table S5). This F-box gene is a predicted ortholog of Arabidopsis TUBBY LIKE PROTEIN3 important for plant responses to ABA, salt, and osmotic stress [59]. Several U-box, RING (really interesting new gene) and HECT (homology to E6-APC terminus) E3 ubiquitin ligase genes are also up- or down-regulated in osmotic stress- or salt-treated Z. xanthoxylum roots (Fig. 3e and f, Additional file 3: Figure S2d and Additional file 2: Table S5) including CL4067.Contig1_All. This gene is a predicted ortholog of Arabidopsis PUB18 (Plant U-box 18) induced by ABA, drought and salt stress, which functions as a negative regulator of ABA-mediated drought stress responses [60]. These findings highlight potentially important roles for E3 ligases as both positive and negative stress response regulators in Z. xanthoxylum roots.
In summary, 459 and 432 protein kinase, transcription factor, and UPS genes were differentially expressed in osmotic-stressed and salt-treated Z. xanthoxylum DEG libraries, respectively. Hierarchical cluster analyses indicate that transcripts for most of these genes are increased at 6 h but not 24 h (Additional file 3: Figure S2). For example, 103 up-regulated and 41 down-regulated transcription factor genes were among the DEGs of Z. xanthoxylum roots treated for 6 h with salt (Additional file 3: Figure S2) but only 9 of these transcription factor genes were among DEGs at 24 h (Additional file 4: Figure S3). Thus, 6 h DEGs are more likely to be early response regulators in Z. xanthoxylum roots under osmotic stress and salt treatments. To test of the reproducibility of our RNA-sequencing data, we used qRT-PCR to independently monitor the transcript abundance of 20 randomly selected DEGs representing protein kinases, transcription factors, and UPS enzymes. These results were generally consistent with the RNA-sequencing data (Additional file 2: Table S6).
Protein-protein interaction networks of Z. xanthoxylum in response to osmotic stress and salt treatments
To further explore functional relationships among regulatory DEGs from Z. xanthoxylum, BLASTp homology searches against the Arabidopsis TAIR 10 genome release were used to identify putative orthologs. Among 459 osmotic stress-responsive regulatory DEGs in Z. xanthoxylum, 359 unique DEG orthologs were identified in Arabidopsis. Similarly, among the 432 salt-responsive regulatory DEGs in Z. xanthoxylum, 339 unique DEG orthologs were identified in Arabidopsis. STRING analysis was performed to present known gene co-expression, genetic interactions, protein interactions, and protein subcellular localization. Cytoscape was used to calculate and display the most over-represented functional categories, demonstrating a central gene cluster enriched in osmotic stress and salt responsive genes (Fig. 4a and b; Additional file 5: Figure S4 and Additional file 6: Figure S5). In the osmotic stress-responsive network, 220 DEG proteins, including 89 protein kinases, 55 transcription factors, and 76 UPS enzymes are represented (Fig. 4a, Additional file 5: Figure S4 and Additional file 2: Table S7). In the salt-responsive network, 204 DEG proteins, including 85 protein kinases, 42 transcription factors, and 77 UPS enzymes are represented (Fig. 4b, Additional file 6: Figure S5 and Additional file 2: Table S7). These highly integrated networks incorporated both conserved and novel gene products associated with exposure to osmotic stress and salt conditions in Z. xanthoxylum. For example, Unigene22615_All matches to Arabidopsis MKK9, a known MAPK determinant of salt tolerance [61]. When protein kinases, transcription factors, and UPS enzymes directly related to MKK9 are examined, these proteins together with relationships among them form a sub-network showing predicted regulatory events surrounding Unigene22615_All in response to osmotic-stress and salt treatment of Z. xanthoxylum, respectively (Fig. 4c and d). On the other hand, Unigene57160_All blast hits to functionally unresolved Arabidopsis transcription factor gene MYB40. Network analysis between MYB40 and protein kinase, transcription factors, and UPS enzymes reveals potentially novel regulatory events (Fig. 4e and f). For example, MYB40 may regulate plant response to osmotic and salt stress by interacting with GATA transcription factors, regulating the transcription level of GATA genes, or GATAs may function upstream of MYB40 (Fig. 4e and f). These relationships provide a theoretical basis for investigating MYB40 function in stress tolerance. Network analysis also reveals an extensive ABA signaling network in Z. xanthoxylum roots in response to osmotic-stress and salt treatments (Fig. 5 and Additional file 2: Table S1). Under osmotic stress, the majority of ABA pathway genes are up-regulated, including 2 ABA biosynthesis and catabolism genes, 1 transport gene, 1 receptor gene, 1 SnRK gene, 1 ABI transcription factor gene, and various other ABA responsive and UPS genes (Fig. 5b). In total, 30 interrelated proteins shed light on a complex ABA-dependent network (Fig. 5a and b). Analysis of conserved and novel gene products within these networks provides a foundation for future study (Figs. 4 and 5 and Additional file 2: Table S7).

Protein-protein interaction analysis of Arabidopsis DEG orthologs. a osmotic stress-responsive DEG network and (b) salt-responsive DEG network. For high resolution images, see Additional file 5: Figure S4 and Additional file 6: Figure S5. c,d predicted MKK9 (Unigene22615_All) module showing regulatory proteins and relationships among them (nodes and edges) for Z. xanthoxylum roots in response to (c) osmotic stress and (d) salt, respectively. e, f predicted MYB40 (Unigene57160_All) module showing regulatory proteins and relationships among them (nodes and edges) for Z. xanthoxylum roots in response to (e) osmotic stress and (f) salt, respectively. Green nodes and edges (lines), kinases and interactions among them; magenta nodes and edges, transcription factors and interactions among them; blue nodes and edges, UPS enzymes and interactions among them; gray edges, interactions among kinases, transcription factors, and UPS enzymes except those among the same category of proteins

Module for ABA signaling pathway in osmotic stress responses of Z. xanthoxylum roots. a Functional network based on known Arabidopsis ABA-dependent signaling components, cross-referenced against the complete DEG functional network. Node colors represent different classes of signaling components: orange, ABA biosynthesis; red, receptors; yellow-green, PP2Cs, green, kinases; dark magenta, transporters; magenta, transcription factors; and blue, UPS enzymes. Border colors of nodes represent expression as log2 fold-change. Magenta, increased expression; the deeper the color, the higher the expression. Green, decreased expression; the deeper the color, the lower the expression. Edge (line) colors, interactions of proteins in the core ABA signaling pathway (red), kinases (green), UPS enzymes (blue), transcription factors (magenta), and proteins except in the same class (cyan). Thickness of edges, normalized link closeness; the thicker the edge, the closer the link. b ABA signaling and biosynthetic pathway components. Shading, colors represent different classes of signaling components: orange, ABA biosynthesis; red, receptors; yellow-green, PP2Cs, green, kinases; dark magenta, transporters; magenta, transcription factors; and blue, UPS enzymes
Validation of stress-related functions for novel ZxNAC regulatory genes
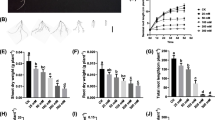
To further analyze the biological relevance of our dataset, novel Z. xanothxylum regulatory DEGs from our study were tested as potential osmotic and salt stress determinants in Arabidopsis. Eight candidate genes were selected for functional characterization including two protein kinases genes [Unigene9800_All (ZxSERK1) and CL11556.Contig3_All (ZxMRH1)] and six transcription factor genes [(Unigene16368_All (ZxNAC083), CL6534.Contig1_All (ZxNAC035), Unigene57160_All (ZxMYB40), CL9880.Contig2_All (ZxWRKY29), Unigene9789_All (ZxWRKY69) and Unigene2222_All (ZxWRKY22)]. Results are presented for ZxNAC083 and ZxNAC035, named after their putative orthologs in Arabidopsis. Independent qRT-PCR analysis confirmed that both genes are highly induced in osmotic-stressed and salt-treated Z. xanthoxylum roots (Additional file 7: Figure S6a, b and Additional file 2: Table S6). Transgenic Arabidopsis plants expressing these genes under the control of a strong constitutive Cauliflower Mosaic Virus 35S promoter were next generated. Two independent transgenic lines per construct were selected for phenotypic analyses under normal, drought, and salt-stress conditions (Additional file 7: Figure S6c, d). The drought tolerance of wild type Col-0 and transgenic lines was first compared by withholding water for 7 days [62]. Under this treatment, wild-type plants began to wilt but the ZxNAC083 and ZxNAC035 transgenic plants grew well (Additional file 8: Figure S7). To further analyze growth and physiological parameters, period drought stress was applied by withholding water for 2 weeks followed by normal watering for 7 days [63]. The wild-type treated Arabidopsis plants had etiolated and wilted leaves, whereas the transgenic lines grew well (Fig. 6a, b, g, and h). To quantify effects on growth, the dry weight of stems was measured. Under normal watering conditions, transgenic plants had slightly shorter stems with a lower dry weight compared to wild-type plants (Fig. 6a, c, g, and i). However, wild-type plant growth was more inhibited by drought compared to transgenic plants (Fig. 6b, c, h, and i). The dry weight of stems from transgenic plants under control and drought stress conditions was similar (Fig. 6c and i). Physiological parameters including relative water content, chlorophyll content, and net photosynthetic rate were also measured. Under well-watered conditions, wild-type and transgenic plants exhibited no significant differences (Fig. 6d and j). After period drought treatment, the relative water content of transgenic plants was higher than wild-type Arabidopsis, suggesting that ZxNAC083 and ZxNAC035 can regulate osmotic homeostasis in plants under drought conditions (Fig. 6d and j). Water deficit can lead to a decrease in chlorophyll content thus lowering net photosynthetic rate [63, 64]. The chlorophyll content of transgenic plants was maintained at more normal levels compared to Col-0 plants (Fig. 6e and k). Transgenic plants also showed a higher net photosynthetic rate compared to Col-0 under drought treatment (Fig. 6f and l).

Phenotypes and physiological parameters of Col-0, ZxNAC083 and ZxNAC035 overexpression transgenic plants under normal conditions and period drought stress. Two independent lines were analyzed for each transgenic. Representative images are shown. a, g Col-0, ZxNAC083 and ZxNAC035 overexpression transgenic plants under normal conditions. b, h Col-0, ZxNAC083 and ZxNAC035 overexpression transgenic plants under period drought stress (2-weeks water withheld, 1-week recovery). c, d, e, f Physiological parameters of Col-0 and ZxNAC083 overexpression transgenic lines under normal conditions and period drought stress. c Dry weight of stems, (d) relative water content, (e) chlorophyll content, and (f) net photosynthetic rate. i, j, k, l Physiological parameters of Col-0 and ZxNAC035 overexpression transgenic plants under normal conditions and period drought stress. i Dry weight of stems, (j) relative water content, (k) chlorophyll content, and (l) net photosynthetic rate. Data are mean ± SD of three replicates. c,i Asterisks, significant difference. p < 0.05 (t-test). (d, e, f, j, k, l) Asterisks, significant difference. p < 0.05 (Duncan’s test)
Finally, we compared the salinity tolerance of wild type and transgenic lines. Plants were irrigated with 100 mM NaCl solution for 2 weeks. After treatment, the rosette leaves of Col-0 were faded and withered whereas transgenic plants grew well (Fig. 7b and h). Col-0 plants had longer stems with a greater dry weight compared to transgenic plants under normal conditions (Fig. 7a and g). However, wild-type plant growth was more severely inhibited by salt stress compared to transgenic plants (Fig. 7c and i). Physiological parameters including relative water content, chlorophyll content and net photosynthetic rate were also measured. Salinity can decrease soil water potential leading to physiological drought. While the relative water content of wild-type and transgenic plants was similar under normal conditions, ZxNAC083 and ZxNAC035 overexpression lines showed significantly higher relative water content compared to wild-type plants after salt treatment (Fig. 7d and j). High relative water content improves chlorophyll stability and photosynthetic system integrity under physiological drought. Under salt stress, both chlorophyll content and net photsynthetic rate of ZxNAC83 and ZxNAC035 transgenic lines were significantly higher than Col-0 wild-type showing a protective effect (Fig. 7e, f, k, and l).

Phenotypes and physiological parameters of Col-0, ZxNAC083 and ZxNAC035 overexpression transgenic plants under normal condition and salt stress. Two independent lines were analyzed for each transgenic. Representative images are shown. a, g Col-0, ZxNAC083 and ZxNAC035 overexpression transgenic plants under normal condition. b, h Col-0, ZxNAC083 and ZxNAC035 overexpression transgenic plants under salt stress (2-weeks, 100 mM NaCl). c, d, e, f Physiological parameters of Col-0, ZxNAC083 overexpression transgenic plants under normal conditions and salt stress. c Dry weight of stems, d relative water content, e chlorophyll content, and (f) net photosynthetic rate. i, j, k, l Physiological parameters of Col-0, ZxNAC035 overexpression transgenic plants under normal conditions and salt stress. i Dry weight of stems, (j) relative water content, (k) chlorophyll content, and (l) net photosynthetic rate. Data are mean ± SD of three replicates. c, i Asterisks, significant difference. p < 0.05 (t-test). (d, e, f, j, k, l) Asterisks, significant difference. p < 0.05 (Duncan’s test)
These combined data demonstrate that overexpression of ZxNAC083 and ZxNAC035 enhances Arabidopsis drought and salt tolerance. These data provide powerful evidence that regulatory genes identified in this study include authentic osmotic and salt stress determinants in Z. xanthoxylum. Many of these genes have unresolved functions in stress tolerance.
Conclusions
This study provides a first comprehensive analysis of regulatory gene transcripts in Z. xanthoxylum under osmotic stress and salt treatments. These data reveal a complex network of conserved and unique regulatory genes induced in Z. xanthoxylum roots with potential roles in abiotic stress tolerance. Functional analysis of these genes is a rich resource for crop improvement.
Methods
Data acquisition and differential expression analysis
The transcriptomic data and digital gene expression libraries (C6, C24, D6, D24, S6, and S24) analyzed in this study were generated as previously described [20]. Seeds of Z. xanthoxylum used in this work were collected from wild plants in Alxa League of Inner-Mongolia Autonomous Region, China, and were stored in the Key Laboratory of Grassland Livestock Industry Innovation, Ministry of Agriculture and Rural Affairs, Lanzhou, China. The raw data are submitted to the NCBI Sequence Read Archive (PRJNA512400). Each transcriptomic library was constructed from seedling roots, using 5–6 seedlings per sample. Libraries C6 and C24 correspond to treatment with a mock solution (1/2 strength Hoagland nutrient solution) for 6 h and 24 h, respectively; D6 and D24 correspond to treatment with an osmotic stress solution (1/2 strength Hoagland nutrient solution with added sorbitol, equivalent to − 0.5 MPa osmotic potential, sorbitol concentration ≈ 165 mM) for 6 h and 24 h, respectively; and S6 and S4 correspond to treatment with a salt solution (1/2 strength Hoagland nutrient solution with 50 mM NaCl) for 6 h and 24 h, respectively. Libraries were constructed using a tag-based digital gene expression protocol [20]. The resulting libraries were sequenced in parallel using an Illumina HiSeq™ 2000 sequencing platform (BGI Shenzhen). Low quality tags from each library were removed (e.g. tags with unknown nucleotide “N”, “empty tags” with no tag sequence between the adaptors, and tags with only one copy number). The resulting “clean” tags were mapped to our transcriptome reference database [20]. To establish differential expression, the number of clean tags for each gene was calculated and normalized to the number of transcripts per million clean tags [65]. To compare differences in gene expression, the tag frequency in each library was statistically analyzed according to Audic and Claverie [66]. The false discovery rate (FDR) was used to determine the threshold p-value in multiple tests [65, 67]. Genes that displayed FDR<0.001 and absolute value of the log2 ratio>1 were selected as significantly different.
DEGs regulatory pathways analysis, hierarchical clustering, and validation by qRT-PCR
The Blast2GO program (https://www.blast2go.com/) was employed to obtain Gene Ontology (GO) annotations for Unigenes. Regulatory pathways were investigated by matching Z. xanthophyllum genes to putative orthologs in the Kyoto Encyclopedia of Genes and Genomes (KEGG) protein database (www.genome.jp/dbget/). Hierchical cluster was performed using Heml software (http://hemi.biocuckoo.org/down.php). Default options were used and Pearson correlations were performed on gene expression data.
RNA sequencing data were verified by measuring the transcript abundance of randomly-selected predicted regulatory genes using total RNA extracted from treated roots treated as described above [20]. First-strand cDNA was synthesized from 2 μg of DNase-treated RNA according to the manufacturer’s instructions (TaKaRa Biotechnology, China). qRT-PCR was performed in triplicate on three bioreplicates using Power SYBR™ Green Master Mix (TaKaRa Biotechnology, China) on a StepOne Real-Time PCR Thermocycler (Applied Biosystems). To normalize sample variance, ZxACTIN (GenBank accession no. EU019550) was used as the internal control gene. The relative expression level of each gene were determined using the 2-ΔΔCt method [65]. Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast) was used for primer design. A standard curve and efficiency test were performed for each set of primers (Additional file 2: Table S8).
Functional data mining in Arabidopsis and network analysis
Z. xanthoxylum DEGs were matched to putative orthologs in Arabidopsis by BLASTp analysis (E-value of 1e− 6) against the reference Arabidopsis TAIR10 genome annotation (http://www.arabidopsis.org). Protein-protein interaction analysis of protein function, protein subcellular localization, and gene co-expression of putative Arabidopsis orthologs was carried out in STRING [68] (http://www.string-db.org/). STRING protein-protein interaction images were re-modulated in Cytoscape [69] (https://cytoscape.org/).
Generating overexpression lines
The coding regions of Unigene16368_All (ZxNAC083) and CL6534.Contig1_All (ZxNAC035) were amplified from seedling total RNA using a SMART RACE cDNA Amplification Kit (TaKaRa Biotechnology, China). The resulting cDNA products were cloned into pDONR™/Zeo using a Gateway® BP reaction (ThermoFisher Scientific, China) then inserted into binary vector pBIB-BASTA-35S-GWR-GFP using a Gateway® LR reaction (ThermoFisher Scientific, China). The resulting constructs were introduced into Agrobacterium tumefaciens strain GV3101 and used to transform wild-type Arabidopsis Col-0 plants by floral dipping (seeds of wild-type Arabidopsis Col-0 were obtained from Ministry of Education Key Laboratory of Cell Activities and Stress Adaptation, Lanzhou, China) [70]. Transgenic overexpression lines were validated using semi-quantitative RT-PCR to measure ZxNAC083 and ZxNAC035 transcript levels. AtACTIN2 was used as the internal control gene. Primer sequences are provided in Additional file 2: Table S8.
Phenotypic and physiological assessments of transgenic plants
Phenotypic assays were performed on Arabidopsis wild-type Col-0 and T3 homozygous transgenic overexpression lines using two independent lines per construct (ZxNAC083-OX48, OX65 and ZxNAC035-OX57, OX59). 4-week-old plants grown on soil in pots (8 × 8 × 8 cm3) were subjected to drought or salt stress treatments as described with minor changes [71]. For period drought stress treatments, water was withheld for 2 weeks followed by normal watering for 7 days to permit recovery [71]. Under normal conditions, each pot was given 150 mL of water every 2 days. For salt stress treatments, each pot was given 150 mL of 100 mM NaCl solution every 2 days for 2 weeks. The dry weight of stems (shoots excluding rosette leaves), relative water content, chlorophyll content, and net photosynthetic rate were determined as previously described [19]. Seedlings were grown in a greenhouse at 20–22 °C with relative humidity 65–75% under long days (16 h light/8 h dark, photon flux density 100–120 μmol·m− 2·s− 1)..
Abbreviations
- ABA:
-
Abscisic acid
- AIB:
-
ABA-inducible bHLH-type transcription factor
- AP2/ERF:
-
APETALA2 and ethylene-responsive element binding proteins
- ARF:
-
Auxin Response Factor
- AUX/IAA:
-
Auxin/Indole-3-Acetic Acid
- BAK1 :
-
BRASSINOSTEROID INSENSITIVE 1-ASSOCIATED RECEPTOR KINASE 1
- BAM3 :
-
BARELY ANY MERISTEM 3
- bHLH:
-
Basic helix-loop-helix
- bZIP:
-
Basic leucine zipper
- CDPKs:
-
Calcium-dependent protein kinases
- DEGs:
-
Differentially expressed genes
- FDR:
-
False discovery rate
- GH3:
-
Gretchen Hagen 3
- GO:
-
Gene Ontology
- GRAS:
-
GAI/RGA/SCR
- HECT:
-
Homology to E6-APC terminus
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LRR-RLKs:
-
Leucine-rich repeat receptor-like kinases
- MAPKs:
-
Mitogen-activated protein kinases
- MYB:
-
Myeloblastosis
- MYC2:
-
Myelocytomatosis protein 2
- NAC:
-
NAM/ATAF/CUC
- PP2C:
-
Protein phosphatase 2C
- PUB18:
-
Plant U-box 18
- PYL/PYR/RCAR:
-
PYRABACTIN RESISTANCE1/PYR1-LIKE/REGULATORY COMPONENTS OF ABA RECEPTORS
- RING:
-
Really interesting new gene
- ROS:
-
Reactive oxygen species
- SAUR:
-
Small Auxin-Up RNA
- SnRKs:
-
Sucrose non-fermenting like kinases
- TIR1/AFB:
-
Transport Inhibitor Response1/Auxin Signaling F-box
- UPS:
-
Ubiquitin proteasome system
- WRKY:
-
WRKY-domain
- Z. xanthoxylum :
-
Zygophyllum xanthoxylum
References
Golldack D, Lüking I, Yang O. Plant tolerance to drought and salinity: stress regulating transcription factors and their functional significance in the cellular transcriptional network. Plant Cell Rep. 2011;30:1383–91.
Martìnez J, Ledent J, Bajji M, Kinet J, Lutts S. Effect of water stress on growth, Na+ and K+ accumulation and water use efficiency in relation to osmotic adjustment in two populations of Atriplex halimus L. Plant Growth Regul. 2003;41:63–73.
Chaves MM, Flexas J, Pinheiro C. Photosynthesis under drought and salt stress: regulation mechanisms from whole plant to cell. Ann Bot. 2009;103:551–60.
Takeda S, Matsuoka M. Genetic approaches to crop improvement: responding to environmental and population changes. Nature Rev Genet. 2008;9:444–57.
Vij S, Tyagi AK. Emerging trends in the functional genomics of the abiotic stress response in crop plants. Plant Biotechnol J. 2007;5:361–80.
Sanchez DH, Pieckenstain FL, Szymanski J, Erban A, Bromke M, Hannah MA, et al. Comparative functional genomics of salt stress in related model and cultivated plants identifies and overcomes limitations to translational genomics. PLoS One. 2011. https://doi.org/10.1371/journal.pone.0017094.
Shinozaki K, Yamaguchi-Shinozaki K, Seki M. Regulatory network of gene expression in the drought and cold stress responses. Curr Opin Plant Biol. 2003;6:410–7.
Mittler R, Blumwald E. Genetic engineering for modern agriculture: challenges and perspectives. Ann Rev Plant Biol. 2010;61:443–62.
Varshney RK, Bansal KC, Aggarwal PK, Datta SK, Craufurd PQ. Agricultural biotechnology for crop improvement in a variable climate: hope or hype? Trends Plant Sci. 2011;16:363–71.
Century K, Reuber TL, Ratcliffe OJ. Regulating the regulators: the future prospects for transcription-factor-based agricultural biotechnology products. Plant Physiol. 2008;147:20–9.
Zhu JK. Abiotic stress signaling and responses in plants. Cell. 2016;167:313–24.
Wang H, Wang H, Shao H, Tang X. Recent advances in utilizing transcription factors to improve plant abiotic stress tolerance by transgenic technology. Trends Plant Sci. 2016. https://doi.org/10.3389/fpls.2016.00067.
Shabala S. Learning from halophytes: physiological basis and strategies to improve abiotic stress tolerance in crops. Ann Bot. 2013;112:1209–21.
Flowers TJ, Colmer TD. Plant salt tolerance: adaptations in halophytes. Ann Bot. 2015;115:327–31.
Tuberosa R, Salvi S. Genomics-based approaches to improve drought tolerance of crops. Trends Plant Sci. 2006;11:405–12.
Pei SF, Hua FU, Cen YM, Jing-bin LI. Influence of Zygophyllum xanthoxylum shrubs on soil fertility in enclosure and grazing conditions. J Desert Res. 2004;24:763–7.
Zhou XR, Zhou ZY, Wu CX. The research of the breeding characters of Zygophyllum xanthoxylum. Prata Sci. 2006;23:38–41.
Wu GQ, Xi JJ, Wang Q, Bao AK, Ma Q, Zhang JL, et al. The ZxNHX gene encoding tonoplast Na+/H+ antiporter from the xerophyte Zygophyllum xanthoxylum plays important roles in response to salt and drought. J Plant Physiol. 2011;168:758–67.
Ma Q, Yue LJ, Zhang JL, Wu GQ, Bao AK, Wang SM. Sodium chloride improves photosynthesis and water status in the succulent xerophyte Zygophyllum xanthoxylum. Tree Physiol. 2012;32:4–13.
Ma Q, Bao AK, Chai WW, Wang WY, Zhang JL, Li YX, et al. Transcriptomic analysis of the succulent xerophyte Zygophyllum xanthoxylum in response to salt treatment and osmotic stress. Plant Soil. 2016;402:343–61.
Ma Q, Hu J, Zhou XR, Yuan HJ, Kumar T, Luan S. ZxAKT1 is essential for K+ uptake and K+/Na+ homeostasis in the succulent xerophyte Zygophyllum xanthoxylum. Plant J. 2017;90:48–60.
Yuan HJ, Ma Q, Wu GQ, Wang P, Hu J, Wang SM. ZxNHX controls Na+ and K+ homeostasis at the whole-plant level in Zygophyllum xanthoxylum through feedback regulation of the expression of genes involved in their transport. Ann Bot. 2015;115:495–507.
Garg R, Shankar R, Thakkar B, Kudapa H, Krishnamurthy L, Mantri N, et al. Transcriptome analyses reveal genotype-and developmental stage-specific molecular responses to drought and salinity stresses in chickpea. Sci Rep. 2016. https://doi.org/10.1038/srep19228.
Yoshida T, Mogami J, Yamaguchi-Shinozaki K. ABA-dependent and ABA-independent signaling in response to osmotic stress in plants. Curr Opin Plant Biol. 2014;21:133–9.
Cutler SR, Rodriguez PL, Finkelstein RR, Abrams SR. Abscisic acid: emergence of a core signaling network. Annu Rev Plant Biol. 2010;61:651–79.
Tognetti VB, Mühlenbock P, Van Breusegem F. Stress homeostasis - the redox and auxin perspective. Plant Cell Environ. 2012;35:321–33.
Iglesias MJ, Terrile MC, Bartoli CG, D'Ippólito S, Casalongué CA. Auxin signaling participates in the adaptative response against oxidative stress and salinity by interacting with redox metabolism in Arabidopsis. Plant Mol Biol. 2010;74:215–22.
Yamaguchi-Shinozaki K, Shinozaki K. Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Ann Rev Plant Biol. 2006;57:781–803.
Osakabe Y, Yamaguchi-Shinozaki K, Shinozaki K, Tran LS. Sensing the environment: key roles of membrane-localized kinases in plant perception and response to abiotic stress. J Exp Bot. 2013;64:445–58.
Ye Y, Ding Y, Jiang Q, Wang F, Sun J, Zhu C. The role of receptor-like protein kinases (RLKs) in abiotic stress response in plants. Plant Cell. 2017;36:235–42.
Chae L, Sudat S, Dudoit S, Zhu T, Luan S. Diverse transcriptional programs associated with environmental stress and hormones in the Arabidopsis receptor-like kinase gene family. Mol Plant. 2009;2:84–107.
Chen J, Yu F, Liu Y, Du C, Li X, Zhu S, et al. FERONIA interacts with ABI2-type phosphatases to facilitate signaling cross-talk between abscisic acid and RALF peptide in Arabidopsis. Proc Nati Acad Sci U S A. 2016;113:5519–27.
Osakabe Y, Mizuno S, Tanaka H, Maruyama K, Osakabe K, Todaka D, et al. Overproduction of the membrane-bound receptor-like protein kinase 1, RPK1, enhances abiotic stress tolerance in Arabidopsis. J Biol Chem. 2010;285:9190–201.
Li J, Wen J, Lease KA, Doke JT, Tax FE, Walker JC. BAK1, an Arabidopsis LRR receptor-like protein kinase, interacts with BRI1 and modulates brassinosteroid signaling. Cell. 2002;110:213–22.
Torii KU, Mitsukawa N, Oosumi T, Matsuura Y, Yokoyama R, Whittier RF, et al. The Arabidopsis ERECTA gene encodes a putative receptor protein kinase with extracellular leucine-rich repeats. Plant Cell. 1996;8:35–746.
De Young BJ, Bickle KL, Schrage KJ, Muskett P, Patel K, Clark SE. The CLAVATA1-related BAM1, BAM2 and BAM3 receptor kinase-like proteins are required for meristem function in Arabidopsis. Plant J. 2006;45:1–16.
Frye CA, Innes RW. An Arabidopsis mutant with enhanced resistance to powdery mildew. Plant Cell. 1998;10:947–56.
He K, Gou X, Yuan T, Lin H, Asami T, Yoshida S, et al. BAK1 and BKK1 regulate brassinosteroid-dependent growth and brassinosteroid-independent cell-death pathways. Curr Biol. 2007;17:1109–15.
Lee JS, Hnilova M, Maes M, Lin YC, Putarjunan A, Han SK, et al. Competitive binding of antagonistic peptides fine-tunes stomatal patterning. Nature. 2015;522:439–43.
Krasensky J, Jonak C. Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J Exp Bot. 2012;63:1593–608.
Matsuoka D, Nanmori T, Sato K, Fukami Y, Kikkawa U, Yasuda T. Activation of AtMEK1, an Arabidopsis mitogen-activated protein kinase kinase, in vitro and in vivo: analysis of active mutants expressed in E. coli and generation of the active form in stress response in seedlings. Plant J. 2002;29:637–47.
Xu J, Tian YS, Peng RH, Xiong AS, Zhu B, Jin XF, et al. AtCPK6, a functionally redundant and positive regulator involved in salt/drought stress tolerance in Arabidopsis. Planta. 2010;231:1251–60.
De Zelicourt A, Colcombet J, Hirt H. The role of MAPK modules and ABA during abiotic stress signaling. Trends Plant Sci. 2016;21:677–85.
Abass M, Morris PC. The Hordeum vulgare signalling protein MAP kinase 4 is a regulator of biotic and abiotic stress responses. J Plant Physiol. 2013;170:1353–9.
Khan SA, Li MZ, Wang SM, Yin HJ. Revisiting the role of plant transcription factors in the battle against abiotic stress. Int J Mol Sci. 2018. https://doi.org/10.3390/ijms19061634.
Ramamoorthy R, Jiang SY, Kumar N, Venkatesh PN, Ramachandran S. A comprehensive transcriptional profiling of the WRKY gene family in rice under various abiotic and phytohormone treatments. Plant Cell Physiol. 2008;49:865–79.
Zhou QY, Tian AG, Zou HF, Xie ZM, Lei G, Huang J, et al. Soybean WRKY-type transcription factor genes, GmWRKY13, GmWRKY21, and GmWRKY54, confer differential tolerance to abiotic stresses in transgenic Arabidopsis plants. Plant Biotechnol J. 2008;6:486–503.
Yang O, Popova OV, Süthoff U, Lüking I, Dietz KJ, Golldack D. The Arabidopsis basic leucine zipper transcription factor AtbZIP24 regulates complex transcriptional networks involved in abiotic stress resistance. Gene. 2009;436:45–55.
Abe H, Urao T, Ito T, Seki M, Shinozaki K, Yamaguchi-Shinozaki K. Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. Plant Cell. 2003;15:63–78.
Li H, Sun J, Xu Y, Jiang H, Wu X, Li C. The bHLH-type transcription factor AtAIB positively regulates ABA response in Arabidopsis. Plant Mol Biol. 2007;65:655–65.
Wang X, Liu S, Tian H, Wang S, Chen JG. The small ethylene response factor ERF96 is involved in the regulation of the abscisic acid aesponse in Arabidopsis. Front Plant Sci. 2015. https://doi.org/10.3389/fpls.2015.01064.
Ahmad A, Niwa Y, Goto S, Ogawa T, Shimizu M, Suzuki A, et al. bHLH106 integrates functions of multiple genes through their G-box to confer salt tolerance on Arabidopsis. PLoS One. 2015. https://doi.org/10.1371/journal.pone.0126872.
Song L, Huang SC, Wise A, Castanon R, Nery JR, Chen H, et al. A transcription factor hierarchy defines an environmental stress response network. Science. 2016;354:1550.
Kloth KJ, Wiegers GL, Busscher-Lange J, van Haarst JC, Kruijer W, Bouwmeester HJ, et al. AtWRKY22 promotes susceptibility to aphids and modulates salicylic acid and jasmonic acid signalling. J Exp Bot. 2016;67:3383–96.
Hartmann L, Pedrotti L, Weiste C, Fekete A, Schierstaedt J, Göttler J, et al. Crosstalk between two bZIP signaling pathways orchestrates salt-induced metabolic reprogramming in Arabidopsis roots. Plant Cell. 2015;27:2244–60.
Gupta P, Nutan KK, Singla-Pareek SL, Pareek A. Abiotic stresses cause differential regulation of alternative splice forms of Gata transcription factor in rice. Front Plant Sci. 2017. https://doi.org/10.3389/fpls.2017.01944.
Lyzenga WJ, Stone SL. Abiotic stress tolerance mediated by protein ubiquitination. J Exp Bot. 2012;63:599–616.
Yu F, Wu Y, Xie Q. Ubiquitin-proteasome system in ABA signaling: from perception to action. Mol Plant. 2016;9:21–33.
Bao Y, Song WM, Jin YL, Jiang CM, Yang Y, Li B, et al. Characterization of Arabidopsis Tubby-like proteins and redundant function of AtTLP3 and AtTLP9 in plant response to ABA and osmotic stress. Plant Mol Biol. 2014;86:471–83.
Seo DH, Ryu MY, Jammes F, Hwang JH, Turek M, Kang BG, et al. Roles of four Arabidopsis U-box E3 ubiquitin ligases in negative regulation of abscisic acid-mediated drought stress responses. Plant Physiol. 2012;160:556–68.
Xu J, Li Y, Wang Y, Liu H, Lei L, Yang H, Liu G, et al. Activation of MAPK kinase 9 induces ethylene and camalexin biosynthesis and enhances sensitivity to salt stress in Arabidopsis. J Biol Chem. 2008;283:26996–7006.
Reusche M, Thole K, Janz D, Truskina J, Rindfleisch S, Drübert C, et al. Verticillium infection triggers VASCULAR-RELATED NAC DOMAIN7-dependent de novo xylem formation and enhances drought tolerance in Arabidopsis. Plant Cell. 2008;24:3823–37.
Daron B, Wang F, Peng T, Liu JH. PtrA/NINV, an alkaline/neutral invertase gene of Poncirus trifoliata, confers enhanced tolerance to multiple abiotic stresses by modulating ROS levels and maintaining photosynthetic efficiency. BMC Plant Biol. 2016. https://doi.org/10.1186/s12870-016-0761-0.
Kato M, Shimizu S. Chlorophyll metabolism in higher plants. IV. Involvement of peroxidase in chlorophyll degradation. Plant Cell Physiol. 1985;26:1291–301.
Yamauchi N, Watada AE. Effectiveness of various phenolic compounds in degradation of chlorophyll by in vitro peroxidase-hydrogen peroxide system. J Ame Soc Hortic Sci. 1994;63:439–44.
Xue J, Bao YY, Li BL, Cheng YB, Peng ZY, Liu H, et al. Transcriptome analysis of the brown planthopper Nilaparvata lugens. PLoS One. 2010. https://doi.org/10.1371/journal.pone.0014233.
Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997;7:986–95.
Dang ZH, Zheng LL, Wang J, Gao Z, Wu SB, Qi Z. Transcriptomic profiling of the salt-stress response in the wild recretohalophyte Reaumuria trigyna. BMC Genomics. 2013. https://doi.org/10.1186/1471-2164-14-29.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:447–52.
Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38:W214–20.
Clough SJ, Bent AF. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998;16:735–43.
Acknowledgements
Not applicable.
Funding
This work has been jointly supported by the National Natural Science Foundation of China (Grant no. 31730093 and 31601992), and the Fundamental Research Funds for the Central Universities (Grant No. 862920 and 862240).
These funding bodies had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
Availability of data and materials
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
SMW and HY designed the study. HY analyzed the transcriptome data. HY and ML prepared the genetic constructs and transformation. ML carried out the real-time PCR. ML, DL, SAK carried out the phenotypic and physiological experiments. DL and SAK analyzed the physiological experiments data. SMW, HY and SRH drafted the manuscript. All the authors critically revised this manuscript. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Venn diagrams showing DEGs in Z. xanthoxylum roots under osmotic stress and salt treatments. Yellow and green colors, up-regulated and down-regulated transcripts under osmotic stress for (a) 6 h and (b) 24 h, respectively. Red and blue colors, up-regulated and down-regulated transcripts under salt treatment for (a) 6 h and (b) 24 h, respectively. (JPG 257 kb)
Additional file 2:
Table S1. ABA signaling pathway DEGs in Z. xanthoxylum roots under osmotic stress and salt treatment. Table S2. Auxin signaling pathway DEGs in Z. xanthoxylum roots under osmotic stress and salt treatments. Table S3. Selection and categorization of significant Z. xanthoxylum kinase DEGs in response to osmotic stress and salt treatments. Table S4. Selection and categorization of significant Z. xanthoxylum transcription factor DEGs in response to osmotic stress and salt treatments. Table S5. Selection and categorization of significant Z. xanthoxylum UPS enzyme DEGs in response to osmotic stress and salt treatments. Table S6. RNA seq data verification by qRT-PCR measurement of randomly selected Z. xanthoxylum osmotic stress and salt responsive protein kinase, transcription factor, and UPS enzyme genes. Table S7. DEGs matched to predicted Arabidopsis orthologs in the complete gene networks in Z. xanthoxylum roots under osmotic stress and salt treatment. Yes (Y) or No (N) indicate gene representation in the corresponding DEG libraries. Table S8. Primers used in current study . (DOCX 91 kb)
Additional file 3:
Figure S2. Hierarchical cluster analysis of differentially expressed genes encoding (a) receptor like kinases, (b) Ca2+ related kinases, (c) E3 ubiquitin ligases, and (d) transcription factors in Z. xanthoxylum roots under osmotic stress and salt treatments. Unigenes are matched to Arabidopsis orthologs where possible. (JPG 12062 kb)
Additional file 4:
Figure S3. Hierarchical cluster analysis of transcription factor genes that are differentially expressed in Z. xanthoxylum roots at both 6 h and 24 h under salt treatment. Unigenes are matched to Arabidopsis orthologs where possible. (JPG 963 kb)
Additional file 5:
Figure S4. High resolution images of Fig. 4a. (PDF 28 kb)
Additional file 6:
Figure S5. High resolution images of Fig. 4b. (PDF 33 kb)
Additional file 7:
Figure S6. Expression analysis of ZxNAC083 and ZxNAC035. (a-b) qRT-PCR validation of RNA sequencing data in Z. xanthoxylum roots under osmotic stress or salt treatment for 6 h. (c) Semi-quantitative RT-PCR assay showing overexpression of ZxNAC083 in transgenic Arabidopsis plants compared to Col-0 wild-type. ZxNAC083 (20 cycles) and AtActin2 (19 cycles). (d) Semi-quantiative RT-PCR experiment assay showing overexpression of ZxNAC035 in transgenic Arabidopsis plants compared to Col-0 wildtype. ZxNAC035 (25 cycles) and AtActin2 (19 cycles). (JPG 429 kb)
Additional file 8:
Figure S7. Phenotypes of Col-0, ZxNAC083 and ZxNAC035 overexpression transgenic plants under normal conditions and 7-day drought stress at vegetative phases. Two independent lines were analyzed for each transgenic. Representative images are shown. (a,c) Col-0, ZxNAC083 and ZxNAC035 overexpression transgenic plants under normal conditions. (b,d) Col-0, ZxNAC083 and ZxNAC035 overexpression transgenic plants under 7-day drought stress. (JPG 1069 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Yin, H., Li, M., Li, D. et al. Transcriptome analysis reveals regulatory framework for salt and osmotic tolerance in a succulent xerophyte. BMC Plant Biol 19, 88 (2019). https://doi.org/10.1186/s12870-019-1686-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-019-1686-1