
Abstract
Background
Blueberry is an economically important fruit crop in Ericaceae family. The substantial quantities of flavonoids in blueberry have been implicated in a broad range of health benefits. However, the information regarding fruit development and flavonoid metabolites based on the transcriptome level is still limited. In the present study, the transcriptome and gene expression profiling over berry development, especially during color development were initiated.
Results
A total of approximately 13.67 Gbp of data were obtained and assembled into 186,962 transcripts and 80,836 unigenes from three stages of blueberry fruit and color development. A large number of simple sequence repeats (SSRs) and candidate genes, which are potentially involved in plant development, metabolic and hormone pathways, were identified. A total of 6429 sequences containing 8796 SSRs were characterized from 15,457 unigenes and 1763 unigenes contained more than one SSR. The expression profiles of key genes involved in anthocyanin biosynthesis were also studied. In addition, a comparison between our dataset and other published results was carried out.
Conclusions
Our high quality reads produced in this study are an important advancement and provide a new resource for the interpretation of high-throughput data for blueberry species whether regarding sequencing data depth or species extension. The use of this transcriptome data will serve as a valuable public information database for the studies of blueberry genome and would greatly boost the research of fruit and color development, flavonoid metabolisms and regulation and breeding of more healthful blueberries.
Similar content being viewed by others


Background
Blueberry (Vaccinium corymbosum) is an economically important small fruit crop and a member of Ericaceae family which includes many species, such as blueberry, cranberry (V. macrocarpon), lingonberry (V. vitis-idaea), rhododendron, and more than 400 other species [1, 2]. Three major types of blueberry are harvested commercially including lowbush (Vaccinium. angustifolium), highbush (V. corymbosum), and rabbiteye bluberry (V. ashei or V. virgatum). Although mostly originated in North America, many blueberry species are widely grown in Asia, Europe, South America, Africa, Australia and New Zealand owing in part to their high level of vitro antioxidant capacities [3, 4]. Blueberry is becoming a major crop in China, cultivated widely from temperate area to subtropical region. There are currently three major areas for blueberry cultivation in China, the Jilin and Liaoning provinces, the Shandong provinces and the areas of the Yangtze River [5].
Demand and consumption worldwide of blueberry has greatly increased in recent years for its beneficial influence on human health. These positive effects are generally due to the high levels of flavonoid [6], which have been linked to improve night vision, prevent macular degeneration, and decrease the heart disease [7, 8]. Therefore, it is crucial to elucidate the molecular mechanisms that trigger biosynthesis and accumulation of anthocyanin metabolites during fruit and color development. The blueberry genome is large (600 Mb/haploid genome) and genomic information is limited compared to some plants like grape, for example [9], which restrains the dissection of blueberry. Over the past decades, more attention has been focused on the analysis of plant cold resistance, cultivation, and effects on human health [10, 11]. Since massive amounts of information can be obtained from genome-scale expression data, RNA sequencing has become a powerful technology to profile the transcriptome [12]. To date, RNA sequencing has been reported in bilberry (Vaccinium myrtillus) and cranberry [13, 14]. Recently, transcriptome sequences of blueberry were analyzed during cold acclimation and at different development stages of fruit by ESTs sequencing or RNA sequencing [2, 15]. So far, transcriptome sequences have been generated using next generation sequencing so far from northern highbush [2], half-high [16], and southern highbush blueberry [17]. However, the information is still limited regarding the control of horticultural traits such as the molecular regulation mechanisms of blueberry maturation and flavonoid metabolism.
In order to gain new insights into molecular mechanism at transcriptome level, we performed transcriptome sequencing and gene expression profiling for the northern highbush blueberry variety ‘Sierra’ over berry development with Illumina sequencing technique. A total of more than 13.67 Gbp of data were generated and assembled into 186,962 transcripts and 80,836 unigenes. Large numbers of simple sequence repeats (SSRs) and candidate genes, which are potentially involved in plant growth, metabolic and hormone pathways, were identified. In addition, RNA-Seq expression profiles and functional annotations have been made publicly available (accession number: SRR2910056). We believe that this study provides a new and more powerful resource for interpretation of high-throughput gene expression data for blueberry species.
Results and discussion
Sequence analysis and de novo assembly
The high quality Illumina sequencing data produced from Vaccinium corymbosum ‘Sierra’ during fruit and color development has been deposited in NCBI SRA database under accession number: SRR2910056. Three independent cDNA libraries were constructed from RNA samples of green fruit (G, 30d), pink fruit (P, 50d) and blue fruit (B, 75d), respectively. In total, after removing adaptor sequences, ambiguous nucleotides and low-quality sequences, 67,689,734 independent reads (13.67 Gbp) with more than 92 % Q30 bases were selected using Illumina HiSeq™ 2000 from the three samples (Table 1). The GC content of the three samples was approximately 46 %.
Using the Trinity de novo assembly program [18], we assembled the short read sequences from the three samples into 186,962 transcripts. An overview of the assembled transcripts and unigenes are exhibited in Tables 2 and 3. The N50 value of the three sample assemblies was 1688 bp, and 66, 855 (36.95 %) transcripts were longer than 1 kb. The transcripts were subjected to cluster and assembly analyses, resulting in 80,836 unigenes (N50 value = 1204 bp), among which 15,457 (19.13 %) genes were greater than 1 kb. This is about 10 times greater than that in a previous report where a 17,134 ESTs resulted in a total of 8500 unigenes from two samples by Sanger sequencing [15]. Li et al. reported 57,331 unigenes in half-high blueberry from their two fruit samples (pulp and skin) [16]. Gupta et al. produced around 60,000 gene models from five stages of berry fruit development and ripening [17]. Therefore, we believe that the 13.67 Gbp of data generated and 186,962 transcripts and 80,836 unigenes assembled in this study are an important advancement and new resource for interpretation of high-throughput data for blueberry species whether regarding sequencing data depth or species extension. It is noteworthy that we have compared our dataset with the previously published Illumina dataset [17]. Compared with the previously published dataset, our dataset has a higher amount of data (Nucleotide), better data quality (Q20% or Q30%), and better assembly results (Average length and N50) (Additional file 1: File S1). There is no doubt that we now have a more powerful information database with higher capacity for gene expression analysis in Vaccinium, especially for fruit development and maturation, flavonoid metabolism and regulation and breeding for more healthful blueberries.
Similarity analysis
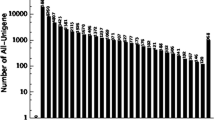
In order to better evaluate the similarity between blueberry and other organisms, blueberry unigenes were submitted to the non-redundant (Nr) NCBI database for BLASTx similarity analysis. The results showed that Vaccinium corymbosum unigenes hit a wide range of plant species including Vitis vinifera, Populus trichocarpa, Prunus avium, Ricinus communis, Fragaria vesca, Jatropha curcas, Solanum lycopersicum, Solanum lycopersicum, Camellia sinensis, Gossypium hirsutum, Brassica rapa, Vaccinium macrocarpon, etc. (Fig. 1). Among them, interestingly, 10,280 (12.72 %) unigenes showed a significant homology with sequences of Vitis vinifera, and 1654 (2.05 %) and 1183 (1.46 %) unigene sequences had a high similarity with those of Populus trichocarpa and Prunus avium, respectively. The high similarity of unigenes between Vaccinium corymbosum and Vitis vinifera suggests the possibility of using Vitis vinifera transcriptomes and genomes as a reference sequence for Vaccinium corymbosum. In contrast, only 242 (0.29 %) unigenes had a high similarity with sequences of Vaccinium macrocarpon, probably owing to the insufficient number of sequences in Genebank.

Species distribution of the top BLAST hits in Nr database. Values were number of unigenes in Vaccinium corymbosum homologous with those in other species such as Vitis vinifera, Populus trichocarpa, Prunus avium, Ricinus communis, Fragaria vesca, Jatropha curcas, Solanum lycopersicum, Solanum lycopersicum, Camellia sinensis, Gossypium hirsutum, Brassica rapa, Vaccinium macrocarpon
Sequence annotation
To annotate the assembled sequences, several complementary approaches were utilized. The unigenes were annotated by aligning with the deposited ones in diverse protein databases (Nr, Nt, Swiss-Prot, KEGG, COG, GO and TrEMBL) and the best one was selected from the matches with an E-value of less than 10−5. The overall functional annotation is described in Table 4. Among the 80,836 unigenes, 10,263 (12.70 %) and 21,870 (27.05 %) unigenes had significant matches in the COG and GO database, 26,902 (33.28 %) in the Nt database, and 21,447 (26.53 %) in the Swiss-Prot database. The 34,006 (42.07 %) unigenes were annotated in the Nr, Nt, Swiss-Prot, KEGG, COG, GO and TrEMBL databases.
GO, COG, KEGG annotation
Gene Ontology (GO) enrichment analysis was carried out to classify gene functions of the unigenes identified. The majority of the GO terms (68,355, 49.58 %) were assigned to biological process, and 35,683 (25.89 %) and 33,840 (24.54 %) were assigned to the molecular function and the cellular component, respectively (Fig. 2). It was noteworthy that cells and organelles are highly represented in category of cellular components, while binding and catalytic activity are highly typical in molecular functions category. For biological processes, genes implicated in metabolic processes and cellular processes are the most represented category followed by the response to stimulus and biological regulation (Fig. 3). These data suggest that the active synthesis of substance and energy and metabolic process are present in a wide variety of organelles and associated with fruit development and in response to stress during fruit ripening.

Functional annotation of assembled sequences based on gene ontology (GO) categorization. A total of 80,836 unigenes with BLAST match to known proteins were assigned to three main categories: 68,355 (49.58 %) were assigned to biological process, and 35,683 (25.89 %) and 33,840 (24.54 %) were assigned to the molecular function and the cellular component, respectively

Clusters of orthologous groups (COG) classification. In total, 80,836 unigenes were assigned to the COG classification according to the Nr database and grouped into 25 COG classifications
Furthermore, all blueberry unigenes were searched against the COG database for functional prediction and classification. Overall, 80,836 unigenes were assigned to the COG classification (Fig. 3). The largest group was the cluster for general function prediction (2502, 17.84 %), followed by translation, ribosomal structure and biogenesis (1297, 9.25 %), replication, recombination and repair (1262, 9.00 %), transcription (1160, 8.27 %), posttranslational modification, protein turnover and chaperones (1071, 7.64 %), signal transduction mechanisms (942, 6.72 %), and carbohydrate transport and metabolism (868, 6.19 %). However, small clusters were for cell motility and nuclear structure (13 and 2 unigenes, respectively). In addition, no unigene was assigned to extracellular structures.
To characterize the active biological pathways in blueberry, KEGG pathway tool was used as an alternative approach to analyze the pathway annotations of unigene sequences. The 7616 unigenes were assigned to 119 biological pathways (Additional file 2: File S2). These predicted pathways are responsible for growth and development probably via compound biosynthesis, degradation, utilization, and assimilation. These results suggest that a large number of metabolic activities are occurring during fruit development and coloring of blueberry.
EST-SSR discovery
As highly informative markers, SSRs have developed into powerful molecular markers for comparative genetic mapping and genotyping among species within genera [19, 20]. To date, SSRs are most widely applied in genetics, evolution and breeding.
To explore EST-SSR profiles in the unigenes of blueberry, 15,457 unigene sequences were submitted to search for SSRs. A total of 6429 sequences containing 8796 SSRs were identified from 15,457 unigenes, with 1763 unigene sequences containing more than one SSR (Table 5 and Additional file 3: File S3). Di-nucleotide motifs and tri-nucleotide motifs were the most abundant with 75.11 % (4426) and 23.81 % (1403), respectively. The most abundant repeat type was AG/CT (4069), followed by AAG/CTT (388), AGG/CCT (227), and ACC/GGT (225). Because SSRs within genes are likely to be subjected to stronger selective pressure than other genomic regions, these SSRs probably represent different putative functions [21]. Therefore, the unigenes yielded from blueberry are a larger resource for SSR mining than ever and the SSR profiles which we explored are a powerful platform for research in genetics, evolution and molecular marker-assistant breeding, etc.
Analysis of gene regulation in anthocyanin biosynthesis pathway using the assembled unigenes
In this study, we analyzed all unigene annotation in blueberry. Some genes responsible for anthocyanin biosynthesis were screened for further analysis. Anthocyanins are a large class of flavonoids which are responsible for the colors of flowers, fruits and other tissues [22]. A simple anthocyanin biosynthesis pathway is shown in Fig. 4. In most of the pathway, more than one unique sequence was annotated as encoding the same enzyme, especially the final step involving 3-glycoside formation by UFGT (UDP Glc-favonoid 3-O-glucosyl transferase) (Additional file 4: File S4).

A schematic representation of anthocyanin biosynthesis pathway
The relative expression levels of candidate unigenes and the content of anthocyanins during blueberry fruit and color development
To validate the results from the bioinformatics analysis, the expression profile of 9 differentially expressed genes of anthocyanin biosynthesis pathway such as PAL, C4H, 4CL, CHS, CHI, F3H, DFR, ANS, and UFGT were tested by RT-qPCR using cDNA templates from different fruit growth stages (Fig. 5). The number of unigenes assumed to be involved in anthocyanin biosynthesis pathway is shown in Additional file 4: File S4 and the specific primers used for RT-qPCR reactions are listed in Table 6. Among them, five candidate genes covered over 10 unigenes. The transcripts of some genes such as CHS, CHI, F3H, DFR, DFR, ANS, and UFGT increased with fruit coloring (stage 4) and reached the highest level at red fruit stage (stage 5), then slightly declined with fruit maturation (stage 5 and 6). CHS showed significant increase in expression at the stage of fruit coloring (stage 4), about a 19-fold increase compared with green fruit (stage 3), whereas only 3 -fold increase was observed for ANS at the stage of red fruit (stage 4). On the contrary, the expression level of PAL, C4H, and 4CL changed little with fruit development. Jaakola et al. reported the expression level of CHS, F3H, DFR, and ANS, detected by northern-blotting, to increase dramatically at stage 5 (red fruit) in bilberry [16]. This is consistent with our results. Zifkin et al. characterized the expression level of DFR, ANS, ANR, and LAR in blueberry and found that the expression of DFR, ANS increased at the S6 stage (red fruit) [16].

Expression levels of the color-related genes and the content of anthocyanins in blueberry. Total RNA was extracted from green, pink and blue fruits, respectively. Each value is the mean ± SE of three independent biological determinations
The anthocyanin content was next quantified at different developmental stages to test if anthocyanins accumulate in accordance with the pattern of gene expression. As expected, at green berry stage, anthocyanins were detected at low levels, only 0.5 mg/g FW. In accordance with the deep coloration, in mature fruits (blue fruit) the levels of anthocyanin peaked dramatically. Analysis of expression and anthocyanin accumulation indicated that there was a significant correlation between the expression profile of candidate genes and the accumulation of anthocyanins.
Conclusions
In the present study, the transcriptome and gene expression profiling over fruit development and coloring of blueberry were initiated by means of Illumina sequencing. A total of approximately 13.67 Gbp of data were obtained and assembled into 186,962 transcripts and 80,836 unigenes from three stages of blueberry fruit. A large number of simple sequence repeats (SSRs) and candidate genes were identified. A total of 6429 sequences containing 8796 SSRs were identified from 15,457 unigenes, and 1763 unigenes containing more than one SSR. Compared with the previously published data, our dataset has a higher amount of data (Nucleotide), better data quality (Q20 or Q30%), and better assembly results (Average length and N50). Therefore, our high quality reads produced in this study are an important advancement and a new resource for interpretation of high-throughput data for blueberry species regarding from sequencing data depth or species extension.
Methods
Plant materials and RNA extraction
Blueberry (Vaccinium corymbosum ‘Sierra’) fruits were harvested from an organic blueberry farm (Tianshuo Farm in Hebei Province, China) during the 2013 to 2015 growing season. The tissues at three different developmental stages including green fruit (G, 30d), pink fruit (P, 50d) and blue fruit (B, 75d), were randomly sampled in the field from 4- or 5-year-old healthy blueberry plants. The plants had been propagated by tissue culture, thus all came from the same mother plant. All samples intended for RNA extraction were flash-frozen in liquid nitrogen immediately after collection and stored at −80 °C until use. Fifty fruits were collected at each time point and combined for RNA extraction. RNA was extracted using the Plant total RNA Kit (TIANGEN, Beijing, China). The purified RNA quality and quantity was evaluated using a spectrophotometer (Thermo Scientific, Waltham, MA, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
mRNA-seq library construction for Illumina sequencing
The mRNA-seq library was constructed following the Illumina’s mRNA-seq Sample Preparation Kit (Illumina Inc., San Diego, CA, USA) from three stages of fruit development, separately. Briefly, the mRNA was purified from total RNA samples using oligo (dT) magnetic beads, and then broken into small pieces by divalent cation under an elevated temperature. Taking these RNA fragments as templates, the first strand cDNA was synthesized by reverse transcriptase and random primers, while the second strand cDNA was synthesized using DNA Polymerase I and RNase H. The cDNA fragments were blunt-ended and ligated to sequencing adaptors. The fragments (200 ± 25 bp) were then separated by agarose gel electrophoresis and selected for PCR amplification. Finally, the mRNA-seq libraries were sequenced by the Illumina HiSeq™ 2000 sequencing.
Sequence data analysis and assembly
The high-quality clean read data for assembly was separated from adapters and low-quality. Reads with more than 10 % Q30 bases were removed. De novo assembly of transcriptome was done using the Trinity method with an optimized k-mer length of 25 [18]. The contigs were clustered and further assembled according to paired-end information. The longest transcripts in the cluster units were defined as unigenes. EMBOSS Getorf Software was used to predict coding regions (http://emboss.bioinformatics.nl/cgi-bin/emboss/getorf).
Sequence annotation
The assembled sequences were searched against the public protein databases, such as the NCBI Nr and Nt databases (National Center for Biotechnology Information (NCBI) nonredundant protein (Nr) database, non-redundant nucleotide sequence (Nt) database), Swissprot, KEGG (Kyoto Encyclopedia of Genes and Genomes, http://www.genome.jp/kegg/kegg2.html), COG, (Cluster of Orthologous Groups of proteins) and TrEMBL using BLASTX with an E-value ≤ 10−5. Gene ontologies (GO) were assigned to each unigene using Blast2GO (Conesa et al., 2005, http://wego.genomics.org.cn/cgi-bin/wego/index.pl).
EST-SSR detection
The EST-SSRs were detected among the 15,457 blueberry unigenes which were longer than 1 kb using the Simple Sequence Repeat Identification Tool (SSRIT, http://www.gramene.org/db/markers/ssrtool). The parameters were adjusted to identify perfect di-, tri-, tetra-, penta- and hexa- nucleotide motifs with a minimum of six, five, four, four and four repeats, respectively [23, 24].
Gene validation and expression analysis by quantitative RT-qPCR
The expression of potential candidate genes of anthocyanin biosynthesis pathway in blueberry such as PAL, C4H, 4CL, CHS, CHI, F3H, DFR, ANS and UFGT were examined by RT-qPCR. Specific primers used for RT-qPCR reactions were listed in Table 6 according to the open reading frames of the target genes. Real-time qPCR reactions were carried out in a StepOnePlus Real-time PCR system (ABI, USA) using SuperRealPreMix (SYBR Green) kit (TIANGEN BIOTECH, Beijing, China) according to the manufacturer’s instructions. UBC28 gene was used as an internal control for normalization, and each sample was assayed in triplicate. Relative transcript levels were calculated and normalized as described previously [25].
Extraction and assay of anthocyanin
The pH differential method was used [26] with minor modification. The fruits at different growth stages were harvested from multiple healthy plants and fully ground in liquid nitrogen. Briefly, aliquots of 1.0 g ground fruit tissue was dissolved in 20 mL of 60 % methanol (pH 3.0), and then kept at 40 °C for 2 h. The extracts was collected through vacuum pump filter followed by decompression concentration in the rotary evaporation apparatus. Next, aliquots of 1 ml of extract was taken in duplicate and diluted to 25 ml in buffer A (0.2 mol.L−1KCL:0.2 mol.L−1 HCl = 25:67,v:v,pH1.0) and buffer B (1 mol.L−1 NaAc:1 mol.L−1 HCL:H2O = 100:60:90,v:v,pH4.5), respectively. The absorbance value at 520 nm was measured via UV-600 ultraviolet and visible spectrophotometers.
The content of anthocyanins was calculated using the formula. The formula is C (mg/g) = (A0 ‐ A1) × V × N × M/(e × m). Among them, A0 and A1 was the light absorption value of anthocyanins at pH 1.0 and pH 4.5, respectively. V means the total volume of the extract, N is the dilution ratio, M is 449 for the standard molecular weight marker anthocyanins, e is 29,600 for standard extinction coefficient, m is 1 g of the sample weight. The assays were repeated three times along with three independent repetitions of the biological experiments and the means of the three biological experiments were calculated for the total anthocyanin levels in each of the samples.
Abbreviations
- 4CL:
-
4-coumarate CoA ligase
- ANS:
-
Anthocyanidin synthase
- C4H:
-
Cinnamate 4-hydroxylase
- CHI:
-
Chalcone isomerase
- CHS:
-
Chalcone synthase
- COG:
-
Clusters of orthologous groups of protein
- DFR:
-
Dihydroflavonol 4-reductase
- F3H:
-
Flavonoid 3-hydroxylase
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- Nr:
-
Non-redundant protein database
- Nt:
-
Non-redundant nucleotide database
- PAL:
-
Phenylalanine ammonialyase
- qRT-PCR:
-
Quantitative real-time PCR
- RPKM:
-
Reads per kb per million reads
- Swiss-Prot:
-
Annotated protein sequence database
- TrEMBL:
-
Computer-annotated supplement to Swiss-Prot database
- UFGT:
-
UDP-Glucose flavonoid 3-O-glucosyl transferase
References
Galletta G, Ballington J. Blueberries, cranberries and lingonberries. Fruit Breeding. 1996;2:1–107.
Rowland LJ, Alkharouf N, Darwish O, Ogden EL, Polashock JJ, Bassil NV, Main D. Generation and analysis of blueberry transcriptome sequences from leaves, developing fruit, and flower buds from cold acclimation through deacclimation. BMC Plant Biol. 2012;12(1):46–64.
Lehnert D. Blueberry production is skyrocketing worldwide. The Fruit Growers News Internet site: http://www.fruitgrowersnews.com/pages/arts.php. 2008.
Prior RL, Gu L. Occurrence and biological significance of proanthocyanidins in the American diet. Phytochemistry. 2005;66(18):2264–80.
Brazelton C. World blueberry acreage & production. North American Blueberry Council Available online: http://www.chilealimentos.com/2013/phocadownload/Aprocesados_congelados/nabc_2012-world-blueberry-acreage-production.pdf. (accessed on 4 May 2015) 2013.
Rasmussen SE, Frederiksen H, Struntze Krogholm K, Poulsen L. Dietary proanthocyanidins: occurrence, dietary intake, bioavailability, and protection against cardiovascular disease. Mol Nutr Food Res. 2005;49(2):159–74.
Cho E, Seddon JM, Rosner B, Willett WC, Hankinson SE. Prospective study of intake of fruits, vegetables, vitamins, and carotenoidsand risk of age-related maculopathy. Arch Ophthalmol. 2004;122(6):883–92.
Kalt W, Joseph JA, Shukitt-Hale B. Blueberries and human health: a review of current research. J Am Pomolog Soc. 2007;61(3):151–60.
Costich D, Ortiz R, Meagher T, Bruederle L, Vorsa N. Determination of ploidy level and nuclear DNA content in blueberry by flow cytometry. Theor Appl Genet. 1993;86(8):1001–6.
Naik D, Dhanaraj AL, Arora R, Rowland LJ. Identification of genes associated with cold acclimation in blueberry (Vaccinium corymbosum L.) using a subtractive hybridization approach. Plant Sci. 2007;173(2):213–22.
Montalba R, Arriagada C, Alvear M, Zúñiga GE. Effects of conventional and organic nitrogen fertilizers on soil microbial activity, mycorrhizal colonization, leaf antioxidant content, and Fusarium wilt in highbush blueberry (Vaccinium corymbosum L.). Sci Hortic. 2010;125(4):775–8.
Vijay N, Poelstra JW, Künstner A, Wolf JB. Challenges and strategies in transcriptome assembly and differential gene expression quantification. A comprehensive in silico assessment of RNA‐seq experiments. Mol Ecol. 2013;22(3):620–34.
Jaakola L, Määttä K, Pirttilä AM, Törrönen R, Kärenlampi S, Hohtola A. Expression of genes involved in anthocyanin biosynthesis in relation to anthocyanin, proanthocyanidin, and flavonol levels during bilberry fruit development. Plant Physiol. 2002;130(2):729–39.
Sun H, Liu Y, Gai Y, Geng J, Chen L, Liu H, Kang L, Tian Y, Li Y. De novo sequencing and analysis of the cranberry fruit transcriptome to identify putative genes involved in flavonoid biosynthesis, transport and regulation. BMC Genomics. 2015;16(652):1–17.
Zifkin M, Jin A, Ozga JA, Zaharia LI, Schernthaner JP, Gesell A, Abrams SR, Kennedy JA, Constabel CP. Gene expression and metabolite profiling of developing highbush blueberry fruit indicates transcriptional regulation of flavonoid metabolism and activation of abscisic acid metabolism. Plant Physiol. 2012;158(1):200–24.
Li X, Sun H, Pei J, Dong Y, Wang F, Chen H, Sun Y, Wang N, Li H, Li Y. De novo sequencing and comparative analysis of the blueberry transcriptome to discover putative genes related to antioxidants. Gene. 2012;511(1):54–61.
Gupta V, Estrada AD, Blakley I, Reid R, Patel K, Meyer MD, Andersen SU, Brown AF, Lila MA, Loraine AE. RNA-Seq analysis and annotation of a draft blueberry genome assembly identifies candidate genes involved in fruit ripening, biosynthesis of bioactive compounds, and stage-specific alternative splicing. GigaScience. 2015;4(5):1–22.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52.
Varshney RK, Graner A, Sorrells ME. Genic microsatellite markers in plants: features and applications. TRENDS Biotechnol. 2005;23(1):48–55.
Varshney RK, Sigmund R, Börner A, Korzun V, Stein N, Sorrells ME, Langridge P, Graner A. Interspecific transferability and comparative mapping of barley EST-SSR markers in wheat, rye and rice. Plant Sci. 2005;168(1):195–202.
Li Y-C, Korol AB, Fahima T, Nevo E. Microsatellites within genes: structure, function, and evolution. Mol Biol Evol. 2004;21(6):991–1007.
Ray H, Yu M, Auser P, Blahut-Beatty L, McKersie B, Bowley S, Westcott N, Coulman B, Lloyd A, Gruber MY. Expression of anthocyanins and proanthocyanidins after transformation of alfalfa with maize Lc. Plant Physiol. 2003;132(3):1448–63.
Zeng S, Xiao G, Guo J, Fei Z, Xu Y, Roe BA, Wang Y. Development of a EST dataset and characterization of EST-SSRs in a traditional Chinese medicinal plant, Epimedium sagittatum (Sieb. Et Zucc.) Maxim. BMC Genomics. 2010;11(1):94–105.
Wei W, Qi X, Wang L, Zhang Y, Hua W, Li D, Lv H, Zhang X. Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genomics. 2011;12(1):451–64.
Willems E, Leyns L, Vandesompele J. Standardization of real-time PCR gene expression data from independent biological replicates. Anal Biochem. 2008;379(1):127–9.
Shaobo W, Yongfeng D, Binghua Y. Determination of anthocyanin content in black bean peel by pH-differential method. Chem Analysis Meterage. 2008;17(1):46–7.
Acknowledgments
For the access to experimental materials we would like to thank Tianshuo Farm in Hebei Province, China, providing blueberry fruit for the experiment. For the technical assistance during the comparison between our dataset and previously published data, we would like to thank Biobreeding Biotechnology Corporation (Shaanxi, China). Likewise, we would like to express great gratitude to the anonymous peer review and critical revise for the manuscript improvement.
Funding
This work was supported by a grant from Agricultural Ministry of China (Grant no. 2014ZX08009-003-002) and the National Natural Science Foundation of China (Grant No. 31270663 to L.Y.Z.).
Availability of data and materials
The data supporting the results of this article is included within the article. All sequence data is available in the Short Read Archive system of the NCBI under accessions SRR2910056. (http://www.ncbi.nlm.nih.gov/sra/).
Authors’ contributions
LL analyzed the data and wrote the manuscript; HZ performed RT-qPCR experiment and revised the manuscript; XC repeated the RT-qPCR experiment; TZ screened internal controls for normalization in the RT-qPCR experiment; ZL and YL sampled the fruits at different stages and were responsible for the detection of anthocyanin content; LZ designed the experiment and revised the manuscript. All authors read and approved the final manuscript.
Competing interest
The authors declare that they have no competing interest.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
File S1. Summary of the sequence assembly for five cultivars of blueberry. (XLSX 12 kb)
Additional file 2:
File S2. Pathway assignment based on KEGG. (XLSX 11 kb)
Additional file 3:
File S3. Repeats of EST-SSRs in blueberry. (XLSX 18 kb)
Additional file 4:
File S4. List of unigenes that cover 9 selected genes respectively. (XLSX 12 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, L., Zhang, H., Liu, Z. et al. Comparative transcriptome sequencing and de novo analysis of Vaccinium corymbosum during fruit and color development. BMC Plant Biol 16, 223 (2016). https://doi.org/10.1186/s12870-016-0866-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-016-0866-5