
Abstract
In this narrative review, we examine biological processes linking psychological stress and cognition, with a focus on how psychological stress can activate multiple neurobiological mechanisms that drive cognitive decline and behavioral change. First, we describe the general neurobiology of the stress response to define neurocognitive stress reactivity. Second, we review aspects of epigenetic regulation, synaptic transmission, sex hormones, photoperiodic plasticity, and psychoneuroimmunological processes that can contribute to cognitive decline and neuropsychiatric conditions. Third, we explain mechanistic processes linking the stress response and neuropathology. Fourth, we discuss molecular nuances such as an interplay between kinases and proteins, as well as differential role of sex hormones, that can increase vulnerability to cognitive and emotional dysregulation following stress. Finally, we explicate several testable hypotheses for stress, neurocognitive, and neuropsychiatric research. Together, this work highlights how stress processes alter neurophysiology on multiple levels to increase individuals’ risk for neurocognitive and psychiatric disorders, and points toward novel therapeutic targets for mitigating these effects. The resulting models can thus advance dementia and mental health research, and translational neuroscience, with an eye toward clinical application in cognitive and behavioral neurology, and psychiatry.
Similar content being viewed by others

Background
Stress is a physiological response that engages the hypothalamic–pituitary–adrenal (HPA) axis and locus coeruleus-norepinephrine (LC-NE) system to address salient stimuli (i.e., stressors) that are either potential or actual threats to our physical and/or psychological well-being. Psychological stressors trigger negative effects on mental health known as psychological stress [e.g., 1, 2]. The magnitude and prevalence of psychological stress effects are well documented. For example, in the United States, work-related stress has an economic impact of up to $190 billion annually [3, 4], and the results of the 2022 survey performed for the American Psychological Association showed that stress is altering all aspects of life [5]. Accordingly, the World Health Organization highlighted the global importance of interventions for stress management and well-being programmes in its Comprehensive Mental Health Action Plan 2013–2030 [6].
Substantial research indicates that psychological stress, which can involve extreme and/or chronic experiences of aversive feelings, can evoke neural responses associated with pain perception [e.g., 7, 8], mentalizing, psychiatric disorders, and cognitive deteriorations [e.g., 1, 2, 9, 10]. Stress is also a risk factor for both acute and progressive cerebral pathologies such as stroke and neurodegenerative disease [e.g., 9, 11,12,13,14], which reciprocal interplay to accelerate cognitive manifestation of dementia [e.g., 15, 16]. Neurodegenerative diseases and psychiatric disorders are often highly comorbid [e.g., 17,18,19].
To better understand stress-induced neural dynamics implicated in cognitive impairment, as well as the etiopathogenetic mechanisms that can be shared by stress-related neurocognitive diseases and psychiatric disorders, we present a comprehensive narrative review of their interconnected cellular processes. The conceptual framework for this review is centered on the idea that psychological stress can alter the brain on multiple levels (e.g., epigenetics, neurotransmitters) that are relevant for affect, behavior, and cognition. First, we briefly revisit the general neurobiology of the stress response to define neurocognitive stress reactivity and the mechanisms of habituation and sensitization. Next, we review the main mechanisms shared by stress responses, cognitive processing, and neuropsychiatric alteration on the level of epigenetic regulation, synaptic transmission, sex hormone co-signalling, photoperiodic plasticity, and psychoneuroimmunology. The review is structured as an array of the sections on each level, as we describe how stress can contribute to cognitive decline and behavioral alteration. We show mechanistic parallelisms between stress response and neuropathology with the aim of proposing new experimental directions and specific scientific hypotheses that should be tested. Ultimately, understanding links between stress, cognitive impairment, and psychiatric disorders will enable researchers to generate testable hypotheses that can in turn lead to novel insights and potentially to new therapeutic targets.
Neuropsychobiology of the stress responses
Summary of the concepts: Stress is a reaction to a stressor in attempt to protect and maintain physiological stability through the process of allostasis that helps adaptation. However, exposure to chronic or severe stress can cause habituation and sensitization associated with harmful effects of altered allostasis.
Takeaway: Stress responses are functions of allostatic status and cognitive appraisal.
The concept of allostasis
Exposure to psychological stressors evokes neural responses directed to protect and maintain physiological stability (i.e., homeostasis) through a process known as allostasis [e.g., 20,21,22]. Allostasis is determined by the synergetic activities of the HPA axis (“neuroendocrine limb”) and the LC-NE system (“cognitive limb”) [e.g., 1, 9, 23,24,25,26]. Allostasis is initiated with the release of corticotropin-releasing hormone (CRH) from the hypothalamus and urocortin from the brainstem, which together form the corticotropin-releasing factor (CRF) family in humans. CRH release stimulates secretion of adrenocorticotropic hormone (ACTH) from the adenohypophysis, which, in turn, stimulates the adrenal cortex to produce cortisol as a stress hormone [e.g., 27,28,29,30]. The cortisol response to stress follows a triphasic pattern during a successful adaptation to stressors [e.g., 21, 22, 31]. First, there is an alarm reaction, then resistance, followed by resolution of the cortisol response due to negative feedback-inhibition over the HPA activity that results in decreased sensitivity to ACTH, and, in turn, suppression of cortisol production. This physiological pattern of cortisol response is moderated by sensitivity of the glucocorticoid receptors (GR).
However, when stress-induced neuroendocrine responses are ongoing or elevated, allostatic load can turn into allostatic overload to which there is a cost of poor health outcomes, including psychiatric disorders [e.g., 32]. The allostatic load can result from exposure to a chronic or repeated stressor (e.g., interpersonal stress), cumulative challenges (e.g., work-related stress and low socioeconomic status) or prior adverse experiences (e.g., maltreatment in childhood). That is why in patients, the same clinical diagnosis does not imply the same underlying health condition. Assessments of allostatic load, such as the evaluation of biopsychosocial determinants of health, may help with advancing personalised clinical interventions [e.g., 33].
Severe or chronic stress can result in allostatic failure defined as a stress response that surpasses the essential needs and, in turn, results in harmful effects (i.e., maladaptation) [e.g., 9, 10, 21, 24, 25, 31,32,33,34,35]. Importantly, stress severity—and, hence, the level of adaptation—is principally linked to a person’s cognitive appraisal of the situation (i.e., how the stressor is perceived; for details, please see our recent work [1]). This means that neurobiological responses to a psychological stressor are largely modulated by neuropsychological feedback, with an individual’s cognitive and behavioral changes in a response to a psychological stressor being called neurocognitive stress reactivity [1].
The neurocognitive responses play a crucial role in early-life stress/adverse childhood experiences. Specifically, the neurodevelopmental aspects increase brain vulnerability to cognitive and emotional dysregulation, which is a risk for childhood stress psychopathology (for details, see [2]). The neurocognitive impact of stress reactivity is also prominent in various neuropsychiatric conditions. For example, when coping with stress, individuals with borderline personality disorder or post-traumatic stress disorder (PTSD) can present with reduced pain perception related to dissociation which, in turn, can be linked to self-harm and suicide attempts [e.g., 36,37,38; see also 39]. Accordingly, the assessment of somatosensory function can serve as a clinical intervention tool for screening psychologically vulnerable populations whose stress-coping mechanisms may increase their risk for suicide attempts [e.g., 40, 41].
Habituation and sensitization in response to stress
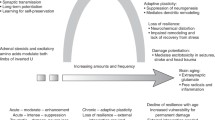
At the neuroendocrine level, maladaptive stress responses depend in part on an individual’s sensitivity to ACTH and the type of stressor experienced. Specifically, the stressor can be novel (i.e., acute exposure to a new aversive stimulus), homotypic (i.e., chronic/repeated exposure to the same/similar aversive stimuli, e.g., chronic bullying), or heterotypic (i.e., exposure to different aversive stimuli, e.g., financial or psychosocial challenges following caregiver burden). Sensitivity to ACTH can be: (a) increased for a novel stressor, (b) initially increased and then decreased during chronic exposure to a homotypic stressor, or (c) diminished in response to a homotypic stressor [42,43,44]. In addition, exposure to homotypic stressors can lead to habituation of the HPA axis response (as can be seen in decreased glucocorticoid response) and enhance post-stress recovery of the HPA axis (i.e., post-stress return to baseline). Yet, prior exposure to homotypic stressors can cause sensitization of the HPA axis response (as can be seen in increased glucocorticoid response) and worsen the recovery of the HPA axis following exposure to a novel stressor [e.g., 34, 45,46,47]. The outcomes of maladaptive stress-induced hyperactivity of the HPA axis seen in prolonged hypercortisolemia and/or altered GR function are related to depression and anxiety [e.g., 9, 48, 49]. Additionally, behavioral sensitization appears to persist longer than the HPA axis sensitization and is often a latent phenomenon revealed by a novel stressor [e.g., 47, also see 50]. In other words, stress adaptation declines in the context of chronic stress, which reduces a person’s ability to recover after a novel stressor exposure and can increase their susceptibility to mood disorders and other stress-related health problems [e.g., 1, 2].
At the same time, stress resilience is associated with CRF-mediated regulation of cognitive and behavioral activity [9, 21, 27,28,29, 31, 44, 51,52,53,54,55]. Amygdalar CRF signalling has anxiogenic-like effects [e.g., 9, 29] and mediates defensive responses such as increased vigilance, ability to discriminate salient stimuli, and active escape [e.g., 56]. Research has shown that stress-induced increases in the expression of amygdalar CRF-1 receptors are associated with deficient hippocampal-dependent memory and learning, as demonstrated in the impaired visual discrimination task [e.g., 29, 54, 55; see also 23], whereas hippocampal CRF signalling can promote synaptic remodelling that is potentially supportive of memory consolidation in acute stress [e.g., 57]. To complicate matters, chronic stress also promotes connectivity between the amygdala and striatum. The consequences can be seen in diminished cognitive flexibility caused by shifts from hippocampus-dependent memory to striatum-dependent memory [e.g., 58,59,60,61,62] and behavioral maladaptation due to amygdalar CRF-mediated excitation [e.g., 9, 29, 43, 55]. In addition, upon involvement of the nucleus accumbens (ventral striatum) that is associated with reward-related behavior, reinstatement of drug addiction via glutamate synaptic plasticity may occur [63]. This effect can be explained by the fact that in the nucleus accumbens, β-adrenoreceptors promote memory consolidation of positive and negative arousing experiences [64, 65]. It is thus not surprising that CRF-1 receptor antagonists are being considered as a promising pharmacotherapy for depression, anxiety, cognitive/neurodegenerative, and stress disorders [e.g., 66,67,68; see also 69,70,71].
Effects of stress on neuronal mechanisms shared with cognitive and emotional processing
Transcriptional and epigenetic effects, and interindividual genetic variation
Summary of the concepts: Psychological stress can induce various epigenetic effects that are associated with the activation of GR. The effects implicated in the pathogenesis of neurocognitive and psychiatric disorders. At the same time, polymorphism in genes associated with serotoninergic signalling has been linked to depression susceptibility, cortisol response to stressors, and greater risk for developing mild cognitive impairment.
Takeaway: Examining the impact of gene × environment interactions is superior to investigating interindividual genetic variation by itself.
Epigenetic regulation: central players
Stress-induced surge in corticosteroid signalling can further initiate epigenetic changes (Figs. 1, 2), which have an influence on cognitive functions, behavior, and mood [e.g., 23, 72, 73; see also 74, 75]. The influence is facilitated by the GRs and mineralocorticoid receptors (MRs) cerebral distribution. GRs are highly expressed in the hippocampus and prefrontal cortex (PFC), which relates to executive functioning [e.g., 21, 28, 51]. Glucocorticoid secretion exhibits circadian and ultradian patterns with levels peaking in the morning and are essential for negative feedback after acute psychological stress [e.g., 76,77,78,79,80,81]. Whereas GRs assist information encoding (i.e., memorization), eradication of inadequate behavioral responses, and stress recovery, MRs, which are mainly engaged in the evening, facilitate processing of sensory information, analysis of environmental information, and execution of proper behavioral responses [28, 35, 48, 72, 82, 83].

Epigenetic Mechanisms of Memory Alteration Following Acute Psychological Stress. Simplified and schematic model of indirect epigenetic mechanisms for fast (< 1 h) nongenomic effects induced by membrane-associate receptors during acute stress. Increased levels of NE bind to BR (namely β2) and rapidly increase Ca2+ influx and cAMP and Ca2+/MK activation, that triggers PKA to modulate synaptic activity (see Fig. 4) and phosphorylate CREB, which activates transcription and gene expression linked to neuronal plasticity, spatial memory, and long-term memory formation. This mechanism supports fear conditioning/learning, whereas severe stress can decrease initially activated Ca2+/MK, which relates to poor memory. Estradiol can activate ERs (mostly type β) linked to fear learning/conditioning via ERK/MAPK pathways which affect post-translational gene regulation via histone acetylation. In the amygdala, independently of sex-hormone levels, short photoperiod reduces melatonin and thus increases estradiol-induced phosphorylation of CREB (males) and ERK aka MAPK (females) linked to aggression. In acute stress, high estradiol levels in proestrus are linked to the prefrontal cortex memory deficit and altered glutamate signalling, potentially due to hyperactivated Ca2+/MK. Ca2+/MK, Ca2+/Calmodulin dependent kinases IIα; CH3CO, acetylation; CREB, cAMP response element-binding protein; ERK, extracellular regulated kinase, aka mitogen-activated protein kinase (MAPK); ER, estrogen receptors; GR, glucocorticoid receptor; H, histone; NE, norepinephrine; PO32, phosphorylation; PKA, protein kinase A

Epigenetic Mechanisms of Memory Alteration Following Chronic Psychological Stress. Simplified and schematic model of epigenetic mechanisms for slow (> 1 h) genomic effects induced by nuclear receptors. During chronic stress, increased nuclear levels of GRs-cortisol complex promote DNA methylation. Methylated DNA sites prevent CREB binding, and vice versa (depicted in double arrows as “synergetic inhibition”). At the same time, CREB activity largely correlates to histone acetylation which is essential for memory consolidation. CH3, methyl group; CH3CO, acetylation; CREB, cAMP response element-binding protein; GR, glucocorticoid receptor; GRE, glucocorticoid response elements; H, histone; PO32, phosphorylation
Epigenetic regulation: factors underlying stress resilience
GRs depend on chaperones, especially heat shock proteins (Hsp) 70 and 90 [e.g., 84, 85]. Chaperones are multimeric complexes that control protein qualities (e.g., folding/unfolding, see Fig. 3) and protect cells by identifying emerging polypeptides from irreversible aggregation in the context of stress [e.g., 86, 87]. The chaperones dysregulated activity can result in misfolded and aggregated proteins like tau deposition, a hallmark of several neurodegenerative diseases [e.g., 86, 88,89,90]. Stress can affect the mechanisms of protein aggregation/clearance, which relates to proteotoxicity that is critical in the development and/or progression of neurodegeneration, including Alzheimer’s disease [e.g., 91,92,93].

Epigenetic Mechanisms Underlying Stress Resilience to Neuropsychiatric Disorders. Stress increases levels of circulating cortisol that easily passes the cellular membrane. In the cellular plasma, [1] GR forms a complex with chaperones [2] Hsp70 (that partially unfolds and inactivates GR) and [3] Hsp90 (that facilitates GR maturation) [84, 87]. [4] Binding with co-chaperone FKBP51 (encoded by gene fkbp5) inhibits GR capacity for nuclear transactivation/signalling and detaches Hsp70 [35, 48, 99, 100]. [5] Encoded by gene fkbp4, co-chaperone FKBP52 competes with FKBP51 and its replacement increases affinity of the GR-Hsp90 complex to bind cortisol [84, 97]. [6] When cortisol binds, chaperone complex releases and thus GR and HSF1 translocate to the nucleus to initiate transcription linked to [7] memory formation [84, 87, 95, 101] and [8] gene expression for chaperones and co-chaperones that promote GR activation in stress-response [86, 94]. [9] In an ultra-short feedback loop, it also promotes gene fkbp5, which has close proximity to GRE. Polymorphism of fkbp5 is associated with interindividual differences in stress reactivity. In addition, fkbp5 expression increases with age due to decreased DNA methylation. This simplified model does not depict all protein/regulators involved in chaperone machinery and signalling pathways. CREB, cAMP response element-binding protein; GR, glucocorticoid receptor; GRE, glucocorticoid response elements; H, histone; HSF, heat shock factor 1
Transcriptional and regulatory effects
The role of GRs in memory relates to their translocation ability in neurons. Specifically, GRs are dynamic molecular structures located mainly in the neural cytoplasm. GRs have the capacity to bind all steroid hormones with a ligand-binding domain that has 12 helices in the form of a “sandwich” [e.g., 94, 95]. Upon cortisol binding, activated GRs release Hsp90 that eases GRs’ nuclear translocation, where it acts as a DNA-binding transcription factor; GRs’ binding to DNA glucocorticoid response elements activates transcription—that is, transactivation effect seen in the increased rate of gene expression [e.g., 35, 48, 72, 84, 86, 87, 94, 96,97,98,99,100; see Fig. 3]. GRs’ nuclear translocation can also induce a transrepression, which is an epigenetic repression of other transcription factors including NF-κB and AP-1 related to the proinflammatory immune response [95, 101]. In addition, nuclear GRs can cause indirect genomic effect via cAMP-dependant protein kinase A that correlates to learning and memory function [e.g., 102; see Figs. 4–6].

Psychological Stress: Noradrenergic Signalling and Synaptic Plasticity. A crucial part in memory formation belongs to synaptic activity/plasticity, particularly in the hippocampus. First, in the axon of a neuron, [1] action potentiation converts electrical stimuli into a chemical message [glutamate] to pass it through the synapse to the dendrite of another neuron. At the dendritic spine, ionotropic channels [2a] NMDA- and [2b] AMPA-type glutamate receptors “receive the message”. [3a] NMDA receptors facilitate Ca2+ entry that triggers Ca2+/Calmodulin dependent kinases Iiα, which results in synaptic incorporation of AMPA receptors—a necessary mechanism for long-term potentiation as a part of memory formation. [3b] Activated AMPA receptors stimulate ERK (aka MAPK). Additionally, in the dorsal hippocampus, activated estrogen receptors can stimulate GluA1 subunit via ERK linked to enhanced neurocognition in females but not in males. During acute stress (depicted by black double arrows), due to emotional arousal (e.g., fear), released norepinephrine binds to β2-adrenoreceptors (G-protein-coupled receptor) and activates protein Gαs; that stimulates adenylate cyclase and cAMP synthesis, which accumulation triggers protein kinase A and ERK/MAPK (depicted by yellow ovals, see Fig. 1 for more details). Protein kinase A activates AMPA receptor by its subunit GluA1 phosphorylation that results in the AMPA receptor’s trafficking and synaptic incorporation. As well, protein kinase A activates Ca2+/Calmodulin dependent kinases IIα directly and via [4] stimulation of L-type Ca2+ channel that increases Ca2+ influx and activates the kinases further (white arrows depict Ca2+ signalling). During severe stress, excessive norepinephrine release can also [5] activate α1-adrenoreceptors and trigger protein kinase C signalling that activates AMPA receptors. Stathmin is a microtubule-stabilizing and ERK-regulated protein that displays cytoprotective function. Specifically, dynamical changes in the microtubule stability are vital for synaptic plasticity and long-term potentiation. Synaptic input, such as during learning/facing a threat, hyperactivates stathmin, which decreases microtubule stability (depolymerization within first hour). SNARE protein complexes interact with serotonin signalling regulated by fkbp5 gene (see Fig. 3), that potentially can determine an individual’s susceptibility to stress and depression. cAMP, cyclic AMP; CREB, cAMP response element-binding protein; ERK, extracellular regulated kinase, aka mitogen-activated protein kinase (MAPK); GR, glucocorticoid receptors; GRE, glucocorticoid response elements; PO32, phosphorylation

Acute Psychological Stress: The Role of Cortisol and Kinases in Memory Formation. During acute stress (depicted by mustard/dark yellow arrows), rapidly increased levels of cortisol can activate GRs that increase surface expression of NMDA and AMPA receptors (nongenomic memory effect). Upregulated Ca2+/Calmodulin dependent kinases IIα pathway activates CREB mechanism (fast indirect epigenetic effect related to neuropsychiatric outcomes such as anxiety and increased risk for post-traumatic stress disorder). At the same time, activated ERK/MAPK inhibits stathmin via phosphorylation; that stabilizes microtubules and, in turn, activates incorporation of the GluA2 subunit (AMPA receptor) to synaptic sites, which is necessary for long-term memory formation yet supports fear conditioning/learning. See Fig. 4 for the path [1]–[4]. cAMP, cyclic AMP; CREB, cAMP response element-binding protein; ERK, extracellular regulated kinase, aka mitogen-activated protein kinase (MAPK); GR, glucocorticoid receptors; GRE, glucocorticoid response elements; PO32, phosphorylation

Chronic Psychological Stress: Proteinopathy and Synaptic Plasticity. During chronic stress (depicted by dark blue arrows), prolonged activation of GRs inhibits Ca2+/Calmodulin dependent kinases IIα and can lead to DNA hypermethylation that supresses gene transcription and protein synthesis (epigenetic memory effect). Severe stress can also reduce the expression of the stathmin that is related to the cellular skeleton, mitosis, and synaptic plasticity, which in turn, relates to poor learning and apoptosis. As well, chronic stress/single prolonged stress can increase SNARE complex formation but alter neurotransmitters fusion that relates to excitotoxicity and pathological accumulation of aggregated proteins. Alteration of the protein-kinase dynamics increases risk for proteinopathy and, in turn, depression and neurodegenerative disorders. See Fig. 4 for the path [1]–[4]. cAMP, cyclic AMP; CREB, cAMP response element-binding protein; ERK, extracellular regulated kinase, aka mitogen-activated protein kinase (MAPK); GR, glucocorticoid receptors; GRE, glucocorticoid response elements; PO32, phosphorylation
Further, stress-activated GRs can induceDNA methylation via mitogen-activated protein kinase (MAPK), also known as extracellular signal-regulated kinases (ERK), that are required for fear memory and mediate stress-related behavioral effects of GRs [e.g., 103, 104; see also 105,106,107,108; Figs. 1–6]. The DNA methylation, which is a chromatin re-modelling by the addition of methyl groups (CH3) to cytosine nucleotide during transactivation (see Figs. 1, 2), serves as a “molecular bridge” between the external (i.e., environment) and internal (i.e., cellular) world [e.g., 109,110,111,112,113,114]. It is regulated by de novo methyltransferases that are crucial for hippocampal memory in contextual fear conditioning, late-phase long-term potentiation, and spatial memory [e.g., 115].
Another important mechanism that can be engaged in stress-associated neuropsychiatric outcomes belongs to the nucleosome modifications. A nucleosome is a “bead” segment of DNA wrapped around histones, which are octamers of duplicated H2A/H2B dimer and H3/H4 tetramer proteins. Histones display a globular structure with protruding “tails” from the nucleosome—N-terminal domains, exclusively in H2A and H2B extra C-terminal domains (Figs. 1, 2). The nucleosome has dynamic phases of “breathing” when it unwraps and rebinds DNA, and thus transiently exposes DNA sites [e.g., 116] so that genes can be regulated post-translationally via (but not limited to) methylation. The “breathing” has a dual role: it condensates DNA to deactivate transcription, and relaxes DNA to activate transcription [e.g., 117, 118]. This process modifies histone tails with “postal codes” (i.e., molecular tags) to be delivered and recognized by “reader” proteins involved in chromatin re-modeling [e.g., 115, 118]. The molecular tagging of the histone tails is an epigenetic mechanism of learning and memory, which is regulated by “eraser” proteins that remove established histone-tail modifications [119]. However, stress can block this process via histone acetylation mechanisms and, in turn, lead to depressive- and anxiety-like behavior, visceral hypersensitivity, and cognitive deterioration [e.g., 120,121,122,123]. The reason is that histone acetylation is a rapid process of histone modification. It is regulated by histone acetyltransferases, which are both the histone “postal codes writers” and “postal codes readers” balanced by histone deacetylase activity that may reverse histone acetylation [e.g., 119]. Histone acetyltransferases contribute to the transcriptional control during memory formation, including contextual fear conditioning, novel object recognition, spatial memory, and long-term potentiation [e.g., 115, 124, 125]. Additionally, histone acetylation and epigenetic mechanisms of de novo DNA methylation are synergetic. For instance, histone deacetylase inhibitor infusion via transcription factor (NGFI-A) binding to the GR promoter reverses DNA methylation, GR expression, and HPA reactions [e.g., 114, 126]. Furthermore, inhibition of DNA methyltransferase blocks promoted histone H3 acetylation in contextual fear conditioning [e.g., 115, 127]. It is thus not surprising that histone modifications are implicated in the pathogenesis of cognitive dysfunction, neuropsychiatric conditions, and neurodegenerative disorders such as PTSD and Alzheimer’s disease [e.g., 128,129,130]. Accordingly, inhibitors of histone deacetylase show neuroprotective properties and show potential as an intervention in the treatment of neurocognitive disorders and PTSD [e.g., 119, 131, 132].
Stress can also alter methylation of hippocampal brain-derived neurotrophic factor (BDNF), which plays an important role in adaptation to chronic stress by promoting neuroplasticity [e.g., 133, 134]. Prolonged cortisol hypersecretion in chronic stress increases methylation of the hippocampal BDNF promoter, which is positively correlated to memory consolidation in contextual fear learning [e.g., 111, 135; also see 136,137,138,139]. Chronic stress also supresses BDNF expression that results in subsequent hippocampal atrophy and deficient acquisition/consolidation of verbal declarative memory [e.g., 140,141,142,143]. Reductions in hippocampal BDNF are linked to desensitization of GRs, higher stress vulnerability, and predisposition to psychiatric comorbidity in stress [e.g., 133, 144,145,146]. The mechanism involves a downregulation of peroxisome proliferator-activated receptor δ, which is related to significantly reduced neurogenesis and behaviors that are consistent with depression [147].
At the same time, stress-related changes in the epigenome can be reversed. For instance, glutamate receptor (NMDA) blockade inhibits BDNF expression and prevents its methylation, which in turn supresses altered memory formation [e.g., 111]. Moreover, the induction of gene-specific demethylation (e.g., by genetic removal of a regulator of active DNA demethylation or with DNA methyltransferase inhibitors) can improve late-phase hippocampal potentiation, spatial memory, and contextual fear memory consolidation [e.g., 115; see also 10]. At least one systematic review and meta-analysis revealed that patients with PTSD have elevated serum BDNF levels compared to healthy individuals [148]. Despite equivocation in the literature, some clinical studies have related the BDNF Val66Met polymorphism to modulation of stress sensitivity [e.g., 149]. For instance, the BDNF Val66Met polymorphism has been shown to moderate the relation between PTSD and fear extinction learning [150] and aversive memory bias in women with PTSD but not in psychiatrically healthy women [151]. At the same time, PTSD and dementia have been shown to be bidirectionally linked [e.g., 152], and that PTSD is a potentially modifiable risk factor for dementia [153; see also 69].
Interindividual genetic variations
Although stress can initiate epigenetic changes that modulate susceptibility to neuropsychiatric conditions, a glucocorticoid-dependent encoding gene fkbp5 plays a major role in stress resilience [e.g., 84, 96, 154]. The fkbp5 gene is in close proximity to a glucocorticoid-responsive element [96, 155, 156]. Moreover, the fkbp5 polymorphism correlates with activation of GRs and can thus increase post-traumatic stress susceptibility to psychiatric disorders [99, 101, 157]. The fkbp5 gene, which encodes co-chaperone FKBP51 and regulates serotoninergic signalling [e.g., 158], is associated with depression susceptibility and reduced cortisol response to stress due to GRs hypofunction. For example, fkbp5 rs1360780 T‐allele carrier status is related to GR resistance seen in reduced concentrations of plasma cortisol and ACTH following dexamethasone administration in depressed patients compared to healthy controls [159]. In healthy volunteers, the fkbp5 polymorphism modulates recovery to the Trier Social Stress Test (TSST). Specifically, the fkbp5 variants (rs1360780, rs3800737, and rs4713916) were related to less cortisol recovery and higher anxiety after psychosocial stress, whereas the GR polymorphism (Bcl1 but not N363S) was related to anticipatory cortisol levels [160]. Additionally, there is a male-specific effect in fkbp5 polymorphism (rs3800737) that is linked to the cortisol responses to TSST (i.e., peak response and response area under the curve), whereas variants rs7209436 and rs110402 in CRH receptor gene (CRHR1) are associated with a trait anxiety × baseline cortisol interaction [161].
The polymorphism in fkbp5 has also been shown to predict individual differences in stress-related memory deficits [e.g., 162, 163] and functional activity/connectivity of the amygdala related to emotional (anxiety/depression) responses to threat [e.g., 164,165,166,167,168]. Its interaction with early life stress is also predictive of reduced connectivity between the amygdala and parahippocampal gyrus, caudate, and frontal gyri [169; see also 170]. In healthy youth, for example, fkbp5 genotypes (rs7748266, rs1360780, rs9296158, rs3800373, rs9470080 and rs9394309) predicted relatively increased threat-related dorsal amygdala reactivity in the context of higher self-reported emotional neglect [171]. Genetic variants in fkbp5, as well as CRHR1 and NR3C2, have been also found to be associated with higher HPA activity, and their interaction with early life stress is associated with right amygdala reactivity to threat and anxious arousal [172]. In adults exposed to high levels of childhood trauma, rs3777747, rs4713902, and rs9470080 (main effects) and rs3800373, rs9296158, and rs1360780 (interactive effects with Childhood Trauma Questionnaire score) were related to a greater risk of a lifetime suicidal behavior [173].
There are interindividual and sex-specific peculiarities of the stress-response related to FKBP51 functions. Specifically, FKBP51 interacts with sex hormones (progesterone and androgen) receptors that modifies their sensitivity. As well, FKBP51 competes with FKBP52 regulation of memory-associated synaptic plasticity and transmembrane calcium channels [e.g., 84, 96]. In healthy undergraduate students, exposure to a socially evaluative cold pressor test directly prior to verbal learning reduced immediate verbal recall in fkbp5 risk allele carriers (rs1360780, rs3800373 and rs929615). In contrast, the stress task enhanced 24-h later recall and recognition memory in non-carriers of the risk alleles [163]. In young healthy adults, fkbp5 A-allele carriers had less pronounced autonomic responses to stress and poor working memory performance on the Stroop color-word task (the results of which positively correlated to the degree of self-reported early life adversity), which was associated with poor health behaviors when compared to fkbp5 GG homozygotes [162].
More studies are needed, however, to investigate serotonin (5-HT) signalling and interactive effects of genetic polymorphisms in relation to the processing of specific memory types. Indeed, spatial memory versus emotional memory, or memory formation versus memory retrieval depend on different circuits and signalling pathways, and therefore may be differentially affected across distinct types, stages, and phases of stress. Such inquiry is also needed given that 5-HT receptors such as 5-HT1A, 5-HT2A, and 5-HT7 play an important role in learning and memory [e.g., 174,175,176,177,178,179,180,181,182]. In addition, there is an association between the serotonin transporter-linked promoter region (5HTTLPR) polymorphism and clinical manifestations in neurodegeneration—for example, cognitive impairment variations in Alzheimer’s disease and delusions in Lewy body dementias [e.g., 183,184,185,186]. The evidence suggests that the 5HTTLPR variants modulate the levels of neural activation in the hippocampus and PFC during memory retrieval in acute psychosocial stress [184; see also 187] and can have a negative effect on memory via mediation of the HPA axis in the elderly [e.g., 188]. These data can explain the findings that the 5HTTLPR polymorphism is a risk factor for developing mild cognitive impairment, which precedes Alzheimer’s disease [e.g., 189]. Therefore, it is one of the neurogenetic components of stress resilience and neuropsychiatric outcomes [e.g., 168, 190, 191].
Neurotransmitter signaling
Summary of the concepts: Stress-induced GR levels modulate neuronal plasticity and stress resilience via neurotransmitters:
-
a.
Successful coping under stressful conditions relates to glutamatergic signalling.
-
b.
During stress, emotional arousal can alter Ca2+/Calmodulin-dependent kinases involved in emotional and cognitive processing.
-
c.
The protein-kinase dynamics can play an important role in emotional homeostasis and cognitive reserve that may determine an individual’s susceptibility to neuropsychiatric disease.
-
d.
Stress-induced increase in norepinephrine (NE) levels affect memory in inverted U-shaped relation.
-
e.
Polymorphisms in genes associated with dopamine levels can be a prospective screening tool for stress resilience capacity based on the phenotypic variability of behavior and cognition patterns.
Takeaway: Pharmacotherapy based on neurotransmitter signaling and administrated to block harmful effects of glucocorticoids on working memory could eliminate memory consolidation of emotionally significant experiences.
Glutamate
During stress, hippocampal-dependent memory and learning can be influenced by emotional hyperarousal (i.e., anger, fear, or happiness). This phenomenon involves amygdala-mediated effects on memory consolidation and retention [192; see also 10, 193,194,195,196,197], which is a reinforcement of synaptic networking between neuronal ensembles (engrams) [e.g., 198, 199]. Engrams require dynamic variability (i.e., synaptic plasticity) that occurs during synaptic transmission: as hyperarousal initiates presynaptic input (i.e., a neurotransmitter release) and postsynaptic output (i.e., receptors expression), it boosts engrams [200]. This is how the amygdalar basolateral nucleus redirects attention toward aversive stressors (i.e., attentional bias), promotes encoding and consolidation for aversive memory, consolidates contextual and spatial information, and fortifies memory of novel contexts [e.g., 36, 192, 201,202,203,204,205,206,207,208,209,210,211]. Hyperarousal in stress thus enables time-dependent associative memory, especially for aversive information, which inhibits or eliminates previous memory and facilitates defensive behavior. Both maintenance and inhibition of associative memory depend on synaptic plasticity and magnitude of hyperarousal [e.g., 212]. Although initial emotional experience mediates plasticity, prolongated stress can suppress hippocampal plasticity and thus impair hippocampal processing [e.g., 192, 201, 213,214,215,216,217].
The activation pattern of persistent changes (increase/decrease) in synaptic transmission defines the communication between synapses, which can be in the form of long-term potentiation (upon increase) or long-term depression (upon decrease). Long-term potentiation follows presynaptic glutamate release and subsequent postsynaptic depolarization. The activation pattern in synaptic transmission is mediated by ionotropic (cation) channels, the NMDA- and AMPA-type glutamate receptors, often via AMPA-receptor subunit composition [e.g., 218,219,220].
Glutamate is the major excitatory neurotransmitter of the central nervous system, with exceptionally high neuronal levels. Stress-activated GRs can increase glutamate release via pre- and post-synaptic mechanisms, which is a risk for hyperactivation of the glutamate receptors [i.e., excitotoxicity; 4, 221,222,223]. The AMPA receptors are transmembrane channels composed of subunits GluA1–4 that differ by intracellular C-terminal tails [e.g., 224]. Each AMPA receptor subunit has a site that can bind glutamate; when two sites are occupied with glutamate, the receptor’s pore (cation channel) opens and enlarges with an increase of occupied binding sites [e.g., 225]. Released from the Golgi apparatus into the synaptic membrane, subunits of the AMPA receptors are reserved to initiate long-term potentiation due to their capacity for rapid redistribution from non-synaptic sites to the synapse, which is necessary for synaptic plasticity: if the postsynaptic AMPA receptors are inactive, synapses are silent [e.g., 224]. Distribution of the AMPA receptors on the synaptic surface is bidirectionally regulated by protein interactions. Of particular interest are scaffolding proteins PSD-95 and SAP97 that regulate incorporation (i.e., postsynaptic density) and function of the AMPA receptors [e.g., 226], which is an essential process for both synaptic transmission and plasticity that underlines learning and memory [e.g., 227,228,229].
Chronic stress can reduce expression of the AMPA and NMDA receptors at the synaptic membrane due to their post-translational modification, such as increased ubiquitin–proteasome-dependent degradation of the receptor’s subunits [e.g., 223; see also 10]. However, stress can also increase the hippocampal expression of GluA2 subunits linked to enhanced spatial learning and memory [e.g., 230, 231]. Researchers have also shown that higher fear-memory retrieval in adult rats is associated with the amygdala-driven preferential upregulation of PSD-95 and GluA2, contrary to reduced fear-memory retrieval in juvenile rats that is linked to the upregulation of kinases (PKMζ and PI3K), GluA2/3 and GluA1 in the dorsal hippocampus [232]. Stress-activated GRs can also alter glutamatergic signalling via increased GluA1 phosphorylation (at Ser 831) that synchronizes with increased phosphorylation of the main NMDA receptor subunits (NR-1 and NR-2B) as integral functioning of the AMPA and NMDA receptors [233]; see also 234]. Successful coping under stressful conditions is determined by integral AMPA/NMDA receptor function [233,234,235,236,237,238], whereas altered glutamatergic signalling is strongly associated with psychiatric disorders and Alzheimer’s disease [e.g., 239, 240].
Calcium
Physiological and psychological arousal is modulated by the integration of interoceptive and exteroceptive inputs and is an essential factor for attention and cognitive processing. Calcium (Ca2+) influx is necessary for behavioural arousal states, and is mediated by NMDA receptors which facilitate Ca2+ entry which triggers Ca2+/Calmodulin-dependent kinases (Ca2+/MK) to shape synaptic structure by the AMPA-receptors’ redistribution—that is, translocation into the synaptic membrane (see Figs. 4, 5, 6). This receptor’s redistribution promotes actin cytoskeleton involved in the dendrite (its spine) and axon development that is necessary for hippocampus-dependent memory [e.g., 241, 242]. Thus, hippocampus-dependent learning and memory require Ca2+ influx, which is supported by several types of channels: (1) neural, high-voltage-activating aka N-type or Non-L involved in certain forms of LTP [e.g., 243]; (2) residual, immediate-voltage-activating aka R-type such as Cav2.3 α1E subunit involved in spatial memory formation [e.g., 244]; and (3) transient, low-voltage-activating aka T-type such as Cav3.2 subunit involved in context-associated memory retrieval [e.g., 245]. The Ca2+ channels that are most highly distributed in dendrites are long-lasting, high-voltage-activating—that is, “triggered by strong and sustained depolarizations,” aka L-type Ca2+ channels, which regulate “gene expression, synaptic efficacy, and cell survival” [246; see also 247].
Emotional arousal at the time of memory consolidation that promotes memory maintenance relates to intracellular Ca2+ influx via L-type of Ca2+ channels, and Ca2+ concentration is regulated mainly by Ca2+/MK in the amygdala, hippocampus, and PFC. Electrochemically, it is seen at the arrival of an action potential that triggers Ca2+ entry at the presynaptic neuronal terminal, which further activates vesicles with neurotransmitters (e.g., NE, glutamate, GABA, dopamine, serotonin) to release into the synaptic cleft (see Fig. 4). Released neurotransmitters join the postsynaptic receptors, resulting in the synaptic transmission that can be excitatory (mainly glutamate-mediated and formed in dendritic protrusions, aka spines) or inhibitory (mainly GABA-mediated) membrane potential [e.g., 218; see also 248,249,250]. During memory retrieval, Ca2+/MK activate proteasome-mediated postsynaptic protein degradation—and thus labialize/weaken memory due to reduced synaptic efficacy—whereas during memory reconsolidation, Ca2+/MK reroute synthetized de novo proteins to more active synapses [e.g., 251].
In the context of stress, activated GRs can regulate mitochondrial function of Ca2+ holding capacity by translocating into mitochondria as a complex with the anti-apoptotic protein Bcl-2. This mechanism was found to be correlated to neuronal plasticity and stress resilience in an inverted “U”-shape depending on corticosterone levels. Whereas low doses improve neural viability/resilience, high doses (i.e., as it is observed in intense stress) augment kainic acid-induced toxicity of cortical neurons [252, see also 253], which partially explains the toxic neural effect of intense and chronic stress.
Chronic stress is also associated with dysfunction of the Ca2+/MK cascade, which is seen in increased intracellular Ca2+ concentration and reduced Ca2+/MK IIα expression in the medial PFC [e.g., rat models of PTSD, 254]. Intracellular Ca2+ elevation for a prolonged period is toxic and is associated with Ca2+-dependent cellular mechanisms that alter dendritic spine density and neuronal morphology of the pyramidal neurons in the medial PFC [255; see also 256].
In addition, Ca2+/MK IIα is linked to cAMP response element-binding protein (CREB) activation (Figs. 1–6), which in turn inversely correlates to the amygdala-induced anxiety-like behavior [257, also see 258,259,260,261] and altered CREB-BDNF signaling pathway relates to cognitive decline and Aβ toxicity in Alzheimer’s disease [260]. Antidepressants can regulate hippocampal Ca2+/MK IIα an adaptive way: namely, whereas short-term treatment inhibited the kinase activation in a concentration-dependent manner, chronic treatment up-regulated Ca2+/MK IIα [262]. Furthermore, in a mouse model of chronic social defeat stress, anxiolytic effects have been associated with the ERK/CREB/BDNF signaling pathway [263]. Inhibited ERK activity in the hippocampus is a promising therapeutical target for depression [e.g., 264, 265]. In support of this possibility, treatment with resveratrol to prevent stress-induced cognitive deficits relates to the upregulation of CREB/BDNF expression in the hippocampus in vivo and in vitro (i.e., in a rat model of chronic unpredictable mild stress; [266, see also 267, 268].
Protein-kinase dynamics
Stress resilience related to glutamatergic transmission involves several protein complexes, most notable of which is the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex that regulates vesicular neurotransmitter release (i.e., fusion) in a final step of presynaptic vesicle traffic [e.g., 269,270,271, see Figs. 4, 5, 6]. The activity of SNARE complex potentially relates to hippocampal “protein adaptation” and emotional homeostasis, and thus may determine an individual’s susceptibility to stress and depression [e.g., 271,272,273]. Further, chronic antidepressant treatment (i.e., with fluoxetine, reboxetine, or desipramine) has been found to increase neurogenesis and reduce stress-induced presynaptic glutamate release linked to altered memory. Moreover, the mechanism underlying this effect is potentially associated with assembly of SNARE complex [e.g., 221,222,223, 274,275,276,277,278].
Accumulating evidence suggests that the kinases (e.g., Ca2+/MK IIα) and proteins (e.g., SNARE protein complexes) play an important role in emotional homeostasis and cognitive reserve and may thus determine an individual’s susceptibility to neuropsychiatric disorders. We hypothesize that stress resilience thus relates to protein-kinase dynamics linked to cellular toxicity and apoptosis and, in turn, neurodegeneration. Specifically, in acute and intense stress, increased NE influx with emotional arousal appears to skew the interplay toward altered Ca2+/MK IIα expression/activation that can affect the CREB pathway (Fig. 5), which relates to fear conditioning, anxiety, and attentional tunneling, and is potentially a risk factor for PTSD. In the context of severe chronic stress, the protein-kinase dynamic is potentially skewed toward proteinopathy of SNARE protein complex (related to glutamate-induced excitotoxicity, reviewed above) and stathmin (related to the cellular skeleton and mitosis) that impair synapses (Fig. 6).
Acute stress and Ca2+/MK IIα
In a rat model of PTSD, single prolonged stress (SPS) exposure altered free intracellular Ca2+ levels (initially increased but then decreased), increased calmodulin expression, and decreased Ca2+/MK IIα expression in the medial PFC (day 1 after SPS) [254]. Similar findings were obtained for the basolateral amygdala [279] and hippocampus [280]. Calmodulin and Ca2+/MK IIα expressions have also been shown to initially increased but then decreased after SPS in the dorsal raphe nucleus, which was assumed to be associated with the activation of 5-HT1A receptor (related to neuronal inhibition via suppression of adenylyl cyclase, Ca2+/MK IIα, and AMPA receptors) [281; see also 282,283,284].
Severe chronic stress and proteinopathy
Recent findings on hippocampal presynaptic membrane dysfunction in rat models of PTSD demonstrated that SPS disabled synaptic vesicle fusion (i.e., reduced expression of synaptotagmin-1, the calcium-ion sensor for fusion), extended axon (i.e., increased expression in proteins, e.g., tau and β-tubulin, but decreased expression in p-tau and stathmin), and potentially increased SNARE complex formation (i.e., increased VAMP, STX1A, and Munc18-1 expression) [273]. The formation of SNARE protein complexes interacts with serotonin signalling regulated by fkbp5 gene associated with depression and low cortisol response to stress. In addition, SNARE protein complexes apparently play an important role in pathological accumulation of aggregated proteins, for instance, α-synuclein in Parkinson’s disease [e.g., 285].
Another important protein is stathmin (aka oncoprotein 18) that regulates stability of the cellular cytoskeleton (i.e., destabilizes microtubules which have assembling-disassembling dynamics) and cycle (e.g., cell proliferation and accumulation) [286, 287, see also 288]. Stathmin also displays a cytoprotective function (e.g., during cellular/osmotic stress) [289], which is associated with cAMP-dependent protein kinase signaling [e.g., 290]. These are significant synaptic factors, and stathmin mutation/reductions relate to impaired memory, fear recognizing/processing/learning, and altered behavior [e.g., 291,292,293,294,295,296]. Altered expression in stathmin genes is associated with anxiety, poor learning, fear memories, and PTSD in animal models of stress [e.g., 297, 298; see also 299]. In one study, SPS reduced expression of stathmin in the hippocampus, medial PFC, and amygdala [294]. At the same time, stathmin alteration is implicated in neurodegeneration, such as Alzheimer’s disease [e.g., 297, 299, 300], while stress can escalate apoptosis [e.g., 301]. The pathways of apoptosis can include but are not limited to: (1) enzymes facilitating target proteins (e.g., receptors); (2) MAPK pathway; and (3) GRs [e.g., 302]. Additionally, recall that chaperones, which, when dysregulated by stress, can also lead to abnormal protein properties. As a result, accumulated protein aggregation into toxic non-native oligomers or fibrils/plaques (i.e., proteotoxicity) increases the risk for neurodegenerative disease. Conversely, behavioral stress-resilience (i.e., better coping) has been found to be associated with lower tau levels in older, amyloid positive adults without cognitive deficits [91].
Thus, increased levels of cortisol during acute stress can increase Ca2+/MK IIα expression /activation (Fig. 5), whereas chronic stress can decrease Ca2+/MK IIα expression/activation (Fig. 6). The stress response is regulated by 5-HT signaling via the ERK pathway associated with anxiety and PTSD due to 5-HT1 and 5-HT2A receptors’ competitive interaction [e.g., 178, 284]. This complex cellular dysfunction in enzymes and proteins can explain the molecular pathway intersection between depression and anxiety, as well as stress-induced neurodegeneration, especially when accounting for the fact that Ca2+/MK regulate apoptosis [e.g., 303], and similar to MAPK/ERK, are linked to tauopathies [e.g., 304,305,306].
Noradrenaline/norepinephrine
In acute stress, activation of the noradrenergic (LC-NE) system supports attention to the stressors [e.g., 1]. However, the noradrenergic signaling cascade can alter memory through activated cAMP-dependent protein kinase A via β-adrenoceptor activation, which is a fast response in contrast to a classical activation of GRs [e.g., 51, 206, 211, 307, 308]. Belonging to group A of the G protein-coupled receptor (GPCR) family, the β-adrenoceptor has a signature seven-transmembrane configuration [e.g., 309] in which cAMP/protein kinase A signalling pathways are shared with GR signalling [51, 205, 206, 310; see Figs. 1 and 4]. Activation of β-adrenoceptors with elevated levels of NE leads to phosphorylation of AMPA receptor’s subunit GluA1 at the Ser 818 site which promotes the AMPA receptor’s functions essential for long-term potentiation [e.g., 310]. Subsequent synaptic incorporation of Ca2+-permeable receptors (AMPA receptors containing GluA1, but missing GluA2) increases synaptic functionality [e.g., 220]. The distinctions are that amygdalar β-adrenoreceptors signalling activates AMPA receptors required for memory acquisition and working memory during threat, and then ERK/MAPK signalling facilitates fear memory consolidation through gene transcription and protein synthesis [e.g., 311; see also 312]. Remarkably, glucocorticoids influence deficit of working memory and enhancement of memory consolidation via a common activation of the noradrenergic signaling pathway within the medial PFC. An inverted U-shaped relationship between NE levels and working memory has been observed [e.g., 313].
Further, the LC neuronal activity regulates attention and low tonic activity relates to drowsiness and disengagement, whereas moderate-to-high tonic activity relates to arousal/hyperarousal [e.g., 314; see also 1]. In states of arousal without stress (i.e., an alert state without hypercortisolemia), released NE optimizes working memory via α2A-receptors. However, during a stress state, elevated NE levels impair PFC function and memory performance via α1- and β1-receptors [e.g., 315]. There is an inverted U-shaped dose–response relation that depends on the severity of the stressor and the condition that is observed in: (1) impaired memory for stressful life events with cortisol suppression, (2) timing-dependent learning improvement with moderate increase in cortisol concentration, and (3) impaired consolidation and recall in hypercortisolemia [e.g., 51, 211]. Therefore, pharmacotherapy (e.g., GRs and α1-/β-adrenoceptor antagonists, or protein kinase A inhibitors) administrated to block harmful effects of glucocorticoids on working memory can eliminate memory consolidation of emotionally significant experiences [e.g., 51, 315].
Dopamine
Stress-induced activation of the HPA axis increases dopamine neurotransmission in the PFC and activation of the downstream signalling cascades that negatively impact mood and cognition [e.g., 316; see also 317,318,319,320]. Indeed, it has been shown that emotional stimuli can influence working memory processing, manipulation, and maintenance that depend on the dorsolateral PFC, which is inversely coactive with the ventrolateral PFC [e.g., 321] and involve dopaminergic D1-receptors signalling. Like the NE effect, dopamine levels alter working memory in a U-shaped manner [e.g., 315, 322]. Furthermore, we consider polymorphism in genes associated with dopamine levels to be a prospective screening tool for stress resilience capacity based on the dopamine-related phenotypic variability of behavior and cognition patterns.
For example, the Val158Met polymorphism of catechol-O-methyltransferase (COMT, which is related to dopamine catabolism in the PFC and is regulated by estrogen) contributes to the complex Sex × Gene × Environment interactions affecting dopamine-dependent neurocognition and anxiety [e.g., 323, 324]. The Val158Met polymorphism has been found to influence decision-making following stressful life events [325]. As compared to the Val allele, the Met allele is linked to better working memory performance (including verbal, visuospatial, and novel social tasks) and poorer executive functioning, which is related to reduced dopamine levels in the PFC in children, adolescents, and adults [326, 327; see also 328]. COMT Val-allele load (COMT Val > Met), in turn, has been shown to be related to the dorsolateral PFC activity during working performance, such as encoding of new information, as well as temporal updating operations, but not in its subsequent retrieval. The distinctions are that high working memory load activates the PFC-parietal-striatal network, where activity in the right dorsolateral PFC is lesser in Val homozygotes than in ValMet individuals, but intermediate in Met homozygotes [329]. Relatively better performance in working memory is associated with decreased coactivity between the dorsolateral PFC and ACC for Val/Val genotype, but increased coactivity between the dorsolateral PFC-amygdala/hippocampus for Met/Met genotype. This association correlates to the regional cerebral blood flow in the amygdala and hippocampus for Val/Val, the parietal lobe for Val/Met, and the thalamus for Met/Met [330].
Resaerchers have also found that COMT is associated with emotional dysregulation and that dopamine receptor 2 genes can be a promising target of antipsychotic medications [e.g., 331]. Additionally, for monoamine oxidase A, H allele carriers have greater stress effects on the right anterior hippocampus hypoactivity and cortisol hyperresponse as compared to the L allele carriers [332]. At the same time, the monoamine oxidase A polymorphism predicts aggressive and oppositional behavior, with better working memory capacity related to fewer externalizing symptoms in children and adolescents [333]. Polymorphism in genes associated with dopamine levels can also serve as genetic biomarkers for higher risk of developing Alzheimer’s disease [e.g., 334,335,336].
Sex hormone co-signaling
Summary of the concepts: Estrogen and androgen signalling influence memory and behavior, which is linked to stress resilience.
Takeaway: Estrogen effects are complex and play a dual, neuro-protective and neuro-harming role, as hormone levels fluctuate. Moreover, the dual effects are exhibited by androgen.
Estrogen signalling
Sex-differences in stress response and reactivity [e.g., 337,338,339] can be explained by the fact that estrogen influences neurocognition, such as working memory, episodic memory, social memory, spatial memory, selective attention, and memory system bias [e.g., 115, 340,341,342,343,344,345]. Estrogen-facilitated hippocampal activity has been shown to exhibit a linear and inverted U-shaped dose–response effect in young women [e.g., 345]. Post-menopausal reduction of estradiol can be a risk factor for cognitive decline [e.g., 346]. However, estrogen plays a dual role and is either neuro-protective or neuro-harming in the stress response [339, 347; see also 348, 349].
We postulate that the estrogen effect is a function of the estrogen signalling type (nuclear or non-nuclear) × environment.
Nuclear signalling and acute stress
Like GRs, activated estrogen receptors (ERs) can affect memory and behavior through regulation of gene transcription. When activated by binding estradiol, ERs can translocate to a nucleus and connect to estrogen response elements on DNA. This is a nuclear type of ERs activation with the direct genomic effects [e.g., 115, 342]. In other words, ERs dirrectly regulate transcription of the genes with cAMP response elements (CRE) in their promoters (e.g., bdnf and CRF genes) and, in turn, activate or attenuate CREB function that enhances or impairs, respectively, memory formation [e.g., 350; Figs. 1 and 4].
Although elevated estradiol levels can promote rapid hippocampal CREB phosphorylation, during acute stress, the outcomes can be harmful [e.g., 351]. For example, the effect can be seen in the amplified stress response possibly due to the co-activation between ERs, GRs and CRH-1 receptor [e.g., 352], impaired fear extinction [e.g., 353], and PFC-mediated working memory [354]. We predict that estrogen-related cognitive vulnerability is associated with an altered Ca2+/MK IIα—CREB pathway. In fact, estrogen can rapidly hyperactivate Ca2+/MK IIα via nongenomic pathways in the hippocampus [e.g., 254, see also 255].
Non-nuclear signalling and chronic stress
Circulating estradiol can also bind to membrane-specific estrogen receptors (mERα and nERβ) and G protein-coupled ERs (GPERs). Activated mERs can trigger rapid (within seconds to 5 min) nongenomic mechanisms in the dorsal hippocampus that involve metabotropic glutamate receptor GluA1 (aka GluR1) interactions. Next, indirect genomic effect can be seen within hours or days due to subsequently modulated CREB phosphorylation and CRE-dependent gene expression via MAPK [e.g., 355,356,357,358,359]. Furthermore, ER signalling can influence NE signalling [e.g., 360; see also 361]. The mnemonic effect of ERs correlates with D1-receptor dopamine activity [e.g., 234] and depends on a photoperiod (melatonin effect) that mediates estradiol-induced aggression [362, 363; see also 339; Photoperiodic Plasticity section below].
Estrogen has been found to support cognitive resilience to chronic or repeated stress in females [e.g., 364,365,366,367]. For instance, in contrast to male rats, chronic restraint stress (2-h/day for a week) did not impair PFC functions in female rats due to ER-related protective effect seen in the preserved temporal order recognition memory and AMPA/NMDA receptors subunits surface expression (i.e., GluR1/2 and NR1/2A/2B) [367].
The mechanistic parallelism is observed in neurological diseases. Specifically, in a mouse model of Alzheimer's disease, GPER30 activation was found to have an ameliorative effect on object recognition memory [368] and selective activation of non-nuclear ERs normalized the mitochondrial apoptotic pathway via pathway-preferential estrogen (PaPE)-1 involved in MAPK and mTOR [e.g., 369]. In a mouse model of stroke, selective non-nuclear ERs stimulation with PaPE-1 also decreased stroke severity and neuroinflammation in the brain in female mice [370]. Furthermore, ERs signalling is implicated in microvascular mechanisms which serve both cerebro- and cardio-protection via mERα-mediated endothelial effects (i.e., rapid dilatation and repair acceleration) and mERβ- mediated synthesis of nitric oxide that play a hypotensive role [371,372,373,374,375,376; see also 358, 367, 377].
Hence, despite the neuro- and cardio-protective properties of estradiol, evidence indicates that high estrogen levels increase cognitive sensitivity to stress and affect disease risk. We thus hypothesize that in the context of an extreme, acute stress response, nuclear ERs act as transcription factors that enable stress effects related to activated Ca2+/MK IIα (i.e., higher estrogen levels are harmful). This may partly explain why the prevalence of PTSD is twice as high in women than men [e.g., 378,379,380]. We also hypothesize that in chronic stress, non-nuclear ERs regulate the translational status, via histone modifications, to favour neuroprotective effects via PaPE-1 activation (i.e., higher estrogen levels are beneficial). Accordingly, estradiol treatment of women that were exposed to interpersonal violence has been shown to attenuate negative effect of severe PTSD symptoms on fear habituation [e.g., 381]. Moreover, mERs are considered a novel treatment target for age-associated memory decline, stroke, and neurodegenerative diseases [e.g., 382, 383].
Testosterone
Similar to estrogen, androgen displays dual effects. Specifically, testosterone can enable (1) hippocampal synapse formation and spatial memory or (2) inhibition of BDNF and reduction in astroglial density that affects synaptic plasticity in men. In contrast to estrogen, androgen is not associated with social memory but social recognition if a male conspecific [e.g., 384, for additional mnemonic effects, see also 385,386,387,388,389,390,391]. The animal models of stress conditions, such as conditioned fear and inhibitory avoidance tasks, show that androgen’s metabolite 3α-diol can bind to mERβ and enhance hippocampal memory [392, 393]. Androgen is also implicated in observed sex-differences in stress reactivity related to impulsive behavior in rats, with males preferring large and delayed rewards and females preferring small and immediate rewards [394]. Clinically, this androgen effect may help explain why women have been found to be more affected by the frequency of the exposure to stress, whereas men appear to be more affected by the severity of stress [395; see also 339, 396].
Photoperiodic plasticity
Summary of the concepts: Altered circadian rhythm with shortened daylight can increase neurocognitive vulnerability to stress.
Takeaway: Pharmacotherapy aimed at melatonin and serotonin signalling has the potential to attenuate cognitive deterioration associated with altered photoperiodic plasticity.
Changes in photoperiod can modulate stress-induced cognitive and affective disorders, such as those associated with seasonal differences. For example, greater stress vulnerability was observed in a rat model during exposure to short days [e.g., 397]. The underlying mechanism appears to be that shortened photoperiod can alter hippocampal volume and dendritic morphology related to the synaptic plasticity (i.e., decreased apical CA1 spine density and increased basilar CA3 spine density), resulting in poor spatial learning and memory [e.g., 398,399,400]. This mechanism involves activation of the melanocortin-4 receptors that promote aversive memory formation via protein kinase A signalling [e.g., 220]. The melanocortin-4 receptors are highly expressed in the medial amygdala [401]. Moreover, their activation increases synaptic plasticity via the dendritic spine morphology and abundance of the AMPA- receptors [e.g., 220]. The melanocortin-4 receptors activation is linked to rapid anxiogenic and anorectic effects in response to emotional stress [e.g., 401]. Stress-induced anxiety and depression can be aggravated by the melanocortin-4 receptors’ agonists and mitigated by the receptors’ antagonists [e.g., 402]. At the same time, the evidence suggests that reduced melatonin signalling is implicated in PTSD, psychiatric disorders, and neurodegenerative diseases [e.g., 397, 403,404,405]. Specifically, in PTSD, melatonin treatment can mitigate PTSD-like behaviors (related to contextual fear memory) and restore cortisol levels [e.g., 403], as well as improve spatial cognitive impairment via genomic mechanisms that increase CREB protein and mRNA levels in the hippocampus [397]. Melatonin supplementation can also attenuate Alzheimer’s-like tau hyperphosphorylation and β-amyloid aggregation, slow down cognitive deterioration, and improve sleep and sundowning in Alzheimer’s disease [e.g., 405,406,407,408,409,410]. Furthermore, researchers have also demonstrated that melatonin effects relate to serotonin and dopamine co-signalling. For example, antidepressant treatment with agomelatine that displays a synergistic effect on melatonergic MT1/MT2 and serotonergic 5-HT2C receptors (agonist and antagonist, respectively) reduces stress-induced glutamate release in the PFC [e.g., 411, 412] and likely reduces depressive symptoms via circadian rhythms restoration following resynchronized sleep–wake cycle [e.g., 413, 414]. At the same time, circadian rhythmicity is bidirectionally interconnected to the stress system [e.g., 415] and relates to dopamine signalling [e.g., 416].
Psychoneuroimmunologic effects
Summary of the concepts: Psychological stress can trigger neuroimmune and proinflammatory responses that influence mood and cognitive decline. In addition, a compromised immune system increases brain vulnerability to stress and neuropsychiatric conditions.
Takeaway: Stress coping and neuroinflammation are related. Indeed, psychological interventions can improve stress resilience via immunologic/anti-inflammatory mechanisms.
In reviewing stress-induced neuronal pathophysiology, it is important to highlight that brain networks communicate with the immune system to monitor and respond to many kinds of threats, including social, physical, and psychological stressors. Psychological stressors can trigger anticipatory neuroimmune responses that reduce risk for injury and infection by upregulating inflammatory activity in the mere presence of social threat [e.g., 417,418,419]. Therefore, even a “painful” feeling such as shame, but not guilt—which is an understanding that our actions have harmed somebody else—can increase proinflammatory cytokine activity while not affecting cortisol levels in healthy adults [e.g., 420]. Consistent with this model, PTSD has been shown to be associated with immune dysregulation [e.g., 421, 422]. Vice versa, a compromised immune system increases brain vulnerability to stress. For example, after controlling for the effects of age, education, and depression/anxiety, in men with HIV, it has been shown that stressful life events were related to cognitive deficit, in contrast to men who were HIV-negative with the same level of stress perception [423; see also 424]. Stress-associated verbal memory deficits have been found to be larger for HIV-positive as compared to HIV-negative women [425].
Rat models of stress provide further insights into how stress coping and neuroinflammation interconnect. For example, exposure to brief social defeat induces circulating inflammatory cytokines and primes neuroinflammatory responses (with the engagement of the LC-NE system) to a subsequent defeat exposure. Moreover, social defeat enhances neuroinflammation in the central amygdala but reduces it in the dorsal raphe [426]. In a repeated resident-intruder stress model in rats, coping strategy affects psychosocial stress susceptibility associated with neuroinflammation measured by interleukin-1β: whereas passive coping was linked to greater inflammation, active coping and stress resistance was linked to lesser inflammation [427, see also 428, 429].
Furthermore, the LC activity enables scanning attention and the analysis of behavior while actions are “on hold so the challenge can first be inspected” [e.g., 1]. It has also been shown that escape responses [56] and subordinate behavior are promoted by CRF influence yet supressed by opioid influence [428]. Hence, we may say that whereas stress-associated hyperarousal enables passive coping and neuroinflammation, emotional downregulation can facilitate active coping and reduce proinflammatory responses. In fact, research has shown that emotion regulation is associated with inflammatory activity levels [e.g., 430], and adequate coping strategies that alter cognitive appraisals and emotional responses can improve health outcomes [e.g., 431]. Given that neuroinflammation is implicated in neurocognitive/psychiatric disorders and neurodegenerative diseases [e.g., 429, 432,433,434,435], this research highlights the potential for psychological interventions such as cognitive and dialectical behavior therapy to improve vulnerable brain function. Indeed, a meta-analysis showed that psychosocial interventions such as cognitive behavior therapy reliably improve immune system function for at least 6 months following treatment [436].
The active role in neuroinflammatory regulation belongs to the cerebral glia—that is, microglia and astrocytes—as well as the endotheliocytes and peripheral immunocytes. Here, “the primary immune surveillance and macrophage-like activities” are performed by microglia [e.g., 437; see also 438], where neuroinflammatory processes are mediated via the LC-NE signaling system [e.g., 439]. Additionally, stress-induced changes exhibit sex-differences in the corticolimbic microglia. Specifically, dendritic re-modeling is seen mostly in the basolateral amygdala in females, as compared to wider microglial cell activation in males, which involve the basolateral amygdala, dorsal hippocampus, medial PFC, and corticolimbic projections [440].
Evidence also suggests that chronic social stress induces genome-wide transcriptional shifts characterized by increased proinflammatory and reduced anti-viral skewing via β-adrenergic signaling [441, 442]; this resonates with the transcriptional changes evident in PTSD [443]. Particularly, this shift involves the upregulation of target genes for a proinflammatory immune response (e.g., NF-κB and AP-1) and a reciprocal downregulation of target genes for an anti-inflammatory response (e.g., GRs coding gene NR3C1 and interferon response factors). This proinflammatory shift explains why chronic social stressor exposure is related to high levels of morbidity and mortality [441].
At the same time, single nucleotide polymorphisms in the promoters of proinflammatory immune response genes, such as IL6, influence DNA binding affinity that in turn affects the extent to which social threat-activated GATA1 transcription factor activity predicts longevity. In this context, individuals with GATA1-sensitive G allele have been found to have higher levels of DNA binding affinity and IL6 gene expression—which is associated with earlier mortality—as compared to their counterparts with the GATA1-insensitive C allele [441, 444]. To complicate the matter, altered immune functioning/inflammatory responses (e.g., levels of IL-6, TNF-α, and C-reactive protein), as well as the polymorphisms in IL6, IL1β, IL10, and TNFIα, appear to contribute to a potential increased susceptibility to depression, cognitive impairment, and dementia/Alzheimer’s disease [e.g., 445,446,447,448,449,450,451,452]. Accordingly, modulating immunity appears to be a therapeutical target to support cognitive function in stress-related psychiatric conditions [e.g., 453].
Conclusion and application
The Global Burden of Disease Study 2019 revealed that “mental disorders remained among the top ten leading causes of burden worldwide, with no evidence of global reduction in the burden since 1990” [454, p. 137]. Moreover, the authors wrote that “Mental health needs are high but responses are insufficient and inadequate. […] About one in eight people in the world live with a mental disorder” [455, p. iv]; “there remains much to be done to ensure all people achieve the highest standard of mental health and well-being. Action must be taken” [454, p. v]. Such conclusions expose a critical gap between the sizable magnitude of this public health crisis and our lackluster global effort to identify novel therapies that can successfully address the problem. To begin to address this issue, we reviewed research aimed at better understanding how stress affects the brain in a way that hampers integral aspects of mental health, including psychosocial well-being, cognitive functioning, and mental resilience.
This research shows that psychological stress increases the risk for cognitive problems, and the development of serious neurological and psychiatric disorders [e.g., 3, 5,6,7,8, 456]. The exact mechanisms of stress-related cognitive deterioration and neuropsychiatric outcomes have yet to be clarified. To provide a fruitful path forward, we applied a multilevel approach that discerns shared cellular mechanisms underlying stress reactivity and neurocognitive processing (Fig. 7). In doing so, we elucidated the multilevel determinants of stress resilience, which may support clinical interventions. However, to realize the full potential of this work for addressing stress-related disease burden, these models will need to be translated into novel therapeutics that are safe, effective, widely accessible, and affordable.

Determinants of Cognitive Resilience to Psychological Stress. Simplified and schematic model of biological processes linking psychological stress and cognition. Psychological stress can induce multiple neurobiological mechanisms related to cognitive decline and behavioral change. Principal determinants of stress resilience, which may support clinical interventions, are shown: (1) Psychobiology; (2) Epigenetics; (3) Neurotransmitters; (4) Sex hormones; (5) Circadian rhythm; and (6) Psychoneuroimmunology (see the Conclusion and Application section)
Level 1: Psychobiology
Stress reactivity is a function of a person’s cognitive appraisal and allostatic status; moreover, stress-induced habituation and sensitization are associated with harmful effects. Access to psychosocial education and services in community-based settings can enhance individual stress resilience. Psychotherapeutic interventions, such as cognitive behavior therapy, can help people cognitively reappraise their ability to deal with a stressor and, in turn, reduce their perceived stress severity and health risks [e.g., 1].
Level 2: Epigenetics
The genetic polymorphism × sex × environment (e.g., level of social safety) interplay is associated with interindividual differences in stress-responses outcomes, such as morbidity risk and longevity [e.g., 441]. Proactive screening and proper support are necessary for individuals at particular risk of mental illness (e.g., genetic testing for physiological and behavioral traits, supporting marginalized groups, [e.g., 457]).
Level 3: Neurotransmitters
Stress can affect multiple neurotransmitters related to neuronal plasticity and stress resilience. We hypothesize that the stress effects can be determined by the enzyme-protein dynamics. Specifically, anxiety and PTSD following acute and/or severe stress can be driven by altered Ca2+/MK IIα pathway, which is a fast nongenomic response with indirect epigenetic effect involving CREB mechanism (Fig. 5). We also hypothesize that depression and neurodegeneration following chronic stress can be driven by SNARE protein complex accumulation in synaptic membranes linked to excito-/proteo-toxicity and slow genomic effect involving ERK/MAPK (Fig. 6). Pharmacotherapy based on neurotransmitter signaling and administrated to block harmful effects of glucocorticoids can help stress resilience and neurocognitive functioning.
Level 4: Sex hormones
Estrogen and androgen signalling influence memory and behavior, which is linked to stress resilience. We hypothesize that dual, neuro-protective or neuro-harming, role of estrogen effect is a function of the estrogen signalling type (nuclear or non-nuclear) × environment (exposure to acute or chronic stress). Advanced mental health care requires promoted sex/gender-specific medicine.
Level 5: Circadian rhythm
Altered circadian rhythm with shortened daylight can increase neurocognitive vulnerability to stress. Addressing circadian dysrhythmia and associated alterations in melatonin and serotonin signalling can support brain functions.
Level 6: Psychoneuroimmunology
There is a bidirectional association between stress coping and immunity. Interventions aimed at enhancing immunity and emotional intelligence may have a protective impact on neurocognitive resilience to stress.
Additional research is needed to test the hypotheses described. To this end, the present model provides a fruitful avenue for investigating aspects of cellular neuropathophysiology that can help advance our understanding of features of the stress responses that contribute to, and interplay with, psychiatric and neurological disorders. The outlined determinants of stress resilience can thus assist cross-disciplinary clinical and translational neuroscience to promote brain and mind health.
Availability of data and materials
Not applicable.
Change history
19 December 2023
The publisher sincerely apologize to the authors and readers for the inconvenience caused by the errors as far as two reference citations that were missing.
Abbreviations
- 5HTTLPR:
-
Serotonin transporter-linked promoter region
- ACTH:
-
Adrenocorticotropic hormone
- BDNF:
-
Brain-derived neurotrophic factor
- Ca2+/MK:
-
Ca2+/Calmodulin-dependent kinases
- COMT:
-
Catechol-O-methyltransferase
- CRE:
-
CAMP response elements (CRE)
- CREB:
-
CAMP-response element-binding protein
- CRF:
-
Corticotropin-releasing factor
- CRH:
-
Corticotropin-releasing hormone
- ER:
-
Estrogen receptor
- ERK:
-
Extracellular regulated kinase
- GR:
-
Glucocorticoid receptor
- HPA:
-
Hypothalamic–pituitary–adrenal
- Hsp:
-
Heat shock proteins
- LC-NE:
-
Locus coeruleus-norepinephrine
- MAPK:
-
Mitogen-activated protein kinase
- MR:
-
Mineralocorticoid receptor
- PFC:
-
Prefrontal cortex
- PTSD:
-
Post-traumatic stress disorder
- SNARE:
-
Soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- SPS:
-
Single prolonged stress
- TSST:
-
Trier Social Stress Test
References
Palamarchuk IS, Vaillancourt T. Mental resilience and coping with stress: a comprehensive, multi-level model of cognitive processing, decision making, and behavior. Front Behav Neurosci. 2021;15: 719674. https://doi.org/10.3389/fnbeh.2021.719674.
Palamarchuk IS, Vaillancourt T. Integrative brain dynamics in childhood bullying victimization: cognitive and emotional convergence associated with stress psychopathology. Front Integr Neurosci. 2022;16: 782154. https://doi.org/10.3389/fnint.2022.782154.
Goh J, Pfeffer J, Zenios SA. The Relationship between workplace stressors and mortality and health costs in the United States. Manage Sci. 2016;62(2):608–28. https://doi.org/10.1287/mnsc.2014.2115.
Goh J, Pfeffer J, Zenios SA. Reducing the health toll from U.S. workplace stress. Behav Sci Policy. 2019;5(1):1–13.
American Psychological Association. Stress in America™ 2020: a national mental health crisis; 2020. https://www.apa.org/news/press/releases/stress/2020/report-october
World Health Organization. Comprehensive Mental Health Action Plan 2013–2030; 2021. https://www.who.int/publications/i/item/9789240031029
Ahmad AH, Zakaria R. Pain in Times of Stress. Malaysian J Med Sci MJMS. 2015;22(Spec Issue):52–61.
Bodnar RJ. Endogenous opiates and behavior: 2014. Peptides. 2016;75:18–70. https://doi.org/10.1016/j.peptides.2015.10.009.
Ross JA, Gliebus G, Van Bockstaele EJ. Stress induced neural reorganization: a conceptual framework linking depression and Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 2018;85:136–51. https://doi.org/10.1016/j.pnpbp.2017.08.004.
Sanacora G, Yan Z, Popoli M. The stressed synapse 20: pathophysiological mechanisms in stress-related neuropsychiatric disorders. Nat Rev Neurosci. 2022;23(2):86–103. https://doi.org/10.1038/s41583-021-00540-x.
Franks KH, Bransby L, Saling MM, Pase MP. Association of stress with risk of dementia and mild cognitive impairment: a systematic review and meta-analysis. J Alzheimer’s Dis JAD. 2021;82(4):1573–90. https://doi.org/10.3233/JAD-210094.
Henderson KM, Clark CJ, Lewis TT, Aggarwal NT, Beck T, Guo H, Lunos S, Brearley A, Mendes de Leon CF, Evans DA, Everson-Rose SA. Psychosocial distress and stroke risk in older adults. Stroke. 2013;44(2):367–72. https://doi.org/10.1161/STROKEAHA.112.679159.
Kotlęga D, Gołąb-Janowska M, Masztalewicz M, Ciećwież S, Nowacki P. The emotional stress and risk of ischemic stroke. Neurol Neurochir Pol. 2016;50(4):265–70. https://doi.org/10.1016/j.pjnns.2016.03.006.
Stansfeld S, Candy B. Psychosocial work environment and mental health–a meta-analytic review. Scand J Work Environ Health. 2006;32(6):443–62. https://doi.org/10.5271/sjweh.1050.
Hachinski V, Einhäupl K, Ganten D, Alladi S, Brayne C, Stephan BCM, Sweeney MD, Zlokovic B, Iturria-Medina Y, Iadecola C, Nishimura N, Schaffer CB, Whitehead SN, Black SE, Østergaard L, Wardlaw J, Greenberg S, Friberg L, Norrving B, Rowe B, et al. Preventing dementia by preventing stroke: The Berlin Manifesto. Alzheimer’s Dementia. 2019;15(7):961–84. https://doi.org/10.1016/j.jalz.2019.06.001.
Pendlebury ST, Rothwell PM, Oxford Vascular Study. Incidence and prevalence of dementia associated with transient ischaemic attack and stroke: analysis of the population-based Oxford Vascular Study. Lancet Neurol. 2019;18(3):248–58. https://doi.org/10.1016/S1474-4422(18)30442-3.
Bature F, Guinn BA, Pang D, Pappas Y. Signs and symptoms preceding the diagnosis of Alzheimer’s disease: a systematic scoping review of literature from 1937 to 2016. BMJ Open. 2017;7(8): e015746. https://doi.org/10.1136/bmjopen-2016-015746.
Botto R, Callai N, Cermelli A, Causarano L, Rainero I. Anxiety and depression in Alzheimer’s disease: a systematic review of pathogenetic mechanisms and relation to cognitive decline. Neurol Sci. 2022;43(7):4107–24. https://doi.org/10.1007/s10072-022-06068-x.
Wingo TS, Liu Y, Gerasimov ES, Vattathil SM, Wynne ME, Liu J, Lori A, Faundez V, Bennett DA, Seyfried NT, Levey AI, Wingo AP. Shared mechanisms across the major psychiatric and neurodegenerative diseases. Nat Commun. 2022;13(1):4314. https://doi.org/10.1038/s41467-022-31873-5.
McEwen BS. Stress, adaptation, and disease. allostasis and allostatic load. Ann N Y Acad Sci. 1998;840:33–44. https://doi.org/10.1111/j.1749-6632.1998.tb09546.x.
McEwen BS. Brain on stress: How the social environment gets under the skin. Proc Natl Acad Sci USA. 2012;109(Suppl 2):17180–5. https://doi.org/10.1073/pnas.1121254109.
McEwen BS, Gianaros PJ. Stress- and allostasis-induced brain plasticity. Annu Rev Med. 2011;62:431–45. https://doi.org/10.1146/annurev-med-052209-100430.
Godoy LD, Rossignoli MT, Delfino-Pereira P, Garcia-Cairasco N, de Lima Umeoka EH. A comprehensive overview on stress neurobiology: basic concepts and clinical implications. Front Behav Neurosci. 2018;12:127. https://doi.org/10.3389/fnbeh.2018.00127.
Gray JD, Kogan JF, Marrocco J, McEwen BS. Genomic and epigenomic mechanisms of glucocorticoids in the brain. Nat Rev Endocrinol. 2017;13(11):661–73. https://doi.org/10.1038/nrendo.2017.97.
Masten AS, Cicchetti D. Developmental cascades. Dev Psychopathol. 2010;22(3):491–5. https://doi.org/10.1017/S0954579410000222.
Poe GR, Foote S, Eschenko O, Johansen JP, Bouret S, Aston-Jones G, Harley CW, Manahan-Vaughan D, Weinshenker D, Valentino R, Berridge C, Chandler DJ, Waterhouse B, Sara SJ. Locus coeruleus: a new look at the blue spot. Nat Rev Neurosci. 2020;21(11):644–59. https://doi.org/10.1038/s41583-020-0360-9.
Charmandari E, Tsigos C, Chrousos G. Endocrinology of the stress response. Annu Rev Physiol. 2005;67:259–84. https://doi.org/10.1146/annurev.physiol.67.040403.120816.
De Kloet ER, Vreugdenhil E, Oitzl MS, Joëls M. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19(3):269–301. https://doi.org/10.1210/edrv.19.3.0331.
Koob GF, Heinrichs SC. A role for corticotropin releasing factor and urocortin in behavioral responses to stressors. Brain Res. 1999;848(1–2):141–52. https://doi.org/10.1016/s0006-8993(99)01991-5.
Miller GE, Chen E, Zhou ES. If it goes up, must it come down? Chronic stress and the hypothalamic-pituitary-adrenocortical axis in humans. Psychol Bull. 2007;133(1):25–45. https://doi.org/10.1037/0033-2909.133.1.25.
Kvetnansky R, Sabban EL, Palkovits M. Catecholaminergic systems in stress: structural and molecular genetic approaches. Physiol Rev. 2009;89(2):535–606. https://doi.org/10.1152/physrev.00042.2006.
Bizik G, Picard M, Nijjar R, Tourjman V, McEwen BS, Lupien SJ, Juster RP. Allostatic load as a tool for monitoring physiological dysregulations and comorbidities in patients with severe mental illnesses. Harv Rev Psychiatry. 2013;21(6):296–313. https://doi.org/10.1097/HRP.0000000000000012.
Guidi J, Lucente M, Sonino N, Fava GA. Allostatic load and its impact on health: a systematic review. Psychother Psychosom. 2021;90(1):11–27. https://doi.org/10.1159/000510696.
Herman JP, McKlveen JM, Ghosal S, Kopp B, Wulsin A, Makinson R, Scheimann J, Myers B. Regulation of the hypothalamic-pituitary-adrenocortical stress response. Compr Physiol. 2016;6(2):603–21. https://doi.org/10.1002/cphy.c150015.
Juruena MF, Cleare AJ, Bauer ME, Pariante CM. Molecular mechanisms of glucocorticoid receptor sensitivity and relevance to affective disorders. Acta Neuropsychiatrica. 2003;15(6):354–67. https://doi.org/10.1046/j.1601-5215.2003.00051.x.
Ludäscher P, Bohus M, Lieb K, Philipsen A, Jochims A, Schmahl C. Elevated pain thresholds correlate with dissociation and aversive arousal in patients with borderline personality disorder. Psychiatry Res. 2007;149(1–3):291–6. https://doi.org/10.1016/j.psychres.2005.04.009.
Graumann L, Heekerens JB, Duesenberg M, Metz S, Spitzer C, Otte C, Roepke S, Wingenfeld K. Association between baseline dissociation levels and stress-induced state dissociation in patients with posttraumatic-stress disorder, borderline personality disorder, and major depressive disorder. Borderline Personality Disorder Emotion Dysregulation. 2023;10(1):11. https://doi.org/10.1186/s40479-023-00215-2.
Sommer JL, Blaney C, Mota N, Bilevicius E, Beatie B, Kilborn K, Chang U, Sareen J, El-Gabalawy R. Dissociation as a transdiagnostic indicator of self-injurious behavior and suicide attempts: a focus on posttraumatic stress disorder and borderline personality disorder. J Trauma Stress. 2021;34(6):1149–58. https://doi.org/10.1002/jts.22726.
Liew BXW, Valera-Calero JA, Varol U, Nijs J, Arendt-Nielsen L, Plaza-Manzano G, Fernández-de-Las-Peñas C. Distress and sensitization as main mediators of severity in women with fibromyalgia: a structural equation model. Biomedicines. 2022;10(5):1188. https://doi.org/10.3390/biomedicines10051188.
Cummins TM, English O, Minnis H, Stahl D, O’Connor RC, Bannister K, McMahon SB, Ougrin D. Assessment of somatosensory function and self-harm in adolescents. JAMA Netw Open. 2021;4(7): e2116853. https://doi.org/10.1001/jamanetworkopen.2021.16853.
Bannister K, Smith RV, Wilkins P, Cummins TM. Towards optimising experimental quantification of persistent pain in Parkinson’s disease using psychophysical testing. NPJ Parkinson’s Dis. 2021;7(1):28. https://doi.org/10.1038/s41531-021-00173-y.
Aguilera G. Regulation of pituitary ACTH secretion during chronic stress. Front Neuroendocrinol. 1994;15(4):321–50. https://doi.org/10.1006/frne.1994.1013.
Aguilera G. Corticotropin releasing hormone, receptor regulation and the stress response. Trends Endocrinol Metab. 1998;9(8):329–36. https://doi.org/10.1016/s1043-2760(98)00079-4.
Aguilera G, Liu Y. The molecular physiology of CRH neurons. Front Neuroendocrinol. 2012;33(1):67–84. https://doi.org/10.1016/j.yfrne.2011.08.002.
García A, Martí O, Vallès A, Dal-Zotto S, Armario A. Recovery of the hypothalamic-pituitary-adrenal response to stress. Effect of stress intensity, stress duration and previous stress exposure. Neuroendocrinology. 2000;72(2):114–25. https://doi.org/10.1159/000054578.
Ulrich-Lai YM, Figueiredo HF, Ostrander MM, Choi DC, Engeland WC, Herman JP. Chronic stress induces adrenal hyperplasia and hypertrophy in a subregion-specific manner. Am J Physiol Endocrinol Metab. 2006;291(5):E965–73. https://doi.org/10.1152/ajpendo.00070.2006.
Belda X, Fuentes S, Daviu N, Nadal R, Armario A. Stress-induced sensitization: the hypothalamic-pituitary-adrenal axis and beyond. Stress (Amsterdam, Netherlands). 2015;18(3):269–79. https://doi.org/10.3109/10253890.2015.1067678.
Juruena MF, Gadelrab R, Cleare AJ, Young AH. Epigenetics: a missing link between early life stress and depression. Progr Neuro-Psychopharmacol Biol Psychiatry. 2020;109: 110231. https://doi.org/10.1016/j.pnpbp.2020.110231.
Nandam LS, Brazel M, Zhou M, Jhaveri DJ. Cortisol and major depressive disorder-translating findings from humans to animal models and back. Front Psych. 2020;10:974. https://doi.org/10.3389/fpsyt.2019.00974.
McCarty R. Learning about stress: neural, endocrine and behavioral adaptations. Stress. 2016;19(5):449–75. https://doi.org/10.1080/10253890.2016.1192120.
Barsegyan A, Mackenzie SM, Kurose BD, McGaugh JL, Roozendaal B. Glucocorticoids in the prefrontal cortex enhance memory consolidation and impair working memory by a common neural mechanism. Proc Natl Acad Sci USA. 2010;107(38):16655–60. https://doi.org/10.1073/pnas.1011975107.
Hauger RL, Risbrough V, Oakley RH, Olivares-Reyes JA, Dautzenberg FM. Role of CRF receptor signaling in stress vulnerability, anxiety, and depression. Ann N Y Acad Sci. 2009;1179:120–43. https://doi.org/10.1111/j.1749-6632.2009.05011.x.
Kovács KJ. CRH: the link between hormonal-, metabolic- and behavioral responses to stress. J Chem Neuroanat. 2013;54:25–33. https://doi.org/10.1016/j.jchemneu.2013.05.003.
Sandi C, Cordero MI, Ugolini A, Varea E, Caberlotto L, Large CH. Chronic stress-induced alterations in amygdala responsiveness and behavior–modulation by trait anxiety and corticotropin-releasing factor systems. Eur J Neurosci. 2008;28(9):1836–48. https://doi.org/10.1111/j.1460-9568.2008.06451.x.
Shekhar A, Truitt W, Rainnie D, Sajdyk T. Role of stress, corticotrophin releasing factor (CRF) and amygdala plasticity in chronic anxiety. Stress. 2005;8(4):209–19. https://doi.org/10.1080/10253890500504557.
Chudoba R, Dabrowska J. Distinct populations of corticotropin-releasing factor (CRF) neurons mediate divergent yet complementary defensive behaviors in response to a threat. Neuropharmacology. 2023;228: 109461. https://doi.org/10.1016/j.neuropharm.2023.109461.
Vandael D, Wierda K, Vints K, Baatsen P, De Groef L, Moons L, Rybakin V, Gounko NV. Corticotropin-releasing factor induces functional and structural synaptic remodelling in acute stress. Transl Psychiatry. 2021;11(1):378. https://doi.org/10.1038/s41398-021-01497-2.
Lago T, Davis A, Grillon C, Ernst M. Striatum on the anxiety map: small detours into adolescence. Brain Res. 2017;1654(Pt B):177–84. https://doi.org/10.1016/j.brainres.2016.06.006.
Packard MG. Anxiety, cognition, and habit: a multiple memory systems perspective. Brain Res. 2009;1293:121–8. https://doi.org/10.1016/j.brainres.2009.03.029.
Vogel S, Schwabe L. Learning and memory under stress: implications for the classroom. NPJ Sci Learn. 2016;1:16011. https://doi.org/10.1038/npjscilearn.2016.11.
Vogel S, Klumpers F, Schröder TN, Oplaat KT, Krugers HJ, Oitzl MS, Joëls M, Doeller CF, Fernández G. Stress induces a shift towards striatum-dependent stimulus-response learning via the mineralocorticoid receptor. Neuropsychopharmacology. 2017;42(6):1262–71. https://doi.org/10.1038/npp.2016.262.
Zerbes G, Kausche FM, Schwabe L. Stress-induced cortisol modulates the control of memory retrieval towards the dorsal striatum. Eur J Neurosci. 2020. https://doi.org/10.1111/ejn.14942.
Campioni MR, Xu M, McGehee DS. Stress-induced changes in nucleus accumbens glutamate synaptic plasticity. J Neurophysiol. 2009;101(6):3192–8. https://doi.org/10.1152/jn.91111.2008.
Reyes-López J, Nuñez-Jaramillo L, Morán-Guel E, Miranda MI. Differential effects of β-adrenergic receptor blockade in the medial prefrontal cortex during aversive and incidental taste memory formation. Neuroscience. 2010;169(1):195–202. https://doi.org/10.1016/j.neuroscience.2010.04.054.
Wichmann R, Fornari RV, Roozendaal B. Glucocorticoids interact with the noradrenergic arousal system in the nucleus accumbens shell to enhance memory consolidation of both appetitive and aversive taste learning. Neurobiol Learn Mem. 2012;98(2):197–205. https://doi.org/10.1016/j.nlm.2012.06.004.
Hauger RL, Risbrough V, Brauns O, Dautzenberg FM. Corticotropin releasing factor (CRF) receptor signaling in the central nervous system: new molecular targets. CNS Neurol Disord: Drug Targets. 2006;5(4):453–79. https://doi.org/10.2174/187152706777950684.
Holsboer F, Ising M. Central CRH system in depression and anxiety - evidence from clinical studies with CRH1 receptor antagonists. Eur J Pharmacol. 2008;583(2–3):350–7. https://doi.org/10.1016/j.ejphar.2007.12.032.
Kehne JH, Cain CK. Therapeutic utility of non-peptidic CRF1 receptor antagonists in anxiety, depression, and stress-related disorders: evidence from animal models. Pharmacol Ther. 2010;128(3):460–87. https://doi.org/10.1016/j.pharmthera.2010.08.011.
Canet G, Hernandez C, Zussy C, Chevallier N, Desrumaux C, Givalois L. Is AD a stress-related disorder? Focus on the HPA axis and its promising therapeutic targets. Front Aging Neurosci. 2019;11:269. https://doi.org/10.3389/fnagi.2019.00269.
Spierling SR, Zorrilla EP. Don’t stress about CRF: assessing the translational failures of CRF1antagonists. Psychopharmacology. 2017;234(9–10):1467–81. https://doi.org/10.1007/s00213-017-4556-2.
Zhang C, Kuo CC, Moghadam SH, Monte L, Campbell SN, Rice KC, Sawchenko PE, Masliah E, Rissman RA. Corticotropin-releasing factor receptor-1 antagonism mitigates beta amyloid pathology and cognitive and synaptic deficits in a mouse model of Alzheimer’s disease. Alzheimer’s Dementia. 2016;12(5):527–37. https://doi.org/10.1016/j.jalz.2015.09.007.
Juruena MF, Werne Baes CV, Menezes IC, Graeff FG. Early life stress in depressive patients: role of glucocorticoid and mineralocorticoid receptors and of hypothalamic-pituitary-adrenal axis activity. Curr Pharm Des. 2015;21(11):1369–78. https://doi.org/10.2174/1381612821666150105125500.
Kolber BJ, Wieczorek L, Muglia LJ. Hypothalamic-pituitary-adrenal axis dysregulation and behavioral analysis of mouse mutants with altered glucocorticoid or mineralocorticoid receptor function. Stress. 2008;11(5):321–38. https://doi.org/10.1080/10253890701821081.
Harris MA, Cox SR, Brett CE, Deary IJ, MacLullich AM. Stress in childhood, adolescence and early adulthood, and cortisol levels in older age. Stress. 2017;20(2):140–8. https://doi.org/10.1080/10253890.2017.1289168.
Li M, Fu X, Xie W, Guo W, Li B, Cui R, Yang W. Effect of Early Life Stress on the Epigenetic Profiles in Depression. Front Cell Dev Biol. 2020;8:867. https://doi.org/10.3389/fcell.2020.00867.
Furay AR, Bruestle AE, Herman JP. The role of the forebrain glucocorticoid receptor in acute and chronic stress. Endocrinology. 2008;149(11):5482–90. https://doi.org/10.1210/en.2008-0642.
Gjerstad JK, Lightman SL, Spiga F. Role of glucocorticoid negative feedback in the regulation of HPA axis pulsatility. Stress. 2018;21(5):403–16. https://doi.org/10.1080/10253890.2018.1470238.
Laryea G, Muglia L, Arnett M, Muglia LJ. Dissection of glucocorticoid receptor-mediated inhibition of the hypothalamic-pituitary-adrenal axis by gene targeting in mice. Front Neuroendocrinol. 2015;36:150–64. https://doi.org/10.1016/j.yfrne.2014.09.002.
Rao R, Androulakis IP. Allostatic adaptation and personalized physiological trade-offs in the circadian regulation of the HPA axis: a mathematical modeling approach. Sci Rep. 2019;9(1):11212. https://doi.org/10.1038/s41598-019-47605-7.
Scheff JD, Calvano SE, Lowry SF, Androulakis IP. Transcriptional implications of ultradian glucocorticoid secretion in homeostasis and in the acute stress response. Physiol Genomics. 2012;44(2):121–9. https://doi.org/10.1152/physiolgenomics.00128.2011.
Scheimann JR, Mahbod P, Morano R, Frantz L, Packard B, Campbell K, Herman JP. Deletion of glucocorticoid receptors in forebrain GABAergic neurons alters acute stress responding and passive avoidance behavior in female mice. Front Behav Neurosci. 2018;12:325. https://doi.org/10.3389/fnbeh.2018.00325.
Lupien SJ, Fiocco A, Wan N, Maheu F, Lord C, Schramek T, Tu MT. Stress hormones and human memory function across the lifespan. Psychoneuroendocrinology. 2005;30(3):225–42. https://doi.org/10.1016/j.psyneuen.2004.08.003.
Lupien SJ, Maheu F, Tu M, Fiocco A, Schramek TE. The effects of stress and stress hormones on human cognition: implications for the field of brain and cognition. Brain Cogn. 2007;65(3):209–37. https://doi.org/10.1016/j.bandc.2007.02.007.
Criado-Marrero M, Rein T, Binder EB, Porter JT, Koren J 3rd, Blair LJ. Hsp90 and FKBP51: complex regulators of psychiatric diseases. Philos Trans R Soc Lond Ser B Biol Sci. 2018;373(1738):20160532. https://doi.org/10.1098/rstb.2016.0532.
Silverstein AM, Galigniana MD, Chen MS, Owens-Grillo JK, Chinkers M, Pratt WB. Protein phosphatase 5 is a major component of glucocorticoid receptor.hsp90 complexes with properties of an FK506-binding immunophilin. Biol Chem. 1997;272(26):16224–30. https://doi.org/10.1074/jbc.272.26.16224.
Lackie RE, Maciejewski A, Ostapchenko VG, Marques-Lopes J, Choy WY, Duennwald ML, Prado VF, Prado M. The Hsp70/Hsp90 chaperone machinery in neurodegenerative diseases. Front Neurosci. 2017;11:254. https://doi.org/10.3389/fnins.2017.00254.
Nollen EA, Morimoto RI. Chaperoning signaling pathways: molecular chaperones as stress-sensing “heat shock” proteins. J Cell Sci. 2002;115(Pt 14):2809–16.
Luo GR, Le WD. Collective roles of molecular chaperones in protein degradation pathways associated with neurodegenerative diseases. Curr Pharm Biotechnol. 2010;11(2):180–7. https://doi.org/10.2174/138920110790909740.
McAlary L, Plotkin SS, Cashman NR. Emerging developments in targeting proteotoxicity in neurodegenerative diseases. CNS Drugs. 2019;33(9):883–904. https://doi.org/10.1007/s40263-019-00657-9.
Ho PW, Leung CT, Liu H, Pang SY, Lam CS, Xian J, Li L, Kung MH, Ramsden DB, Ho SL. Age-dependent accumulation of oligomeric SNCA/α-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: role for therapeutic activation of chaperone-mediated autophagy (CMA). Autophagy. 2020;16(2):347–70. https://doi.org/10.1080/15548627.2019.1603545.
Arenaza-Urquijo EM, Przybelski SA, Machulda MM, Knopman DS, Lowe VJ, Mielke MM, Reddy AL, Geda YE, Jack CR Jr, Petersen RC, Vemuri P. Better stress coping associated with lower tau in amyloid-positive cognitively unimpaired older adults. Neurology. 2020;94(15):e1571–9. https://doi.org/10.1212/WNL.0000000000008979.
Sierra-Fonseca JA, Gosselink KL. Tauopathy and neurodegeneration: a role for stress. Neurobiol Stress. 2018;9:105–12. https://doi.org/10.1016/j.ynstr.2018.08.009.
Marin MF, Lord C, Andrews J, Juster RP, Sindi S, Arsenault-Lapierre G, Fiocco AJ, Lupien SJ. Chronic stress, cognitive functioning and mental health. Neurobiol Learn Mem. 2011;96(4):583–95. https://doi.org/10.1016/j.nlm.2011.02.016.
Fries GR, Gassen NC, Rein T. The FKBP51 glucocorticoid receptor co-chaperone: regulation, function, and implications in health and disease. Int J Mol Sci. 2017;18(12):2614. https://doi.org/10.3390/ijms18122614.
Kino T, Su YA, Chrousos GP. Human glucocorticoid receptor isoform beta: recent understanding of its potential implications in physiology and pathophysiology. Cell Mol Life Sci. 2009;66(21):3435–48. https://doi.org/10.1007/s00018-009-0098-z.
Criado-Marrero M, Gebru NT, Gould LA, Smith TM, Kim S, Blackburn RJ, Dickey CA, Blair LJ. Early Life stress and high fkbp5 interact to increase anxiety-like symptoms through altered AKT signaling in the dorsal hippocampus. Int J Mol Sci. 2019;20(11):2738. https://doi.org/10.3390/ijms20112738.
Kirschke E, Goswami D, Southworth D, Griffin PR, Agard DA. Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell. 2014;157(7):1685–97. https://doi.org/10.1016/j.cell.2014.04.038.
Mazaira GI, Daneri-Becerra C, Zgajnar NR, Lotufo CM, Galigniana MD. Gene expression regulation by heat-shock proteins: the cardinal roles of HSF1 and Hsp90. Biochem Soc Trans. 2018;46(1):51–65. https://doi.org/10.1042/BST20170335.
Wang Q, Shelton RC, Dwivedi Y. Interaction between early-life stress and FKBP5 gene variants in major depressive disorder and post-traumatic stress disorder: a systematic review and meta-analysis. J Affect Disord. 2018;225:422–8. https://doi.org/10.1016/j.jad.2017.08.066.
Watkins LE, Han S, Harpaz-Rotem I, Mota NP, Southwick SM, Krystal JH, Gelernter J, Pietrzak RH. FKBP5 polymorphisms, childhood abuse, and PTSD symptoms: results from the National Health and Resilience in Veterans Study. Psychoneuroendocrinology. 2016;69:98–105. https://doi.org/10.1016/j.psyneuen.2016.04.001.
Argentieri MA, Nagarajan S, Seddighzadeh B, Baccarelli AA, Shields AE. Epigenetic pathways in human disease: the impact of DNA methylation on stress-related pathogenesis and current challenges in biomarker development. EBioMedicine. 2017;18:327–50. https://doi.org/10.1016/j.ebiom.2017.03.044.
Giese KP, Mizuno K. The roles of protein kinases in learning and memory. Learn Mem. 2013;20(10):540–52. https://doi.org/10.1101/lm.028449.112.
Revest JM, Di Blasi F, Kitchener P, Rougé-Pont F, Desmedt A, Turiault M, Tronche F, Piazza PV. The MAPK pathway and Egr-1 mediate stress-related behavioral effects of glucocorticoids. Nat Neurosci. 2005;8(5):664–72. https://doi.org/10.1038/nn1441.
Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79(1):143–80. https://doi.org/10.1152/physrev.1999.79.1.143.
Cestari V, Rossi-Arnaud C, Saraulli D, Costanzi M. The MAP(K) of fear: from memory consolidation to memory extinction. Brain Res Bull. 2014;105:8–16. https://doi.org/10.1016/j.brainresbull.2013.09.007.
Duvarci S, Nader K, LeDoux JE. Activation of extracellular signal-regulated kinase- mitogen-activated protein kinase cascade in the amygdala is required for memory reconsolidation of auditory fear conditioning. Eur J Neurosci. 2005;21(1):283–9. https://doi.org/10.1111/j.1460-9568.2004.03824.x.
Herry C, Trifilieff P, Micheau J, Lüthi A, Mons N. Extinction of auditory fear conditioning requires MAPK/ERK activation in the basolateral amygdala. Eur J Neurosci. 2006;24(1):261–9. https://doi.org/10.1111/j.1460-9568.2006.04893.x.
Schafe GE, Atkins CM, Swank MW, Bauer EP, Sweatt JD, LeDoux JE. Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of pavlovian fear conditioning. J Neurosci. 2000;20(21):8177–87. https://doi.org/10.1523/JNEUROSCI.20-21-08177.2000.
Bayraktar G, Kreutz MR. Neuronal DNA methyltransferases: epigenetic mediators between synaptic activity and gene expression? Neurosci Rev J Bringing Neurobiol Neurol Psychiatry. 2018;24(2):171–85. https://doi.org/10.1177/1073858417707457.
Cui D, Xu X. DNA methyltransferases, DNA methylation, and age-associated cognitive function. Int J Mol Sci. 2018;19(5):1315. https://doi.org/10.3390/ijms19051315.
Lubin FD, Roth TL, Sweatt JD. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci. 2008;28(42):10576–86. https://doi.org/10.1523/JNEUROSCI.1786-08.2008.
Rudenko A, Tsai LH. Epigenetic modifications in the nervous system and their impact upon cognitive impairments. Neuropharmacology. 2014;80:70–82. https://doi.org/10.1016/j.neuropharm.2014.01.043.
Sultan FA, Day JJ. Epigenetic mechanisms in memory and synaptic function. Epigenomics. 2011;3(2):157–81. https://doi.org/10.2217/epi.11.6.
Szyf M, Bick J. DNA methylation: a mechanism for embedding early life experiences in the genome. Child Dev. 2013;84(1):49–57. https://doi.org/10.1111/j.1467-8624.2012.01793.x.
Frick KM. Epigenetics, oestradiol and hippocampal memory consolidation. J Neuroendocrinol. 2013;25(11):1151–62. https://doi.org/10.1111/jne.12106.
Li Z, Kono H. Distinct roles of histone H3 and H2A tails in nucleosome stability. Sci Rep. 2016;6:31437. https://doi.org/10.1038/srep31437.
Kuo M-H, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. BioEssays. 1998;20(8):615–26. https://doi.org/10.1002/(sici)1521-1878(199808)20:8%3c615::aid-bies4%3e3.0.co;2-h.
Rossetto D, Avvakumov N, Côté J. Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics. 2012;7(10):1098–108. https://doi.org/10.4161/epi.21975.
Peixoto L, Abel T. The role of histone acetylation in memory formation and cognitive impairments. Neuropsychopharmacology. 2013;38(1):62–76. https://doi.org/10.1038/npp.2012.86.
Radley JJ, Kabbaj M, Jacobson L, Heydendael W, Yehuda R, Herman JP. Stress risk factors and stress-related pathology: neuroplasticity, epigenetics and endophenotypes. Stress (Amsterdam, Netherlands). 2011;14(5):481–97. https://doi.org/10.3109/10253890.2011.604751.
Wu J, Liu C, Zhang L, He B, Shi WP, Shi HL, Qin C. Chronic restraint stress impairs cognition via modulating HDAC2 expression. Transl Neurosci. 2021;12(1):154–63. https://doi.org/10.1515/tnsci-2020-0168.
Martins de Carvalho L, Chen WY, Lasek AW. Epigenetic mechanisms underlying stress-induced depression. Int Rev Neurobiol. 2021;156:87–126. https://doi.org/10.1016/bs.irn.2020.08.001.
Guan L, Shi X, Tang Y, Yan Y, Chen L, Chen Y, Gao G, Lin C, Chen A. Contribution of amygdala histone acetylation in early life stress-induced visceral hypersensitivity and emotional comorbidity. Front Neurosci. 2022;16: 843396. https://doi.org/10.3389/fnins.2022.843396.
Maddox SA, Watts CS, Schafe GE. p300/CBP histone acetyltransferase activity is required for newly acquired and reactivated fear memories in the lateral amygdala. Learn Mem. 2013;20(2):109–19. https://doi.org/10.1101/lm.029157.112.
Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87(5):953–9. https://doi.org/10.1016/S0092-8674(00)82001-2.
Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7(8):847–54. https://doi.org/10.1038/nn1276.
Miller CA, Campbell SL, Sweatt JD. DNA methylation and histone acetylation work in concert to regulate memory formation and synaptic plasticity. Neurobiol Learn Mem. 2008;89(4):599–603. https://doi.org/10.1016/j.nlm.2007.07.016.
Bahari-Javan S, Sananbenesi F, Fischer A. Histone-acetylation: a link between Alzheimer’s disease and post-traumatic stress disorder? Front Neurosci. 2014;8:160. https://doi.org/10.3389/fnins.2014.00160.
Lu X, Wang L, Yu C, Yu D, Yu G. Histone acetylation modifiers in the pathogenesis of Alzheimer’s disease. Front Cell Neurosci. 2015;9:226. https://doi.org/10.3389/fncel.2015.00226.
Park J, Lee K, Kim K, Yi SJ. The role of histone modifications: from neurodevelopment to neurodiseases. Signal Transduct Target Ther. 2022;7(1):217. https://doi.org/10.1038/s41392-022-01078-9.
Wagner FF, Zhang YL, Fass DM, Joseph N, Gale JP, Weïwer M, McCarren P, Fisher SL, Kaya T, Zhao WN, Reis SA, Hennig KM, Thomas M, Lemercier BC, Lewis MC, Guan JS, Moyer MP, Scolnick E, Haggarty SJ, Tsai LH, et al. Kinetically selective inhibitors of histone deacetylase 2 (HDAC2) as cognition enhancers. Chem Sci. 2015;6(1):804–15. https://doi.org/10.1039/C4SC02130D.
Bonomi RE, Girgenti M, Krystal JH, Cosgrove KP. A role for histone deacetylases in the biology and treatment of post-traumatic stress disorder: what do we know and where do we go from here? Complex psychiatry. 2022;8(1–2):13–27. https://doi.org/10.1159/000524079.
Arango-Lievano M, Lambert WM, Bath KG, Garabedian MJ, Chao MV, Jeanneteau F. Neurotrophic-priming of glucocorticoid receptor signaling is essential for neuronal plasticity to stress and antidepressant treatment. Proc Natl Acad Sci USA. 2015;112(51):15737–42. https://doi.org/10.1073/pnas.1509045112.
Taliaz D, Loya A, Gersner R, Haramati S, Chen A, Zangen A. Resilience to chronic stress is mediated by hippocampal brain-derived neurotrophic factor. J Neurosci. 2011;31(12):4475–83. https://doi.org/10.1523/JNEUROSCI.5725-10.2011.
Musumeci G, Minichiello L. BDNF-TrkB signalling in fear learning: from genetics to neural networks. Rev Neurosci. 2011;22(3):303–15. https://doi.org/10.1515/RNS.2011.031.
Ou LC, Yeh SH, Gean PW. Late expression of brain-derived neurotrophic factor in the amygdala is required for persistence of fear memory. Neurobiol Learn Mem. 2010;93(3):372–82. https://doi.org/10.1016/j.nlm.2009.12.003.
Roth TL, Lubin FD, Funk AJ, Sweatt JD. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol Psychiat. 2009;65(9):760–9. https://doi.org/10.1016/j.biopsych.2008.11.028.
Takei S, Morinobu S, Yamamoto S, Fuchikami M, Matsumoto T, Yamawaki S. Enhanced hippocampal BDNF/TrkB signaling in response to fear conditioning in an animal model of posttraumatic stress disorder. J Psychiatr Res. 2011;45(4):460–8. https://doi.org/10.1016/j.jpsychires.2010.08.009.
Li XH, Liu NB, Zhang MH, Zhou YL, Liao JW, Liu XQ, Chen HW. Effects of chronic multiple stress on learning and memory and the expression of Fyn, BDNF, TrkB in the hippocampus of rats. Chin Med J. 2007;120(8):669–74.
Bekinschtein P, Cammarota M, Katche C, Slipczuk L, Rossato JI, Goldin A, Izquierdo I, Medina JH. BDNF is essential to promote persistence of long-term memory storage. Proc Natl Acad Sci USA. 2008;105(7):2711–6. https://doi.org/10.1073/pnas.0711863105.
Carrion VG, Wong SS. Can traumatic stress alter the brain? Understanding the implications of early trauma on brain development and learning. J Adolesc Health. 2012;51(2 Suppl):S23–8. https://doi.org/10.1016/j.jadohealth.2012.04.010.
Warner-Schmidt JL, Duman RS. Hippocampal neurogenesis: opposing effects of stress and antidepressant treatment. Hippocampus. 2006;16(3):239–49. https://doi.org/10.1002/hipo.20156.
Yamada K, Nabeshima T. Brain-derived neurotrophic factor/TrkB signaling in memory processes. J Pharmacol Sci. 2003;91(4):267–70. https://doi.org/10.1254/jphs.91.267.
Miao Z, Wang Y, Sun Z. The relationships between stress, mental disorders, and epigenetic regulation of BDNF. Int J Mol Sci. 2020;21(4):1375. https://doi.org/10.3390/ijms21041375.
Mitchelmore C, Gede L. Brain derived neurotrophic factor: epigenetic regulation in psychiatric disorders. Brain Res. 2014;1586:162–72. https://doi.org/10.1016/j.brainres.2014.06.037.
Zheleznyakova GY, Cao H, Schiöth HB. BDNF DNA methylation changes as a biomarker of psychiatric disorders: literature review and open access database analysis. Behav Brain Funct BBF. 2016;12(1):17. https://doi.org/10.1186/s12993-016-0101-4.
Chen F, Yu X, Meng G, Mei Z, Du Y, Sun H, Reed MN, Kong L, Suppiramaniam V, Hong H, Tang S. Hippocampal genetic knockdown of PPARδ causes depression-like behaviors and neurogenesis suppression. Int J Neuropsychopharmacol. 2019;22(6):372–82. https://doi.org/10.1093/ijnp/pyz008.
Mojtabavi H, Saghazadeh A, van den Heuvel L, Bucker J, Rezaei N. Peripheral blood levels of brain-derived neurotrophic factor in patients with post-traumatic stress disorder (PTSD): a systematic review and meta-analysis. PLoS ONE. 2020;15(11): e0241928. https://doi.org/10.1371/journal.pone.0241928.
Notaras M, van den Buuse M. Neurobiology of BDNF in fear memory, sensitivity to stress, and stress-related disorders. Mol Psychiatry. 2020;25(10):2251–74. https://doi.org/10.1038/s41380-019-0639-2.
Felmingham KL, Zuj DV, Hsu K, Nicholson E, Palmer MA, Stuart K, Vickers JC, Malhi GS, Bryant RA. The BDNF Val66Met polymorphism moderates the relationship between posttraumatic stress disorder and fear extinction learning. Psychoneuroendocrinology. 2018;91:142–8. https://doi.org/10.1016/j.psyneuen.2018.03.002.
Hori H, Itoh M, Yoshida F, Lin M, Niwa M, Hakamata Y, Ino K, Imai R, Ogawa S, Matsui M, Kamo T, Kunugi H, Kim Y. The BDNF Val66Met polymorphism affects negative memory bias in civilian women with PTSD. Sci Rep. 2020;10(1):3151. https://doi.org/10.1038/s41598-020-60096-1.
Desmarais P, Weidman D, Wassef A, Bruneau MA, Friedland J, Bajsarowicz P, Thibodeau MP, Herrmann N, Nguyen QD. The interplay between post-traumatic stress disorder and dementia: a systematic review. Am J Geriatr Psychiatry. 2020;28(1):48–60. https://doi.org/10.1016/j.jagp.2019.08.006.
Günak MM, Billings J, Carratu E, Marchant NL, Favarato G, Orgeta V. Post-traumatic stress disorder as a risk factor for dementia: systematic review and meta-analysis. Br J Psychiatry. 2020;217(5):600–8. https://doi.org/10.1192/bjp.2020.150.
Kwon J, Kim YJ, Choi K, Seol S, Kang HJ. Identification of stress resilience module by weighted gene co-expression network analysis in Fkbp5-deficient mice. Mol Brain. 2019;12(1):99. https://doi.org/10.1186/s13041-019-0521-9.
Palma-Gudiel H, Córdova-Palomera A, Leza JC, Fañanás L. Glucocorticoid receptor gene (NR3C1) methylation processes as mediators of early adversity in stress-related disorders causality: a critical review. Neurosci Biobehav Rev. 2015;55:520–35. https://doi.org/10.1016/j.neubiorev.2015.05.016.
Weder N, Zhang H, Jensen K, Yang BZ, Simen A, Jackowski A, Lipschitz D, Douglas-Palumberi H, Ge M, Perepletchikova F, O’Loughlin K, Hudziak JJ, Gelernter J, Kaufman J. Child abuse, depression, and methylation in genes involved with stress, neural plasticity, and brain circuitry. J Am Acad Child Adolesc Psychiatry. 2014;53(4):417-24.e5. https://doi.org/10.1016/j.jaac.2013.12.025.
Hawn SE, Sheerin CM, Lind MJ, Hicks TA, Marraccini ME, Bountress K, Bacanu SA, Nugent NR, Amstadter AB. GxE effects of FKBP5 and traumatic life events on PTSD: a meta-analysis. J Affect Disord. 2019;243:455–62. https://doi.org/10.1016/j.jad.2018.09.058.
Lesiak AJ, Coffey K, Cohen JH, Liang KJ, Chavkin C, Neumaier JF. Sequencing the serotonergic neuron translatome reveals a new role for Fkbp5 in stress. Mol Psychiatry. 2020. https://doi.org/10.1038/s41380-020-0750-4.
Menke A, Klengel T, Rubel J, Brückl T, Pfister H, Lucae S, Uhr M, Holsboer F, Binder EB. Genetic variation in FKBP5 associated with the extent of stress hormone dysregulation in major depression. Genes Brain Behav. 2013;12(3):289–96. https://doi.org/10.1111/gbb.12026.
Ising M, Depping AM, Siebertz A, Lucae S, Unschuld PG, Kloiber S, Horstmann S, Uhr M, Müller-Myhsok B, Holsboer F. Polymorphisms in the FKBP5 gene region modulate recovery from psychosocial stress in healthy controls. Eur J Neurosci. 2008;28(2):389–98. https://doi.org/10.1111/j.1460-9568.2008.06332.x.
Mahon PB, Zandi PP, Potash JB, Nestadt G, Wand GS. Genetic association of FKBP5 and CRHR1 with cortisol response to acute psychosocial stress in healthy adults. Psychopharmacology. 2013;227(2):231–41. https://doi.org/10.1007/s00213-012-2956-x.
Lovallo WR, Enoch MA, Acheson A, Cohoon AJ, Sorocco KH, Hodgkinson CA, Vincent AS, Goldman D. Early-life adversity interacts with FKBP5 genotypes: altered working memory and cardiac stress reactivity in the oklahoma family health patterns project. Neuropsychopharmacology. 2016;41(7):1724–32. https://doi.org/10.1038/npp.2015.347.
Zoladz PR, Dailey AM, Nagle HE, Fiely MK, Mosley BE, Brown CM, Duffy TJ, Scharf AR, Earley MB, Rorabaugh BR. FKBP5 polymorphisms influence pre-learning stress-induced alterations of learning and memory. Eur J Neurosci. 2017;45(5):648–59. https://doi.org/10.1111/ejn.13514.
Hariri AR, Mattay VS, Tessitore A, Kolachana B, Fera F, Goldman D, Egan MF, Weinberger DR. Serotonin transporter genetic variation and the response of the human amygdala. Science. 2002;297(5580):400–3. https://doi.org/10.1126/science.1071829.
Hariri AR, Drabant EM, Munoz KE, Kolachana BS, Mattay VS, Egan MF, Weinberger DR. A susceptibility gene for affective disorders and the response of the human amygdala. Arch Gen Psychiatry. 2005;62(2):146–52. https://doi.org/10.1001/archpsyc.62.2.146.
Heinz A, Braus DF, Smolka MN, Wrase J, Puls I, Hermann D, Klein S, Grüsser SM, Flor H, Schumann G, Mann K, Büchel C. Amygdala-prefrontal coupling depends on a genetic variation of the serotonin transporter. Nat Neurosci. 2005;8(1):20–1. https://doi.org/10.1038/nn1366.
Heinz A, Smolka MN, Braus DF, Wrase J, Beck A, Flor H, Mann K, Schumann G, Büchel C, Hariri AR, Weinberger DR. Serotonin transporter genotype (5-HTTLPR): effects of neutral and undefined conditions on amygdala activation. Biol Psychiat. 2007;61(8):1011–4. https://doi.org/10.1016/j.biopsych.2006.08.019.
Penner-Goeke S, Binder EB. Epigenetics and depression. Dialogues Clin Neurosci. 2019;21(4):397–405. https://doi.org/10.31887/DCNS.2019.21.4/ebinder.
Pagliaccio D, Luby JL, Bogdan R, Agrawal A, Gaffrey MS, Belden AC, Botteron KN, Harms MP, Barch DM. Amygdala functional connectivity, HPA axis genetic variation, and life stress in children and relations to anxiety and emotion regulation. J Abnorm Psychol. 2015;124(4):817–33. https://doi.org/10.1037/abn0000094.
Gillespie CF, Phifer J, Bradley B, Ressler KJ. Risk and resilience: genetic and environmental influences on development of the stress response. Depress Anxiety. 2009;26(11):984–92. https://doi.org/10.1002/da.20605.
White MG, Bogdan R, Fisher PM, Muñoz KE, Williamson DE, Hariri AR. FKBP5 and emotional neglect interact to predict individual differences in amygdala reactivity. Genes Brain Behav. 2012;11(7):869–78. https://doi.org/10.1111/j.1601-183X.2012.00837.x.
Di Iorio CR, Carey CE, Michalski LJ, Corral-Frias NS, Conley ED, Hariri AR, Bogdan R. Hypothalamic-pituitary-adrenal axis genetic variation and early stress moderates amygdala function. Psychoneuroendocrinology. 2017;80:170–8. https://doi.org/10.1016/j.psyneuen.2017.03.016.
Roy A, Gorodetsky E, Yuan Q, Goldman D, Enoch MA. Interaction of FKBP5, a stress-related gene, with childhood trauma increases the risk for attempting suicide. Neuropsychopharmacology. 2010;35(8):1674–83. https://doi.org/10.1038/npp.2009.236.
Cifariello A, Pompili A, Gasbarri A. 5-HT(7) receptors in the modulation of cognitive processes. Behav Brain Res. 2008;195(1):171–9. https://doi.org/10.1016/j.bbr.2007.12.012.
Elvander-Tottie E, Eriksson TM, Sandin J, Ogren SO. 5-HT(1A) and NMDA receptors interact in the rat medial septum and modulate hippocampal-dependent spatial learning. Hippocampus. 2009;19(12):1187–98. https://doi.org/10.1002/hipo.20596.
Gasbarri A, Pompili A. Serotonergic 5-HT7 receptors and cognition. Rev Neurosci. 2014;25(3):311–23. https://doi.org/10.1515/revneuro-2013-0066.
Leiser SC, Li Y, Pehrson AL, Dale E, Smagin G, Sanchez C. Serotonergic regulation of prefrontal cortical circuitries involved in cognitive processing: a review of individual 5-HT receptor mechanisms and concerted effects of 5-HT receptors exemplified by the multimodal antidepressant vortioxetine. ACS Chem Neurosci. 2015;6(7):970–86. https://doi.org/10.1021/cn500340j.
Murnane KS. Serotonin 2A receptors are a stress response system: implications for post-traumatic stress disorder. Behav Pharmacol. 2019;30(2 and 3-Spec Issue):151–62. https://doi.org/10.1097/FBP.0000000000000459.
Ogren SO, Eriksson TM, Elvander-Tottie E, Addario C, Ekström JC, Svenningsson P, Meister B, Kehr J, Stiedl O. The role of 5-HT(1A) receptors in learning and memory. Behav Brain Res. 2008;195(1):54–77. https://doi.org/10.1016/j.bbr.2008.02.023.
Pehrson AL, Leiser SC, Gulinello M, Dale E, Li Y, Waller JA, Sanchez C. Treatment of cognitive dysfunction in major depressive disorde—-a review of the preclinical evidence for efficacy of selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors and the multimodal-acting antidepressant vortioxetine. Eur J Pharmacol. 2015;753:19–31. https://doi.org/10.1016/j.ejphar.2014.07.044.
Roberts AJ, Hedlund PB. The 5-HT(7) receptor in learning and memory. Hippocampus. 2012;22(4):762–71. https://doi.org/10.1002/hipo.20938.
Sarkisyan G, Hedlund PB. The 5-HT7 receptor is involved in allocentric spatial memory information processing. Behav Brain Res. 2009;202(1):26–31. https://doi.org/10.1016/j.bbr.2009.03.011.
Creese B, Ballard C, Aarsland D, Londos E, Sharp S, Jones E. Determining the association of the 5HTTLPR polymorphism with delusions and hallucinations in Lewy body dementias. Am J Geriatr Psychiatry. 2014;22(6):580–6. https://doi.org/10.1016/j.jagp.2012.11.001.
Creese B, Ballard C, Jones E. Cognitive impairment in studies of 5HTTLPR and psychosis in Alzheimer’s disease: a systematic review. Dement Geriatr Cogn Disord. 2013;35(3–4):155–64. https://doi.org/10.1159/000346733.
D’Onofrio G, Panza F, Sancarlo D, Lauriola M, Dagostino MP, Paroni G, Lozupone M, Mangiacotti A, Bisceglia P, Gravina C, Urbano M, Addante F, Paris F, Cascavilla L, Greco A, Seripa D. Hydroxytryptamine transporter gene-linked polymorphic region (5HTTLPR) is associated with delusions in Alzheimer’s disease. Transl Neurodegen. 2019;8:4. https://doi.org/10.1186/s40035-019-0144-1.
Li S, Tang J, Gao Y, Thiel CM, Wolf OT. The serotonin transporter gene variants modulate acute stress-induced hippocampus and dorsomedial prefrontal cortex activity during memory retrieval. PsyCh J. 2019;8(3):363–77. https://doi.org/10.1002/pchj.297.
Sun X, Li C, Zhong X, Dong D, Ming Q, Gao Y, Xiong G, Cheng C, Zhao H, Wang X, Yao S. Influence of psychosocial stress on activation in human brain regions: moderation by the 5-HTTLPR genetic locus. Physiol Behav. 2020;220: 112876. https://doi.org/10.1016/j.physbeh.2020.112876.
O’Hara R, Schröder CM, Mahadevan R, Schatzberg AF, Lindley S, Fox S, Weiner M, Kraemer HC, Noda A, Lin X, Gray HL, Hallmayer JF. Serotonin transporter polymorphism, memory and hippocampal volume in the elderly: association and interaction with cortisol. Mol Psychiatry. 2007;12(6):544–55. https://doi.org/10.1038/sj.mp.4001978.
Marini S, Bagnoli S, Bessi V, Tedde A, Bracco L, Sorbi S, Nacmias B. Implication of serotonin-transporter (5-HTT) gene polymorphism in subjective memory complaints and mild cognitive impairment (MCI). Arch Gerontol Geriatr. 2011;52(2):e71–4. https://doi.org/10.1016/j.archger.2010.06.006.
Mammarella N, Gatti M, Ceccato I, Di Crosta A, Di Domenico A, Palumbo R. The protective role of neurogenetic components in reducing stress-related effects during spaceflights: evidence from the age-related positive memory approach. Life. 2022;12(8):1176. https://doi.org/10.3390/life12081176.
Migliore L, Coppedè F. Gene-environment interactions in Alzheimer disease: the emerging role of epigenetics. Nat Rev Neurol. 2022;18(11):643–60. https://doi.org/10.1038/s41582-022-00714-w.
Tyng CM, Amin HU, Saad M, Malik AS. The influences of emotion on learning and memory. Front Psychol. 2017;8:1454. https://doi.org/10.3389/fpsyg.2017.01454.
Squire LR. Memory and the hippocampus: a synthesis from findings with rats, monkeys, and humans. Psychol Rev. 1992;99(2):195–231. https://doi.org/10.1037/0033-295x.99.2.195.
Squire LR, Dede AJ. Conscious and unconscious memory systems. Cold Spring Harb Perspect Biol. 2015;7(3): a021667. https://doi.org/10.1101/cshperspect.a021667.
Reber PJ. The neural basis of implicit learning and memory: a review of neuropsychological and neuroimaging research. Neuropsychologia. 2013;51(10):2026–42. https://doi.org/10.1016/j.neuropsychologia.2013.06.019.
Tulving E, Markowitsch HJ. Episodic and declarative memory: role of the hippocampus. Hippocampus. 1998;8(3):198–204. https://doi.org/10.1002/(SICI)1098-1063(1998)8:3%3c198::AID-HIPO2%3e3.0.CO;2-G.
Grady CL. Meta-analytic and functional connectivity evidence from functional magnetic resonance imaging for an anterior to posterior gradient of function along the hippocampal axis. Hippocampus. 2020;30(5):456–71. https://doi.org/10.1002/hipo.23164.
Josselyn SA, Köhler S, Frankland PW. Finding the engram. Nat Rev Neurosci. 2015;16(9):521–34. https://doi.org/10.1038/nrn4000.
Holohean AM, Magleby KL. Neuromuscular junction (NMJ): presynaptic short-term plasticity of neuromuscular transmission. In: Encyclopedia of neuroscience. Elsevier; 2009. p. 629–34.
Grueter BA, Winder DG. Group II and III metabotropic glutamate receptors suppress excitatory synaptic transmission in the dorsolateral bed nucleus of the stria terminalis. Neuropsychopharmacology. 2005;30(7):1302–11. https://doi.org/10.1038/sj.npp.1300672.
Abe K. Modulation of hippocampal long-term potentiation by the amygdala: a synaptic mechanism linking emotion and memory. Jpn J Pharmacol. 2001;86(1):18–22. https://doi.org/10.1254/jjp.86.18.
Ferry B, McGaugh JL. Role of amygdala norepinephrine in mediating stress hormone regulation of memory storage. Acta Pharmacol Sin. 2000;21(6):481–93.
Rabinak CA, Maren S. Associative structure of fear memory after basolateral amygdala lesions in rats. Behav Neurosci. 2008;122(6):1284–94. https://doi.org/10.1037/a0012903.
Roozendaal B. 1999 Curt P. Richter award. Glucocorticoids and the regulation of memory consolidation. Psychoneuroendocrinology. 2000;25(3):213–38. https://doi.org/10.1016/s0306-4530(99)00058-x.
Roozendaal B, Quirarte GL, McGaugh JL. Glucocorticoids interact with the basolateral amygdala beta-adrenoceptor–cAMP/cAMP/PKA system in influencing memory consolidation. Eur J Neurosci. 2002;15(3):553–60. https://doi.org/10.1046/j.0953-816x.2001.01876.x.
Roozendaal B, Okuda S, de Quervain DJ, McGaugh JL. Glucocorticoids interact with emotion-induced noradrenergic activation in influencing different memory functions. Neuroscience. 2006;138(3):901–10. https://doi.org/10.1016/j.neuroscience.2005.07.049.
McGaugh JL, Roozendaal B. Role of adrenal stress hormones in forming lasting memories in the brain. Curr Opin Neurobiol. 2002;12(2):205–10. https://doi.org/10.1016/s0959-4388(02)00306-9.
McGaugh JL. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu Rev Neurosci. 2004;27:1–28. https://doi.org/10.1146/annurev.neuro.27.070203.144157.
Nathan SV, Griffith QK, McReynolds JR, Hahn EL, Roozendaal B. Basolateral amygdala interacts with other brain regions in regulating glucocorticoid effects on different memory functions. Ann N Y Acad Sci. 2004;1032:179–82. https://doi.org/10.1196/annals.1314.015.
Souza-Braga P, Lorena FB, Nascimento B, Marcelino CP, Ravache TT, Ricci E, Bernardi MM, Ribeiro MO. Adrenergic receptor β3 is involved in the memory consolidation process in mice. Braz J Med Biol Res Revista brasileira de pesquisas medicas e biologicas. 2018;51(10): e7564. https://doi.org/10.1590/1414-431X20187564.
Osborne DM, Pearson-Leary J, McNay EC. The neuroenergetics of stress hormones in the hippocampus and implications for memory. Front Neurosci. 2015;9:164. https://doi.org/10.3389/fnins.2015.00164.
Pagani MR, Merlo E. Kinase and phosphatase engagement is dissociated between memory formation and extinction. Front Mol Neurosci. 2019;12:38. https://doi.org/10.3389/fnmol.2019.00038.
Akirav I, Richter-Levin G. Biphasic modulation of hippocampal plasticity by behavioral stress and basolateral amygdala stimulation in the rat. J Neurosci. 1999;19(23):10530–5. https://doi.org/10.1523/JNEUROSCI.19-23-10530.1999.
Akirav I, Richter-Levin G. Mechanisms of amygdala modulation of hippocampal plasticity. J Neurosci. 2002;22(22):9912–21. https://doi.org/10.1523/JNEUROSCI.22-22-09912.2002.
Kim JJ, Diamond DM. The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci. 2002;3(6):453–62. https://doi.org/10.1038/nrn849.
Kim EJ, Pellman B, Kim JJ. Stress effects on the hippocampus: a critical review. Learn Memory (Cold Spring Harbor, NY). 2015;22(9):411–6. https://doi.org/10.1101/lm.037291.114.
Szeszko PR, Lehrner A, Yehuda R. Glucocorticoids and hippocampal structure and function in PTSD. Harv Rev Psychiatry. 2018;26(3):142–57. https://doi.org/10.1097/HRP.0000000000000188.
Vitureira N, Goda Y. Cell biology in neuroscience: the interplay between Hebbian and homeostatic synaptic plasticity. J Cell Biol. 2013;203(2):175–86. https://doi.org/10.1083/jcb.201306030.
Sanderson JL, Scott JD, Dell’Acqua ML. Control of homeostatic synaptic plasticity by AKAP-anchored kinase and phosphatase regulation of Ca2+-permeable AMPA receptors. J Neurosci. 2018;38(11):2863–76. https://doi.org/10.1523/JNEUROSCI.2362-17.2018.
Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–26. https://doi.org/10.1146/annurev.neuro.25.112701.142758.
Musazzi L, Milanese M, Farisello P, Zappettini S, Tardito D, Barbiero VS, Bonifacino T, Mallei A, Baldelli P, Racagni G, Raiteri M, Benfenati F, Bonanno G, Popoli M. Acute stress increases depolarization-evoked glutamate release in the rat prefrontal/frontal cortex: the dampening action of antidepressants. PLoS ONE. 2010;5(1): e8566. https://doi.org/10.1371/journal.pone.0008566.
Nava N, Treccani G, Liebenberg N, Chen F, Popoli M, Wegener G, Nyengaard JR. Chronic desipramine prevents acute stress-induced reorganization of medial prefrontal cortex architecture by blocking glutamate vesicle accumulation and excitatory synapse increase. Int J Neuropsychopharmacol. 2014;18(3):pyu085. https://doi.org/10.1093/ijnp/pyu085.
Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2011;13(1):22–37. https://doi.org/10.1038/nrn3138.
Greger IH, Ziff EB, Penn AC. Molecular determinants of AMPA receptor subunit assembly. Trends Neurosci. 2007;30(8):407–16. https://doi.org/10.1016/j.tins.2007.06.005.
Mayer ML. Glutamate receptor ion channels. Curr Opin Neurobiol. 2005;15(3):282–8. https://doi.org/10.1016/j.conb.2005.05.004.
Schlüter OM, Xu W, Malenka RC. Alternative N-terminal domains of PSD-95 and SAP97 govern activity-dependent regulation of synaptic AMPA receptor function. Neuron. 2006;51(1):99–111. https://doi.org/10.1016/j.neuron.2006.05.016.
Bassani S, Folci A, Zapata J, Passafaro M. AMPAR trafficking in synapse maturation and plasticity. Cell Mol Life Sci. 2013;70(23):4411–30. https://doi.org/10.1007/s00018-013-1309-1.
Choquet D. Linking nanoscale dynamics of AMPA receptor organization to plasticity of excitatory synapses and learning. J Neurosci. 2018;38(44):9318–29. https://doi.org/10.1523/JNEUROSCI.2119-18.2018.
Keifer J, Zheng Z. AMPA receptor trafficking and learning. Eur J Neurosci. 2010;32(2):269–77. https://doi.org/10.1111/j.1460-9568.2010.07339.x.
Conboy L, Sandi C. Stress at learning facilitates memory formation by regulating AMPA receptor trafficking through a glucocorticoid action. Neuropsychopharmacology. 2010;35(3):674–85. https://doi.org/10.1038/npp.2009.172.
Lin M, Hou G, Zhao Y, Yuan TF. Recovery of chronic stress-triggered changes of hippocampal glutamatergic transmission. Neural Plast. 2018;2018:9360203. https://doi.org/10.1155/2018/9360203.
Zanca RM, Sanay S, Avila JA, Rodriguez E, Shair HN, Serrano PA. Contextual fear memory modulates PSD95 phosphorylation, AMPAr subunits, PKMζ and PI3K differentially between adult and juvenile rats. Neurobiol Stress. 2018;10: 100139. https://doi.org/10.1016/j.ynstr.2018.11.002.
Fumagalli F, Caffino L, Vogt MA, Frasca A, Racagni G, Sprengel R, Gass P, Riva MA. AMPA GluR-A receptor subunit mediates hippocampal responsiveness in mice exposed to stress. Hippocampus. 2011;21(9):1028–35. https://doi.org/10.1002/hipo.20817.
Caudal D, Godsil BP, Mailliet F, Bergerot D, Jay TM. Acute stress induces contrasting changes in AMPA receptor subunit phosphorylation within the prefrontal cortex, amygdala and hippocampus. PLoS ONE. 2010;5(12): e15282. https://doi.org/10.1371/journal.pone.0015282.
Lee MT, Peng WH, Kan HW, Wu CC, Wang DW, Ho YC. Neurobiology of depression: chronic stress alters the glutamatergic system in the brain-focusing on AMPA receptor. Biomedicines. 2022;10(5):1005. https://doi.org/10.3390/biomedicines10051005.
Wu QL, Gao Y, Li JT, Ma WY, Chen NH. The role of AMPARs composition and trafficking in synaptic plasticity and diseases. Cell Mol Neurobiol. 2022;42(8):2489–504. https://doi.org/10.1007/s10571-021-01141-z.
Kuniishi H, Yamada D, Wada K, Yamada M, Sekiguchi M. Stress induces insertion of calcium-permeable AMPA receptors in the OFC-BLA synapse and modulates emotional behaviours in mice. Transl Psychiatry. 2020;10(1):154. https://doi.org/10.1038/s41398-020-0837-3Kunishi.
Jumaili WA, Trivedi C, Chao T, Kubosumi A, Jain S. The safety and efficacy of ketamine NMDA receptor blocker as a therapeutic intervention for PTSD review of a randomized clinical trial. Behav Brain Res. 2022;424: 113804. https://doi.org/10.1016/j.bbr.2022.113804.
Andersen JV, Schousboe A, Verkhratsky A. Astrocyte energy and neurotransmitter metabolism in Alzheimer’s disease: integration of the glutamate/GABA-glutamine cycle. Prog Neurobiol. 2022;217: 102331. https://doi.org/10.1016/j.pneurobio.2022.102331.
Reddy-Thootkur M, Kraguljac NV, Lahti AC. The role of glutamate and GABA in cognitive dysfunction in schizophrenia and mood disorders—a systematic review of magnetic resonance spectroscopy studies. Schizophr Res. 2022;249:74–84. https://doi.org/10.1016/j.schres.2020.02.001.
Patriarchi T, Buonarati OR, Hell JW. Postsynaptic localization and regulation of AMPA receptors and Cav1.2 by β2 adrenergic receptor/PKA and Ca2+/CaMKII signaling. EMBO J. 2018;37(20): e99771. https://doi.org/10.15252/embj.201899771.
Reiner A, Levitz J. Glutamatergic signaling in the central nervous system: ionotropic and metabotropic receptors in concert. Neuron. 2018;98(6):1080–98. https://doi.org/10.1016/j.neuron.2018.05.018.
Weiss N, Zamponi GW. Trafficking of neuronal calcium channels. Neuronal Signal. 2017;1(1): NS20160003. https://doi.org/10.1042/NS20160003.
Kubota M, Murakoshi T, Saegusa H, Kazuno A, Zong S, Hu Q, Noda T, Tanabe T. Intact LTP and fear memory but impaired spatial memory in mice lacking Ca(v)2.3 (alpha(IE)) channel. Biochem Biophys Res Commun. 2001;282(1):242–8. https://doi.org/10.1006/bbrc.2001.4572.
Chen CC, Shen JW, Chung NC, Min MY, Cheng SJ, Liu IY. Retrieval of context-associated memory is dependent on the Ca(v)32 T-type calcium channel. PLoS ONE. 2012;7(1): e29384. https://doi.org/10.1371/journal.pone.0029384.
Lipscombe D, Helton TD, Xu W. L-type calcium channels: the low down. J Neurophysiol. 2004;92(5):2633–41. https://doi.org/10.1152/jn.00486.2004.
Noyer L, Lemonnier L, Mariot P, Gkika D. Partners in crime: towards new ways of targeting calcium channels. Int J Mol Sci. 2019;20(24):6344. https://doi.org/10.3390/ijms20246344.
Abbott LF, Nelson SB. Synaptic plasticity: taming the beast. Nat Neurosci. 2000;3(Suppl):1178–83. https://doi.org/10.1038/81453.
Campanac E, Debanne D. Plasticity of neuronal excitability: hebbian rules beyond the synapse. Arch Ital Biol. 2007;145(3–4):277–87.
Debanne D, Daoudal G, Sourdet V, Russier M. Brain plasticity and ion channels. J Physiol Paris. 2003;97(4–6):403–14. https://doi.org/10.1016/j.jphysparis.2004.01.004.
Ebert DH, Greenberg ME. Activity-dependent neuronal signalling and autism spectrum disorder. Nature. 2013;493(7432):327–37. https://doi.org/10.1038/nature11860.
Du J, Wang Y, Hunter R, Wei Y, Blumenthal R, Falke C, Khairova R, Zhou R, Yuan P, Machado-Vieira R, McEwen BS, Manji HK. Dynamic regulation of mitochondrial function by glucocorticoids. Proc Natl Acad Sci USA. 2009;106(9):3543–8. https://doi.org/10.1073/pnas.0812671106.
Lee SR, Kim HK, Song IS, Youm J, Dizon LA, Jeong SH, Ko TH, Heo HJ, Ko KS, Rhee BD, Kim N, Han J. Glucocorticoids and their receptors: insights into specific roles in mitochondria. Prog Biophys Mol Biol. 2013;112(1–2):44–54. https://doi.org/10.1016/j.pbiomolbio.2013.04.001.
Wen Y, Li B, Han F, Wang E, Shi Y. Dysfunction of calcium/calmodulin/CaM kinase IIα cascades in the medial prefrontal cortex in post-traumatic stress disorder. Mol Med Rep. 2012;6(5):1140–4. https://doi.org/10.3892/mmr.2012.1022.
Moench KM, Wellman CL. Stress-induced alterations in prefrontal dendritic spines: Implications for post-traumatic stress disorder. Neurosci Lett. 2015;601:41–5. https://doi.org/10.1016/j.neulet.2014.12.035.
Anderson RM, Glanz RM, Johnson SB, Miller MM, Romig-Martin SA, Radley JJ. Prolonged corticosterone exposure induces dendritic spine remodeling and attrition in the rat medial prefrontal cortex. J Comp Neurol. 2016;524(18):3729–46. https://doi.org/10.1002/cne.24027.
Wand G. The anxious amygdala: CREB signaling and predisposition to anxiety and alcoholism. J Clin Investig. 2005;115(10):2697–9. https://doi.org/10.1172/JCI26436.
Carlezon WA Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28(8):436–45. https://doi.org/10.1016/j.tins.2005.06.005.
Keshavarzi S, Kermanshahi S, Karami L, Motaghinejad M, Motevalian M, Sadr S. Protective role of metformin against methamphetamine induced anxiety, depression, cognition impairment and neurodegeneration in rat: The role of CREB/BDNF and Akt/GSK3 signaling pathways. Neurotoxicology. 2019;72:74–84. https://doi.org/10.1016/j.neuro.2019.02.004.
Amidfar M, de Oliveira J, Kucharska E, Budni J, Kim YK. The role of CREB and BDNF in neurobiology and treatment of Alzheimer’s disease. Life Sci. 2020;257: 118020. https://doi.org/10.1016/j.lfs.2020.118020.
Zhang LL, Wang JJ, Liu Y, Lu XB, Kuang Y, Wan YH, Chen Y, Yan HM, Fei J, Wang ZG. GPR26-deficient mice display increased anxiety- and depression-like behaviors accompanied by reduced phosphorylated cyclic AMP responsive element-binding protein level in central amygdala. Neuroscience. 2011;196:203–14. https://doi.org/10.1016/j.neuroscience.2011.08.069.
Tiraboschi E, Giambelli R, D’Urso G, Galietta A, Barbon A, de Bartolomeis A, Gennarelli M, Barlati S, Racagni G, Popoli M. Antidepressants activate CaMKII in neuron cell body by Thr286 phosphorylation. NeuroReport. 2004;15(15):2393–6. https://doi.org/10.1097/00001756-200410250-00018.
Jiang N, Wang H, Lv J, Wang Q, Lu C, Li Y, Liu X. Dammarane sapogenins attenuates stress-induced anxiety-like behaviors by upregulating ERK/CREB/BDNF pathways. Phytotherapy Research: PTR. 2020;34(10):2721–9. https://doi.org/10.1002/ptr.6713.
Jiang N, Lv JW, Wang HX, Lu C, Wang Q, Xia TJ, Bao Y, Li SS, Liu XM. Dammarane sapogenins alleviates depression-like behaviours induced by chronic social defeat stress in mice through the promotion of the BDNF signalling pathway and neurogenesis in the hippocampus. Brain Res Bull. 2019;153:239–49. https://doi.org/10.1016/j.brainresbull.2019.09.007.
Wang JQ, Mao L. The ERK pathway: molecular mechanisms and treatment of depression. Mol Neurobiol. 2019;56(9):6197–205. https://doi.org/10.1007/s12035-019-1524-3.
Shen J, Xu L, Qu C, Sun H, Zhang J. Resveratrol prevents cognitive deficits induced by chronic unpredictable mild stress: Sirt1/miR-134 signalling pathway regulates CREB/BDNF expression in hippocampus in vivo and in vitro. Behav Brain Res. 2018;349:1–7. https://doi.org/10.1016/j.bbr.2018.04.050.
Sawai T, Bernier F, Fukushima T, Hashimoto T, Ogura H, Nishizawa Y. Estrogen induces a rapid increase of calcium-calmodulin-dependent protein kinase II activity in the hippocampus. Brain Res. 2002;950(1–2):308–11. https://doi.org/10.1016/s0006-8993(02)03186-4.
Yang S, Wang J. Estrogen activates AMP-activated protein kinase in human endothelial cells via ERβ/Ca(2+)/calmodulin-dependent protein kinase kinase β pathway. Cell Biochem Biophys. 2015;72(3):701–7. https://doi.org/10.1007/s12013-015-0521-z.
Bombardier JP, Munson M. Three steps forward, two steps back: mechanistic insights into the assembly and disassembly of the SNARE complex. Curr Opin Chem Biol. 2015;29:66–71. https://doi.org/10.1016/j.cbpa.2015.10.003.
Yoon TY, Munson M. SNARE complex assembly and disassembly. Curr Biol CB. 2018;28(8):R397–401. https://doi.org/10.1016/j.cub.2018.01.005.
Wang T, Li L, Hong W. SNARE proteins in membrane trafficking. Traffic (Copenhagen, Denmark). 2017;18(12):767–75. https://doi.org/10.1111/tra.12524.
Südhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science (New York, NY). 2009;323(5913):474–7. https://doi.org/10.1126/science.1161748.
Guan Y, Chen X, Zhao B, Shi Y, Han F. What happened in the hippocampal axon in a rat model of posttraumatic stress disorder. Cell Mol Neurobiol. 2020. https://doi.org/10.1007/s10571-020-00960-w.
Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20(24):9104–10. https://doi.org/10.1523/JNEUROSCI.20-24-09104.2000.
Li Q, Wang P, Huang C, Chen B, Liu J, Zhao M, Zhao J. N-acetyl serotonin protects neural progenitor cells against oxidative stress-induced apoptosis and improves neurogenesis in adult mouse hippocampus following traumatic brain injury. J Mol Neurosci MN. 2019;67(4):574–88. https://doi.org/10.1007/s12031-019-01263-6.
Zhao H, Mohamed NE, Chan SJ, Tan CT, Tao R, Yu VC, Wong PT. Absence of stress response in dorsal raphe nucleus in modulator of apoptosis 1-deficient mice. Mol Neurobiol. 2019;56(3):2185–201. https://doi.org/10.1007/s12035-018-1205-7.
Bonanno G, Giambelli R, Raiteri L, Tiraboschi E, Zappettini S, Musazzi L, Raiteri M, Racagni G, Popoli M. Chronic antidepressants reduce depolarization-evoked glutamate release and protein interactions favoring formation of SNARE complex in hippocampus. J Neurosci. 2005;25(13):3270–9. https://doi.org/10.1523/JNEUROSCI.5033-04.2005.
Milanese M, Tardito D, Musazzi L, Treccani G, Mallei A, Bonifacino T, Gabriel C, Mocaer E, Racagni G, Popoli M, Bonanno G. Chronic treatment with agomelatine or venlafaxine reduces depolarization-evoked glutamate release from hippocampal synaptosomes. BMC Neurosci. 2013;14:75. https://doi.org/10.1186/1471-2202-14-75.
Xiao B, Han F, Shi YX. Dysfunction of Ca2+/CaM kinase IIalpha cascades in the amygdala in post-traumatic stress disorder. Int J Mol Med. 2009;24(6):795–9. https://doi.org/10.3892/ijmm_00000294.
Liu H, Han F, Shi Y. Effect of calreticulin on Ca2+/CaM kinaseIIα and endoplasmic reticulum stress in hippocampal in a rat model of post-traumatic stress disorder. Neurochem Res. 2013;38(7):1407–14. https://doi.org/10.1007/s11064-013-1038-8.
Xie H, Han F, Shi X. Single-prolonged stress induce changes of CaM/CaMKIIα in the rats of dorsal raphe nucleus. Neurochem Res. 2012;37(5):1043–9. https://doi.org/10.1007/s11064-012-0705-5.
Xiang M, Jiang Y, Hu Z, Yang Y, Botchway BOA, Fang M. Stimulation of anxiety-like behavior via ERK pathway by competitive serotonin receptors 2A and 1A in post-traumatic stress disordered mice. Neurosignals. 2017;25(1):39–53. https://doi.org/10.1159/000481791.
Garcia-Garcia AL, Newman-Tancredi A, Leonardo ED. 5-HT(1A) [corrected] receptors in mood and anxiety: recent insights into autoreceptor versus heteroreceptor function. Psychopharmacology. 2014;231(4):623–36. https://doi.org/10.1007/s00213-013-3389-x.
Cai X, Gu Z, Zhong P, Ren Y, Yan Z. Serotonin 5-HT1A receptors regulate AMPA receptor channels through inhibiting Ca2+/calmodulin-dependent kinase II in prefrontal cortical pyramidal neurons. J Biol Chem. 2002;277(39):36553–62. https://doi.org/10.1074/jbc.M203752200.
Cuddy LK, Wani WY, Morella ML, Pitcairn C, Tsutsumi K, Fredriksen K, Justman CJ, Grammatopoulos TN, Belur NR, Zunke F, Subramanian A, Affaneh A, Lansbury PT Jr, Mazzulli JR. Stress-induced cellular clearance is mediated by the SNARE Protein ykt6 and disrupted by α-synuclein. Neuron. 2019;104(5):869-884.e11. https://doi.org/10.1016/j.neuron.2019.09.001.
Belletti B, Baldassarre G. Stathmin: a protein with many tasks. New biomarker and potential target in cancer. Expert Opin Ther Targets. 2011;15(11):1249–66. https://doi.org/10.1517/14728222.2011.620951.
Rubin CI, Atweh GF. The role of stathmin in the regulation of the cell cycle. J Cell Biochem. 2004;93(2):242–50. https://doi.org/10.1002/jcb.20187.
Lyu J, Yang EJ, Zhang B, Wu C, Pardeshi L, Shi C, Mou PK, Liu Y, Tan K, Shim JS. Synthetic lethality of RB1 and aurora A is driven by stathmin-mediated disruption of microtubule dynamics. Nat Commun. 2020;11(1):5105. https://doi.org/10.1038/s41467-020-18872-0.
Ng DC, Zhao TT, Yeap YY, Ngoei KR, Bogoyevitch MA. c-Jun N-terminal kinase phosphorylation of stathmin confers protection against cellular stress. J Biol Chem. 2010;285(37):29001–13. https://doi.org/10.1074/jbc.M110.128454.
Yip YY, Yeap YY, Bogoyevitch MA, Ng DC. cAMP-dependent protein kinase and c-Jun N-terminal kinase mediate stathmin phosphorylation for the maintenance of interphase microtubules during osmotic stress. J Biol Chem. 2014;289(4):2157–69. https://doi.org/10.1074/jbc.M113.470682.
Han F, Jiang J, Ding J, Liu H, Xiao B, Shi Y. Change of Rin1 and stathmin in the animal model of traumatic stresses. Front Behav Neurosci. 2017;11:62. https://doi.org/10.3389/fnbeh.2017.00062.
Martel G, Nishi A, Shumyatsky GP. Stathmin reveals dissociable roles of the basolateral amygdala in parental and social behaviors. Proc Natl Acad Sci USA. 2008;105(38):14620–5. https://doi.org/10.1073/pnas.0807507105.
Martel G, Uchida S, Hevi C, Chévere-Torres I, Fuentes I, Park YJ, Hafeez H, Yamagata H, Watanabe Y, Shumyatsky GP. Genetic demonstration of a role for stathmin in adult hippocampal neurogenesis, spinogenesis, and NMDA receptor-dependent memory. J Neurosci. 2016;36(4):1185–202. https://doi.org/10.1523/JNEUROSCI.4541-14.2016.
Shan W, Han F, Xu Y, Shi Y. Stathmin regulates spatiotemporal variation in the memory loop in single-prolonged stress rats. J Mol Neurosci. 2020;70(4):576–89. https://doi.org/10.1007/s12031-019-01459-w.
Shumyatsky GP, Malleret G, Shin RM, Takizawa S, Tully K, Tsvetkov E, Zakharenko SS, Joseph J, Vronskaya S, Yin D, Schubart UK, Kandel ER, Bolshakov VY. stathmin, a gene enriched in the amygdala, controls both learned and innate fear. Cell. 2005;123(4):697–709. https://doi.org/10.1016/j.cell.2005.08.038.
Uchida S, Martel G, Pavlowsky A, Takizawa S, Hevi C, Watanabe Y, Kandel ER, Alarcon JM, Shumyatsky GP. Learning-induced and stathmin-dependent changes in microtubule stability are critical for memory and disrupted in ageing. Nat Commun. 2014;5:4389. https://doi.org/10.1038/ncomms5389.
Uchida S, Shumyatsky GP. Deceivingly dynamic: learning-dependent changes in stathmin and microtubules. Neurobiol Learn Mem. 2015;124:52–61. https://doi.org/10.1016/j.nlm.2015.07.011.
Nguyen TB, Prabhu VV, Piao YH, Oh YE, Zahra RF, Chung YC. Effects of stathmin 1 gene knockout on behaviors and dopaminergic markers in mice exposed to social defeat stress. Brain Sci. 2019;9(9):215. https://doi.org/10.3390/brainsci9090215.
Dent EW. Of microtubules and memory: implications for microtubule dynamics in dendrites and spines. Mol Biol Cell. 2017;28(1):1–8. https://doi.org/10.1091/mbc.E15-11-0769.
Penazzi L, Bakota L, Brandt R. Microtubule dynamics in neuronal development, plasticity, and neurodegeneration. Int Rev Cell Mol Biol. 2016;321:89–169. https://doi.org/10.1016/bs.ircmb.2015.09.004.
Chen X, Jiang Y, Wang J, Liu Y, Xiao M, Song C, Bai Y, Yinuo Han N, Han F. Synapse impairment associated with enhanced apoptosis in post-traumatic stress disorder. Synapse. 2020;74(2): e22134. https://doi.org/10.1002/syn.22134.
Jia Y, Han Y, Wang X, Han F. Role of apoptosis in the post-traumatic stress disorder model-single prolonged stressed rats. Psychoneuroendocrinology. 2018;95:97–105. https://doi.org/10.1016/j.psyneuen.2018.05.015.
Swulius MT, Waxham MN. Ca(2+)/calmodulin-dependent protein kinases. Cell Mol Life Sci CMLS. 2008;65(17):2637–57. https://doi.org/10.1007/s00018-008-8086-2.
Ferrer I, Blanco R, Carmona M, Puig B. Phosphorylated mitogen-activated protein kinase (MAPK/ERK-P), protein kinase of 38 kDa (p38-P), stress-activated protein kinase (SAPK/JNK-P), and calcium/calmodulin-dependent kinase II (CaM kinase II) are differentially expressed in tau deposits in neurons and glial cells in tauopathies. J Neural Transm. 2001;108(12):1397–415. https://doi.org/10.1007/s007020100016.
Ferrer I, Blanco R, Carmona M, Ribera R, Goutan E, Puig B, Rey MJ, Cardozo A, Viñals F, Ribalta T. Phosphorylated map kinase (ERK1, ERK2) expression is associated with early tau deposition in neurones and glial cells, but not with increased nuclear DNA vulnerability and cell death, in Alzheimer disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Brain Pathol. 2001;11(2):144–58. https://doi.org/10.1111/j.1750-3639.2001.tb00387.x.
Ferrer I, Gomez-Isla T, Puig B, Freixes M, Ribé E, Dalfó E, Avila J. Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer’s disease and tauopathies. Curr Alzheimer Res. 2005;2(1):3–18. https://doi.org/10.2174/1567205052772713.
Gibbs ME, Summers RJ. Role of adrenoceptor subtypes in memory consolidation. Prog Neurobiol. 2002;67(5):345–91. https://doi.org/10.1016/s0301-0082(02)00023-0.
Gibbs ME, Hutchinson DS, Summers RJ. Noradrenaline release in the locus coeruleus modulates memory formation and consolidation; roles for α- and β-adrenergic receptors. Neuroscience. 2010;170(4):1209–22. https://doi.org/10.1016/j.neuroscience.2010.07.052.
Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3(9):639–50. https://doi.org/10.1038/nrm908.
Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287(5461):2262–7. https://doi.org/10.1126/science.287.5461.2262.
Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22(2):153–83. https://doi.org/10.1210/edrv.22.2.0428.
Schiff HC, Johansen JP, Hou M, Bush DE, Smith EK, Klein JE, LeDoux JE, Sears RM. β-adrenergic receptors regulate the acquisition and consolidation phases of aversive memory formation through distinct, temporally regulated signaling pathways. Neuropsychopharmacology. 2017;42(4):895–903. https://doi.org/10.1038/npp.2016.238.
Baldi E, Bucherelli C. The inverted “u-shaped” dose-effect relationships in learning and memory: modulation of arousal and consolidation. Nonlinearity Biol Toxicol Med. 2005;3(1):9–21. https://doi.org/10.2201/nonlin.003.01.002.
Sara SJ, Bouret S. Orienting and reorienting: the locus coeruleus mediates cognition through arousal. Neuron. 2012;76(1):130–41. https://doi.org/10.1016/j.neuron.2012.09.011.
Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. 2009;10(6):410–22. https://doi.org/10.1038/nrn2648.
Bahari Z, Meftahi GH, Meftahi MA. Dopamine effects on stress-induced working memory deficits. Behav Pharmacol. 2018;29(7):584–91. https://doi.org/10.1097/FBP.0000000000000429.
Gamo NJ, Lur G, Higley MJ, Wang M, Paspalas CD, Vijayraghavan S, Yang Y, Ramos BP, Peng K, Kata A, Boven L, Lin F, Roman L, Lee D, Arnsten AF. Stress impairs prefrontal cortical function via D1 dopamine receptor interactions with hyperpolarization-activated cyclic nucleotide-gated channels. Biol Psychiat. 2015;78(12):860–70. https://doi.org/10.1016/j.biopsych.2015.01.009.
Mizoguchi K, Yuzurihara M, Ishige A, Sasaki H, Chui DH, Tabira T. Chronic stress induces impairment of spatial working memory because of prefrontal dopaminergic dysfunction. J Neurosci. 2000;20(4):1568–74. https://doi.org/10.1523/JNEUROSCI.20-04-01568.2000.
Puig MV, Antzoulatos EG, Miller EK. Prefrontal dopamine in associative learning and memory. Neuroscience. 2014;282:217–29. https://doi.org/10.1016/j.neuroscience.2014.09.026.
Reneaux M, Gupta R. Prefronto-cortical dopamine D1 receptor sensitivity can critically influence working memory maintenance during delayed response tasks. PLoS ONE. 2018;13(5): e0198136. https://doi.org/10.1371/journal.pone.0198136.
Ray RD, Zald DH. Anatomical insights into the interaction of emotion and cognition in the prefrontal cortex. Neurosci Biobehav Rev. 2012;36(1):479–501. https://doi.org/10.1016/j.neubiorev.2011.08.005.
Cools R, D’Esposito M. Inverted-U-shaped dopamine actions on human working memory and cognitive control. Biol Psychiat. 2011;69(12):e113–25. https://doi.org/10.1016/j.biopsych.2011.03.028.
Barkus C, Korn C, Stumpenhorst K, Laatikainen LM, Ballard D, Lee S, Sharp T, Harrison PJ, Bannerman DM, Weinberger DR, Chen J, Tunbridge EM. Genotype-dependent effects of COMT inhibition on cognitive function in a highly specific, novel mouse model of altered COMT Activity. Neuropsychopharmacology. 2016;41(13):3060–9. https://doi.org/10.1038/npp.2016.119.
Papaleo F, Erickson L, Liu G, Chen J, Weinberger DR. Effects of sex and COMT genotype on environmentally modulated cognitive control in mice. Proc Natl Acad Sci USA. 2012;109(49):20160–5. https://doi.org/10.1073/pnas.1214397109.
He Q, Xue G, Chen C, Lu ZL, Chen C, Lei X, Liu Y, Li J, Zhu B, Moyzis RK, Dong Q, Bechara A. COMT Val158Met polymorphism interacts with stressful life events and parental warmth to influence decision making. Sci Rep. 2012;2:677. https://doi.org/10.1038/srep00677.
Dumontheil I, Jensen SK, Wood NW, Meyer ML, Lieberman MD, Blakemore SJ. Preliminary investigation of the influence of dopamine regulating genes on social working memory. Soc Neurosci. 2014;9(5):437–51. https://doi.org/10.1080/17470919.2014.925503.
Dumontheil I, Kilford EJ, Blakemore SJ. Development of dopaminergic genetic associations with visuospatial, verbal and social working memory. Dev Sci. 2020;23(2): e12889. https://doi.org/10.1111/desc.12889.
Berryhill ME, Wiener M, Stephens JA, Lohoff FW, Coslett HB. COMT and ANKK1-Taq-Ia genetic polymorphisms influence visual working memory. PLoS ONE. 2013;8(1): e55862. https://doi.org/10.1371/journal.pone.0055862.
Miskowiak KW, Kjaerstad HL, Støttrup MM, Svendsen AM, Demant KM, Hoeffding LK, Werge TM, Burdick KE, Domschke K, Carvalho AF, Vieta E, Vinberg M, Kessing LV, Siebner HR, Macoveanu J. The catechol-O-methyltransferase (COMT) Val158Met genotype modulates working memory-related dorsolateral prefrontal response and performance in bipolar disorder. Bipolar Disord. 2017;19(3):214–24. https://doi.org/10.1111/bdi.12497.
Heim AF, Coyne MJ, Kamboh MI, Ryan C, Jennings JR. The catechol-o-methyltransferase Val158 Met polymorphism modulates organization of regional cerebral blood flow response to working memory in adults. Int J Psychophysiol. 2013;90(2):149–56. https://doi.org/10.1016/j.ijpsycho.2013.06.023.
Castellini G, Merola GP, Baccaredda Boy O, Pecoraro V, Bozza B, Cassioli E, Rossi E, Bessi V, Sorbi S, Nacmias B, Ricca V. Emotional dysregulation, alexithymia and neuroticism: a systematic review on the genetic basis of a subset of psychological traits. Psychiatr Genet. 2023;33(3):79–101. https://doi.org/10.1097/YPG.0000000000000335.
Sun X, Ming Q, Zhong X, Dong D, Li C, Xiong G, Cheng C, Cao W, He J, Wang X, Yi J, Yao S. The MAOA gene influences the neural response to psychosocial stress in the human brain. Front Behav Neurosci. 2020;14:65. https://doi.org/10.3389/fnbeh.2020.00065.
Ziermans T, Dumontheil I, Roggeman C, Peyrard-Janvid M, Matsson H, Kere J, Klingberg T. Working memory brain activity and capacity link MAOA polymorphism to aggressive behavior during development. Transl Psychiatry. 2012;2(2): e85. https://doi.org/10.1038/tp.2012.7.
Babić Leko M, Nikolac Perković M, Klepac N, Štrac DŠ, Borovečki F, Pivac N, Hof PR, Šimić G. IL-1β, IL-6, IL-10, and TNFα single nucleotide polymorphisms in human influence the susceptibility to Alzheimer’s disease pathology. J Alzheimers Dis. 2020;75(3):1029–47. https://doi.org/10.3233/JAD-200056.
Borroni B, Bianchi M, Premi E, Alberici A, Archetti S, Paghera B, Cerini C, Papetti A, Padovani A. The brain-derived neurotrophic factor Val66Met polymorphism is associated with reduced hippocampus perfusion in frontotemporal lobar degeneration. J Alzheimers Dis. 2012;31(2):243–51. https://doi.org/10.3233/JAD-2012-120226.
Huey ED, Fremont R, Manoochehri M, Gazes Y, Lee S, Cosentino S, Tierney M, Wassermann EM, Momeni P, Grafman J. Effect of functional BDNF and COMT polymorphisms on symptoms and regional brain volume in frontotemporal dementia and corticobasal syndrome. J Neuropsychiatry Clin Neurosci. 2020;32(4):362–9. https://doi.org/10.1176/appi.neuropsych.19100211.
Goldfarb EV, Seo D, Sinha R. Sex differences in neural stress responses and correlation with subjective stress and stress regulation. Neurobiol Stress. 2019;11: 100177. https://doi.org/10.1016/j.ynstr.2019.100177.
Stevens JS, Hamann S. Sex differences in brain activation to emotional stimuli: a meta-analysis of neuroimaging studies. Neuropsychologia. 2012;50(7):1578–93. https://doi.org/10.1016/j.neuropsychologia.2012.03.011.
Hidalgo V, Pulopulos MM, Salvador A. Acute psychosocial stress effects on memory performance: Relevance of age and sex. Neurobiol Learn Mem. 2019;157:48–60. https://doi.org/10.1016/j.nlm.2018.11.013.
Koss WA, Haertel JM, Philippi SM, Frick KM. Sex differences in the rapid cell signaling mechanisms underlying the memory-enhancing effects of 17β-estradiol. eNeuro. 2018. https://doi.org/10.1523/ENEURO.0267-18.2018.
Frick KM, Kim J. Mechanisms underlying the rapid effects of estradiol and progesterone on hippocampal memory consolidation in female rodents. Horm Behav. 2018;104:100–10. https://doi.org/10.1016/j.yhbeh.2018.04.013.
Frick KM, Tuscher JJ, Koss WA, Kim J, Taxier LR. Estrogenic regulation of memory consolidation: a look beyond the hippocampus, ovaries, and females. Physiol Behav. 2018;187:57–66. https://doi.org/10.1016/j.physbeh.2017.07.028.
Schwabe MR, Taxier LR, Frick KM. It takes a neural village: circuit-based approaches for estrogenic regulation of episodic memory. Front Neuroendocrinol. 2020;59: 100860. https://doi.org/10.1016/j.yfrne.2020.100860.
Prakapenka AV, Peña VL, Strouse I, Northup-Smith S, Schrier A, Ahmed K, Bimonte-Nelson HA, Sirianni RW. Intranasal 17β-estradiol modulates spatial learning and memory in a rat model of surgical menopause. Pharmaceutics. 2020;12(12):1225. https://doi.org/10.3390/pharmaceutics12121225.
Bayer J, Gläscher J, Finsterbusch J, Schulte LH, Sommer T. Linear and inverted U-shaped dose-response functions describe estrogen effects on hippocampal activity in young women. Nat Commun. 2018;9(1):1220. https://doi.org/10.1038/s41467-018-03679-x.
Almey A, Milner TA, Brake WG. Estrogen receptors in the central nervous system and their implication for dopamine-dependent cognition in females. Horm Behav. 2015;74:125–38. https://doi.org/10.1016/j.yhbeh.2015.06.010.
Shields GS, Sazma MA, McCullough AM, Yonelinas AP. The effects of acute stress on episodic memory: a meta-analysis and integrative review. Psychol Bull. 2017;143(6):636–75. https://doi.org/10.1037/bul0000100.
Heck AL, Handa RJ. Sex differences in the hypothalamic-pituitary-adrenal axis’ response to stress: an important role for gonadal hormones. Neuropsychopharmacology. 2019;44(1):45–58. https://doi.org/10.1038/s41386-018-0167-9.
Hwang WJ, Lee TY, Kim NS, Kwon JS. The role of estrogen receptors and their signaling across psychiatric disorders. Int J Mol Sci. 2020;22(1):373. https://doi.org/10.3390/ijms22010373.
Kida S, Serita T. Functional roles of CREB as a positive regulator in the formation and enhancement of memory. Brain Res Bull. 2014;105:17–24. https://doi.org/10.1016/j.brainresbull.2014.04.011.
Hokenson RE, Alam YH, Short AK, Jung S, Jang C, Baram TZ. Sex-dependent effects of multiple acute concurrent stresses on memory: a role for hippocampal estrogens. Front Behav Neurosci. 2022;16: 984494. https://doi.org/10.3389/fnbeh.2022.984494.
Hokenson RE, Short AK, Chen Y, Pham AL, Adams ET, Bolton JL, Swarup V, Gall CM, Baram TZ. Unexpected role of physiological estrogen in acute stress-induced memory deficits. J Neurosci. 2021;41(4):648–62. https://doi.org/10.1523/JNEUROSCI.2146-20.2020.
Graham BM, Scott E. Effects of systemic estradiol on fear extinction in female rats are dependent on interactions between dose, estrous phase, and endogenous estradiol levels. Horm Behav. 2018;97:67–74. https://doi.org/10.1016/j.yhbeh.2017.10.009.
Shansky RM, Rubinow K, Brennan A, Arnsten AF. The effects of sex and hormonal status on restraint-stress-induced working memory impairment. Behav Brain Funct BBF. 2006;2:8. https://doi.org/10.1186/1744-9081-2-8.
Luine VN, Frankfurt M. Estrogens facilitate memory processing through membrane mediated mechanisms and alterations in spine density. Front Neuroendocrinol. 2012;33(4):388–402. https://doi.org/10.1016/j.yfrne.2012.07.004.
Boulware MI, Weick JP, Becklund BR, Kuo SP, Groth RD, Mermelstein PG. Estradiol activates group I and II metabotropic glutamate receptor signaling, leading to opposing influences on cAMP response element-binding protein. J Neurosci. 2005;25(20):5066–78. https://doi.org/10.1523/JNEUROSCI.1427-05.2005.
Boulware MI, Heisler JD, Frick KM. The memory-enhancing effects of hippocampal estrogen receptor activation involve metabotropic glutamate receptor signaling. J Neurosci. 2013;33(38):15184–94. https://doi.org/10.1523/JNEUROSCI.1716-13.2013.
Arnal JF, Lenfant F, Metivier R, Flouriot G, Henrion D, Adlanmerini M, Fontaine C, Gourdy P, Chambon P, Katzenellenbogen B, Katzenellenbogen J. Membrane and nuclear estrogen receptor alpha actions: from tissue specificity to medical implications. Physiol Rev. 2017;97(3):1045–87. https://doi.org/10.1152/physrev.00024.2016.
Kim J, Szinte JS, Boulware MI, Frick KM. 17β-Estradiol and agonism of g-protein-coupled estrogen receptor enhance hippocampal memory via different cell-signaling mechanisms. J Neurosci. 2016;36(11):3309–21. https://doi.org/10.1523/JNEUROSCI.0257-15.2016.
Morales A, Gonzalez M, Marin R, Diaz M, Alonso R. Estrogen inhibition of norepinephrine responsiveness is initiated at the plasma membrane of GnRH-producing GT1-7 cells. J Endocrinol. 2007;194(1):193–200. https://doi.org/10.1677/JOE-06-0001.
Moreira-Pais A, Ferreira R, Neves JS, Vitorino R, Moreira-Gonçalves D, Nogueira-Ferreira R. Sex differences on adipose tissue remodeling: from molecular mechanisms to therapeutic interventions. J Mol Med (Berl). 2020;98(4):483–93. https://doi.org/10.1007/s00109-020-01890-2.
Laredo SA, Villalon Landeros R, Trainor BC. Rapid effects of estrogens on behavior: environmental modulation and molecular mechanisms. Front Neuroendocrinol. 2014;35(4):447–58. https://doi.org/10.1016/j.yfrne.2014.03.005.
Milner TA, Lubbers LS, Alves SE, McEwen BS. Nuclear and extranuclear estrogen binding sites in the rat forebrain and autonomic medullary areas. Endocrinology. 2008;149(7):3306–12. https://doi.org/10.1210/en.2008-0307.
Luine V. Estradiol: Mediator of memories, spine density and cognitive resilience to stress in female rodents. J Steroid Biochem Mol Biol. 2016;160:189–95. https://doi.org/10.1016/j.jsbmb.2015.07.022.
Yuen EY, Wei J, Yan Z. Estrogen in prefrontal cortex blocks stress-induced cognitive impairments in female rats. J Steroid Biochem Mol Biol. 2016;160:221–6. https://doi.org/10.1016/j.jsbmb.2015.08.028.
do Nascimento, E. B., Dierschnabel, A. L., de Macêdo Medeiros, A., Suchecki, D., Silva, R. H., & Ribeiro, A. M. Memory impairment induced by different types of prolonged stress is dependent on the phase of the estrous cycle in female rats. Horm Behav. 2019;115: 104563. https://doi.org/10.1016/j.yhbeh.2019.104563.
Wei J, Yuen EY, Liu W, Li X, Zhong P, Karatsoreos IN, McEwen BS, Yan Z. Estrogen protects against the detrimental effects of repeated stress on glutamatergic transmission and cognition. Mol Psychiatry. 2014;19(5):588–98. https://doi.org/10.1038/mp.2013.83.
Kubota T, Matsumoto H, Kirino Y. Ameliorative effect of membrane-associated estrogen receptor G protein coupled receptor 30 activation on object recognition memory in mouse models of Alzheime’’s disease. J Pharmacol Sci. 2016;131(3):219–22. https://doi.org/10.1016/j.jphs.2016.06.005.
Wnuk A, Przepiórska K, Rzemieniec J, Pietrzak B, Kajta M. Selective Targeting of non-nuclear estrogen receptors with PaPE-1 as a New treatment strategy for Alzheimer’s disease. Neurotox Res. 2020;38(4):957–66. https://doi.org/10.1007/s12640-020-00289-8.
Selvaraj UM, Zuurbier KR, Whoolery CW, Plautz EJ, Chambliss KL, Kong X, Zhang S, Kim SH, Katzenellenbogen BS, Katzenellenbogen JA, Mineo C, Shaul PW, Stowe AM. Selective nonnuclear estrogen receptor activation decreases stroke severity and promotes functional recovery in female mice. Endocrinology. 2018;159(11):3848–59. https://doi.org/10.1210/en.2018-00600.
Adlanmerini M, Solinhac R, Abot A, Fabre A, Raymond-Letron I, Guihot AL, Boudou F, Sautier L, Vessières E, Kim SH, Lière P, Fontaine C, Krust A, Chambon P, Katzenellenbogen JA, Gourdy P, Shaul PW, Henrion D, Arnal JF, Lenfant F. Mutation of the palmitoylation site of estrogen receptor α in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc Natl Acad Sci USA. 2014;111(2):E283–90. https://doi.org/10.1073/pnas.1322057111.
Guo X, Razandi M, Pedram A, Kassab G, Levin ER. Estrogen induces vascular wall dilation: Mediation through kinase signaling to nitric oxide and estrogen receptors alpha and beta. J Biol Chem. 2005;280(20):19704–10. https://doi.org/10.1074/jbc.M501244200.
Florian M, Lu Y, Angle M, Magder S. Estrogen induced changes in Akt-dependent activation of endothelial nitric oxide synthase and vasodilation. Steroids. 2004;69(10):637–45. https://doi.org/10.1016/j.steroids.2004.05.016.
Murphy E. Estrogen signaling and cardiovascular disease. Circ Res. 2011;109(6):687–96. https://doi.org/10.1161/CIRCRESAHA.110.236687.
Stirone C, Boroujerdi A, Duckles SP, Krause DN. Estrogen receptor activation of phosphoinositide-3 kinase, akt, and nitric oxide signaling in cerebral blood vessels: rapid and long-term effects. Mol Pharmacol. 2005;67(1):105–13. https://doi.org/10.1124/mol.104.004465.
Zhu Y, Bian Z, Lu P, Karas RH, Bao L, Cox D, Hodgin J, Shaul PW, Thoren P, Smithies O, Gustafsson JA, Mendelsohn ME. Abnormal vascular function and hypertension in mice deficient in estrogen receptor beta. Science (New York, NY). 2002;295(5554):505–8. https://doi.org/10.1126/science.1065250.
Mahmoodzadeh S, Dworatzek E. The role of 17β-estradiol and estrogen receptors in regulation of Ca2+ channels and mitochondrial function in cardiomyocytes. Front Endocrinol. 2019;10:310. https://doi.org/10.3389/fendo.2019.00310.
Breslau N. Gender differences in trauma and posttraumatic stress disorder. J Gender Specific Med JGSM. 2002;5(1):34–40.
Pooley AE, Benjamin RC, Sreedhar S, Eagle AL, Robison AJ, Mazei-Robison MS, Breedlove SM, Jordan CL. Sex differences in the traumatic stress response: PTSD symptoms in women recapitulated in female rats. Biol Sex Differ. 2018;9(1):31. https://doi.org/10.1186/s13293-018-0191-9.
Zlotnick C, Johnson J, Kohn R, Vicente B, Rioseco P, Saldivia S. Epidemiology of trauma, post-traumatic stress disorder (PTSD) and co-morbid disorders in Chile. Psychol Med. 2006;36(11):1523–33. https://doi.org/10.1017/S0033291706008282.
Sartin-Tarm A, Ross MC, Privatsky AA, Cisler JM. Estradiol modulates neural and behavioral arousal in women with posttraumatic stress disorder during a fear learning and extinction task. Biol Psychiatry Cogn Neurosci Neuroimaging. 2020;5(12):1114–22. https://doi.org/10.1016/j.bpsc.2020.04.012.
Wnuk A, Przepiórska K, Pietrzak BA, Kajta M. Emerging evidence on membrane estrogen receptors as novel therapeutic targets for central nervous system pathologies. Int J Mol Sci. 2023;24(4):4043. https://doi.org/10.3390/ijms24044043.
Luo W, Yan Y, Cao Y, Zhang Y, Zhang Z. The effects of GPER on age-associated memory impairment induced by decreased estrogen levels. Front Mol Biosci. 2023;10:1097018. https://doi.org/10.3389/fmolb.2023.1097018.
Karlsson SA, Studer E, Kettunen P, Westberg L. Neural androgen receptors modulate gene expression and social recognition but not social investigation. Front Behav Neurosci. 2016;10:41. https://doi.org/10.3389/fnbeh.2016.00041.
Atwi S, McMahon D, Scharfman H, MacLusky NJ. Androgen modulation of hippocampal structure and function. Neuroscientist Rev J Bringing Neurobiol Neurol psychiatry. 2016;22(1):46–60. https://doi.org/10.1177/1073858414558065.
Bianchi VE. Impact of testosterone on Alzheimer’s disease. World J Men’s Health. 2022;40(2):243–56. https://doi.org/10.5534/wjmh.210175.
Corona G, Guaraldi F, Rastrelli G, Sforza A, Maggi M. Testosterone deficiency and risk of cognitive disorders in aging males. World J Men’s Health. 2021;39(1):9–18. https://doi.org/10.5534/wjmh.200017.
Dubal DB. Sex difference in Alzheimer’s disease: an updated, balanced and emerging perspective on differing vulnerabilities. Handb Clin Neurol. 2020;175:261–73. https://doi.org/10.1016/B978-0-444-64123-6.00018-7.
Hsu CK, Ney LJ, Honan C, Felmingham KL. Gonadal steroid hormones and emotional memory consolidation: a systematic review and meta-analysis. Neurosci Biobehav Rev. 2021;130:529–42. https://doi.org/10.1016/j.neubiorev.2021.09.010.
Pompili A, Iorio C, Gasbarri A. Effects of sex steroid hormones on memory. Acta Neurobiol Exp. 2020;80(2):117–28.
Spritzer MD, Roy EA. Testosterone and adult neurogenesis. Biomolecules. 2020;10(2):225. https://doi.org/10.3390/biom10020225.
Edinger KL, Frye CA. Androgens’ effects to enhance learning may be mediated in part through actions at estrogen receptor-beta in the hippocampus. Neurobiol Learn Mem. 2007;87(1):78–85. https://doi.org/10.1016/j.nlm.2006.07.001.
Edinger KL, Lee B, Frye CA. Mnemonic effects of testosterone and its 5alpha-reduced metabolites in the conditioned fear and inhibitory avoidance tasks. Pharmacol Biochem Behav. 2004;78(3):559–68. https://doi.org/10.1016/j.pbb.2004.04.024.
Hernandez CM, Orsini C, Wheeler AR, Ten Ten Eyck TW, Betzhold SM, Labiste CC, Wright NG, Setlow B, Bizon JL. Testicular hormones mediate robust sex differences in impulsive choice in rats. Elife. 2020;9: e58604. https://doi.org/10.7554/eLife.58604.
Grissom NM, Reyes TM. Let’s call the whole thing off: evaluating gender and sex differences in executive function. Neuropsychopharmacology. 2019;44(1):86–96. https://doi.org/10.1038/s41386-018-0179-5.
Gaillard A, Fehring DJ, Rossell SL. Sex differences in executive control: a systematic review of functional neuroimaging studies. Eur J Neurosci. 2021;53(8):2592–611. https://doi.org/10.1111/ejn.15107.
Lee B, Shim I, Lee H, Hahm D-H. Melatonin ameliorates cognitive memory by regulation of cAMP-response element-binding protein expression and the anti-inflammatory response in a rat model of post-traumatic stress disorder. BMC Neurosci. 2018;19(1):38. https://doi.org/10.1186/s12868-018-0439-7.
Miller MA, Leckie RL, Donofry SD, Gianaros PJ, Erickson KI, Manuck SB, Roecklein KA. Photoperiod is associated with hippocampal volume in a large community sample. Hippocampus. 2015;25(4):534–43. https://doi.org/10.1002/hipo.22390.
Pyter LM, Reader BF, Nelson RJ. Short photoperiods impair spatial learning and alter hippocampal dendritic morphology in adult male white-footed mice (Peromyscus leucopus). J Neurosci. 2005;25(18):4521–6. https://doi.org/10.1523/JNEUROSCI.0795-05.2005.
Walton JC, Chen Z, Weil ZM, Pyter LM, Travers JB, Nelson RJ. Photoperiod-mediated impairment of long-term potention and learning and memory in male white-footed mice. Neuroscience. 2011;175:127–32. https://doi.org/10.1016/j.neuroscience.2010.12.004.
Liu J, Garza JC, Li W, Lu XY. Melanocortin-4 receptor in the medial amygdala regulates emotional stress-induced anxiety-like behaviour, anorexia and corticosterone secretion. Int J Neuropsychopharmacol. 2013;16(1):105–20. https://doi.org/10.1017/S146114571100174X.
Micioni Di Bonaventura E, Botticelli L, Del Bello F, Giorgioni G, Piergentili A, Quaglia W, Romano A, Gaetani S, Micioni Di Bonaventura MV, Cifani C. Investigating the role of the central melanocortin system in stress and stress-related disorders. Pharmacol Res. 2022;185: 106521. https://doi.org/10.1016/j.phrs.2022.106521.
Xu Z, Li W, Sun Y, Jin W, Yu L, Yang J, Wang Q. Melatonin alleviates PTSD-like behaviors and restores serum GABA and cortisol levels in mice. Psychopharmacology. 2023;240(2):259–69. https://doi.org/10.1007/s00213-023-06312-y.
Tonon AC, Pilz LK, Markus RP, Hidalgo MP, Elisabetsky E. Melatonin and Depression: a translational perspective from animal models to clinical studies. Front Psych. 2021;12: 638981. https://doi.org/10.3389/fpsyt.2021.638981.
Chen D, Zhang T, Lee TH. Cellular mechanisms of melatonin: insight from neurodegenerative diseases. Biomolecules. 2020;10(8):1158. https://doi.org/10.3390/biom10081158.
Chen D, Lan G, Li R, Mei Y, Shui X, Gu X, Wang L, Zhang T, Gan CL, Xia Y, Hu L, Tian Y, Zhang M, Lee TH. Melatonin ameliorates tau-related pathology via the miR-504-3p and CDK5 axis in Alzheimer’s disease. Transl Neurodegen. 2022;11(1):27. https://doi.org/10.1186/s40035-022-00302-4.
Lin L, Huang QX, Yang SS, Chu J, Wang JZ, Tian Q. Melatonin in Alzheimer’s disease. Int J Mol Sci. 2013;14(7):14575–93. https://doi.org/10.3390/ijms140714575.
Luo F, Sandhu AF, Rungratanawanich W, Williams GE, Akbar M, Zhou S, Song BJ, Wang X. Melatonin and autophagy in aging-related neurodegenerative diseases. Int J Mol Sci. 2020;21(19):7174. https://doi.org/10.3390/ijms21197174.
Pandi-Perumal SR, BaHammam AS, Brown GM, Spence DW, Bharti VK, Kaur C, Hardeland R, Cardinali DP. Melatonin antioxidative defense: therapeutical implications for aging and neurodegenerative processes. Neurotox Res. 2013;23(3):267–300. https://doi.org/10.1007/s12640-012-9337-4.
Wang JZ, Wang ZF. Role of melatonin in Alzheimer-like neurodegeneration. Acta Pharmacol Sin. 2006;27(1):41–9. https://doi.org/10.1111/j.1745-7254.2006.00260.x.
Racagni G, Riva MA, Molteni R, Musazzi L, Calabrese F, Popoli M, Tardito D. Mode of action of agomelatine: synergy between melatonergic and 5-HT2C receptors. World J Biol Psychiatry. 2011;12(8):574–87. https://doi.org/10.3109/15622975.2011.595823.
Tardito D, Milanese M, Bonifacino T, Musazzi L, Grilli M, Mallei A, Mocaer E, Gabriel-Gracia C, Racagni G, Popoli M, Bonanno G. Blockade of stress-induced increase of glutamate release in the rat prefrontal/frontal cortex by agomelatine involves synergy between melatonergic and 5-HT2C receptor-dependent pathways. BMC Neurosci. 2010;11:68. https://doi.org/10.1186/1471-2202-11-68.
Tardito D, Molteni R, Popoli M, Racagni G. Synergistic mechanisms involved in the antidepressant effects of agomelatine. Eur Neuropsychopharmacol. 2012;22(Suppl 3):S482–6. https://doi.org/10.1016/j.euroneuro.2012.06.016.
Racagni G, Riva MA, Popoli M. The interaction between the internal clock and antidepressant efficacy. Int Clin Psychopharmacol. 2007;22(Suppl 2):S9–14. https://doi.org/10.1097/01.yic.0000277957.75852.c7.
Korshunov KS, Blakemore LJ, Trombley PQ. Dopamine: a modulator of circadian rhythms in the central nervous system. Front Cell Neurosci. 2017;11:91. https://doi.org/10.3389/fncel.2017.00091.
Agorastos A, Nicolaides NC, Bozikas VP, Chrousos GP, Pervanidou P. Multilevel interactions of stress and circadian system: implications for traumatic stress. Front Psych. 2020;10:1003. https://doi.org/10.3389/fpsyt.2019.01003.
Slavich GM. Social safety theory: a biologically based evolutionary perspective on life stress, health, and behavior. Annu Rev Clin Psychol. 2020;16:265–95. https://doi.org/10.1146/annurev-clinpsy-032816-045159.
Slavich GM. Social safety theory: understanding social stress, disease risk, resilience, and behavior during the COVID-19 pandemic and beyond. Curr Opin Psychol. 2022;45: 101299. https://doi.org/10.1016/j.copsyc.2022.101299.
Slavich GM, Roos LG, Mengelkoch S, Webb CA, Shattuck EC, Moriarity DP, Alley JC. Social safety theory: conceptual foundation, underlying mechanisms, and future directions. Health Psychol Rev. 2023;17(1):5–59. https://doi.org/10.1080/17437199.2023.2171900.
Dickerson SS, Gruenewald TL, Kemeny ME. When the social self is threatened: shame, physiology, and health. J Pers. 2004;72(6):1191–216. https://doi.org/10.1111/j.1467-6494.2004.00295.x.
Breen MS, Tylee DS, Maihofer AX, Neylan TC, Mehta D, Binder EB, Chandler SD, Hess JL, Kremen WS, Risbrough VB, Woelk CH, Baker DG, Nievergelt CM, Tsuang MT, Buxbaum JD, Glatt SJ. PTSD blood transcriptome mega-analysis: shared inflammatory pathways across biological sex and modes of trauma. Neuropsychopharmacology. 2018;43(3):469–81. https://doi.org/10.1038/npp.2017.220.
Wang J, Zhao H, Girgenti MJ. Posttraumatic stress disorder brain transcriptomics: convergent genomic signatures across biological sex. Biol Psychiat. 2022;91(1):6–13. https://doi.org/10.1016/j.biopsych.2021.02.012.
Pukay-Martin ND, Cristiani SA, Saveanu R, Bornstein RA. The relationship between stressful life events and cognitive function in HIV-infected men. J Neuropsychiatry Clin Neurosci. 2003;15(4):436–41. https://doi.org/10.1176/jnp.15.4.436.
Hutchinson P, Dhairyawan R. Shame and HIV: strategies for addressing the negative impact shame has on public health and diagnosis and treatment of HIV. Bioethics. 2018;32(1):68–76. https://doi.org/10.1111/bioe.12378.
Rubin LH, Cook JA, Weber KM, Cohen MH, Martin E, Valcour V, Milam J, Anastos K, Young MA, Alden C, Gustafson DR, Maki PM. The association of perceived stress and verbal memory is greater in HIV-infected versus HIV-uninfected women. J Neurovirol. 2015;21(4):422–32. https://doi.org/10.1007/s13365-015-0331-5.
Finnell JE, Moffitt CM, Hesser LA, Harrington E, Melson MN, Wood CS, Wood SK. The contribution of the locus coeruleus-norepinephrine system in the emergence of defeat-induced inflammatory priming. Brain Behav Immun. 2019;79:102–13. https://doi.org/10.1016/j.bbi.2019.01.021.
Wood SK, Wood CS, Lombard CM, Lee CS, Zhang XY, Finnell JE, Valentino RJ. Inflammatory Factors mediate vulnerability to a social stress-induced depressive-like phenotype in passive coping rats. Biol Psychiat. 2015;78(1):38–48. https://doi.org/10.1016/j.biopsych.2014.10.026.
Reyes B, Zhang XY, Dufourt EC, Bhatnagar S, Valentino RJ, Van Bockstaele EJ. Neurochemically distinct circuitry regulates locus coeruleus activity during female social stress depending on coping style. Brain Struct Funct. 2019;224(4):1429–46. https://doi.org/10.1007/s00429-019-01837-5.
Sanford LD, Wellman LL, Adkins AM, Guo ML, Zhang Y, Ren R, Yang L, Tang X. Modeling integrated stress, sleep, fear and neuroimmune responses: relevance for understanding trauma and stress-related disorders. Neurobiol Stress. 2023;23: 100517. https://doi.org/10.1016/j.ynstr.2023.100517.
Moriarity DP, Grehl MM, Walsh RFL, Roos LG, Slavich GM, Alloy LB. A systematic review of associations between emotion regulation characteristics and inflammation. Neurosci Biobehav Rev. 2023;150: 105162. https://doi.org/10.1016/j.neubiorev.2023.105162.
Denson TF, Spanovic M, Miller N. Cognitive appraisals and emotions predict cortisol and immune responses: a meta-analysis of acute laboratory social stressors and emotion inductions. Psychol Bull. 2009;135(6):823–53. https://doi.org/10.1037/a0016909.
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918–34. https://doi.org/10.1016/j.cell.2010.02.016.
Pape K, Tamouza R, Leboyer M, Zipp F. Immunoneuropsychiatry—novel perspectives on brain disorders. Nat Rev Neurol. 2019;15(6):317–28. https://doi.org/10.1038/s41582-019-0174-4.
Scheiblich H, Trombly M, Ramirez A, Heneka MT. Neuroimmune connections in aging and neurodegenerative diseases. Trends Immunol. 2020;41(4):300–12. https://doi.org/10.1016/j.it.2020.02.002.
Najjar S, Pearlman DM, Alper K, Najjar A, Devinsky O. Neuroinflammation and psychiatric illness. J Neuroinflammation. 2013;10:43. https://doi.org/10.1186/1742-2094-10-43.
Shields GS, Spahr CM, Slavich GM. Psychosocial interventions and immune system function: a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiat. 2020;77:1031–43. https://doi.org/10.1001/jamapsychiatry.2020.0431.
DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem. 2016;139(Suppl 2):136–53. https://doi.org/10.1111/jnc.13607.
Borst K, Schwabenland M, Prinz M. Microglia metabolism in health and disease. Neurochem Int. 2019;130: 104331. https://doi.org/10.1016/j.neuint.2018.11.006.
Feinstein DL, Kalinin S, Braun D. Causes, consequences, and cures for neuroinflammation mediated via the locus coeruleus: noradrenergic signaling system. J Neurochem. 2016;139(Suppl 2):154–78. https://doi.org/10.1111/jnc.13447.
Bollinger JL, Collins KE, Patel R, Wellman CL. Behavioral stress alters corticolimbic microglia in a sex- and brain region-specific manner. PLoS ONE. 2017;12(12): e0187631. https://doi.org/10.1371/journal.pone.0187631.
Slavich GM, Cole SW. The emerging field of human social genomics. Clin Psychol Sci. 2013;1(3):331–48. https://doi.org/10.1177/2167702613478594.
Slavich GM, Mengelkoch S, Cole SW. Human social genomics: Concepts, mechanisms, and implications for health. Lifestyle Med (Hoboken NJ). 2023;4(2): e75. https://doi.org/10.1002/lim2.75.
O’Donovan A, Sun B, Cole S, Rempel H, Lenoci M, Pulliam L, Neylan T. Transcriptional control of monocyte gene expression in post-traumatic stress disorder. Dis Mark. 2011;30(2–3):123–32. https://doi.org/10.3233/DMA-2011-0768.
Slavich GM. Psychoneuroimmunology of stress and mental health. In: Harkness KL, Hayden EP, editors. The Oxford handbook of stress and mental health. New York: Oxford University Press; 2020. p. 519–46. https://doi.org/10.1093/oxfordhb/9780190681777.013.24.
Ng A, Tam WW, Zhang MW, Ho CS, Husain SF, McIntyre RS, Ho RC. IL-1β, IL-6, TNF- α and CRP in elderly patients with depression or alzheimer’s disease: systematic review and meta-analysis. Sci Rep. 2018;8(1):12050. https://doi.org/10.1038/s41598-018-30487-6.
Farina MP, Kim JK, Hayward MD, Crimmins EM. Links between inflammation and immune functioning with cognitive status among older Americans in the Health and Retirement Study. Brain Behav Immunity Health. 2022;26: 100559. https://doi.org/10.1016/j.bbih.2022.100559.
Babić Leko M, Nikolac Perković M, Klepac N, Švob Štrac D, Borovečki F, Pivac N, Hof PR, Šimić G. Relationships of cerebrospinal fluid Alzheimer’s disease biomarkers and COMT, DBH, and MAOB single nucleotide polymorphisms. J Alzheimers Dis. 2020;73(1):135–45. https://doi.org/10.3233/JAD-190991.
Hegazy SH, Thomassen JQ, Rasmussen IJ, Nordestgaard BG, Tybjaerg-Hansen A, Frikke-Schmidt R. C-reactive protein levels and risk of dementia-observational and genetic studies of 111,242 individuals from the general population. Alzheimers Dement. 2022;18(11):2262–71. https://doi.org/10.1002/alz.12568.
Long S, Chen Y, Meng Y, Yang Z, Wei M, Li T, Ni J, Shi J, Tian J. Peripheral high levels of CRP predict progression from normal cognition to dementia: a systematic review and meta-analysis. J Clin Neurosci. 2022;107:54–63. https://doi.org/10.1016/j.jocn.2022.11.016.
Fu P, Peng F, Alzheimer’s Disease Neuroimaging Initiative. CSF TNF α levels were associated with conversion from mild cognitive impairment to dementia. PLoS ONE. 2022;17(10): e0274503. https://doi.org/10.1371/journal.pone.0274503.
Beurel E, Toups M, Nemeroff CB. The bidirectional relationship of depression and inflammation: double trouble. Neuron. 2020;107(2):234–56. https://doi.org/10.1016/j.neuron.2020.06.002.
Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, Ferrucci L, Gilroy DW, Fasano A, Miller GW, Miller AH, Mantovani A, Weyand CM, Barzilai N, Goronzy JJ, Rando TA, Effros RB, Lucia A, Kleinstreuer N, Slavich GM. Chronic inflammation in the etiology of disease across the life span. Nat Med. 2019;25:1822–32. https://doi.org/10.1038/s41591-019-0675-0.
Donegan JJ, Girotti M, Weinberg MS, Morilak DA. A novel role for brain interleukin-6: facilitation of cognitive flexibility in rat orbitofrontal cortex. J Neurosci. 2014;34(3):953–62. https://doi.org/10.1523/JNEUROSCI.3968-13.2014.
GBD 2019 Mental Disorders Collaborators. Global, regional, and national burden of 12 mental disorders in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Psychiatry. 2022;9(2):137–50. https://doi.org/10.1016/S2215-0366(21)00395-3.
World mental health report: transforming mental health for all. Geneva: World Health Organization; 2022. Licence: CC BY-NC-SA 3.0 IGO.
Girotti M, Adler SM, Bulin SE, Fucich EA, Paredes D, Morilak DA. Prefrontal cortex executive processes affected by stress in health and disease. Prog Neuropsychopharmacol Biol Psychiatry. 2018;85:161–79. https://doi.org/10.1016/j.pnpbp.2017.07.004.
Blum K, Gondré-Lewis MC, Modestino EJ, Lott L, Baron D, Siwicki D, McLaughlin T, Howeedy A, Krengel MH, Oscar-Berman M, Thanos PK, Elman I, Hauser M, Fried L, Bowirrat A, Badgaiyan RD. Understanding the scientific basis of post-traumatic stress disorder (PTSD): precision behavioral management overrides stigmatization. Mol Neurobiol. 2019;56(11):7836–50. https://doi.org/10.1007/s12035-019-1600-8.
Funding
ISP was supported by the Temerty-Tanz-TDRA Initiative on Dementia and Depression. GMS was supported by grant #OPR21101 from the California Governor’s Office of Planning and Research/California Initiative to Advance Precision Medicine. These organizations had no role in writing, editing, or reviewing this article, or in deciding to submit this article for publication.
Author information
Authors and Affiliations
Contributions
ISP conceptualized the review, searched the literature, developed the hypotheses, wrote the first draft of the manuscript, designed the figures, reviewed and edited the manuscript, and approved the final version of the article. GMS reviewed and edited the manuscript, and approved the final version of the article. TV reviewed and edited the manuscript, and approved the final version of the article. TKR reviewed and edited the manuscript, and approved the final version of the article.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Palamarchuk, I.S., Slavich, G.M., Vaillancourt, T. et al. Stress-related cellular pathophysiology as a crosstalk risk factor for neurocognitive and psychiatric disorders. BMC Neurosci 24, 65 (2023). https://doi.org/10.1186/s12868-023-00831-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12868-023-00831-2