
Abstract
Background
Neuroinflammation plays a prominent role in Alzheimer’s disease (AD), both in pathogenesis and disease progression. It has been shown that TLR/MYD88 signaling is involved in the chronic low-grade sterile inflammation associated with AD. Several studies have evidenced high levels of MYD88 in the brain of patients and animal models of AD, but no study has assessed so far its levels in blood.
Methods
In this study we evaluated the blood mRNA levels of MYD88 in a mouse model of AD, and also the putative effect of Rivastigmine treatment on MYD88 expression. Twenty-eight transgenic APP/TAU mice (AT) and twenty-two control C57/BL6j mice (WT) were included in this study, out of which five transgenic AT and five WT mice were treated with Rivastigmine.
Results
Increased MYD88 transcript in the whole blood from AT mice as compared to WT controls was found, which seems to increase in time due to disease progression and not to aging. This finding suggests that blood leukocytes are primed to develop TLR/MYD-mediated inflammatory processes. Moreover, results indicate that MYD88 blood levels were not modulated by the diseases-specific treatment with Rivastigmine.
Conclusions
Our results suggest that MYD88 might be a promising blood biomarker to monitor AD progression.
Similar content being viewed by others


Background
Sterile inflammation of the central nervous system (CNS), was shown to underlie various neurodegenerative disorders including amyotrophic lateral sclerosis (ALS), Parkinson disease (PD) and Alzheimer’s disease (AD) [1]. Among these diseases, AD has been indicated as the condition where neuroinflammation has the most prominent role, being involved both in pathogenesis and progression [2]. Microglia and astrocytes are the main local immune cells involved in this process by producing pro-inflammatory factors like cytokines and chemokines, along with reactive oxygen species. Brain-infiltrating blood leukocytes and capillary endothelial cells also contribute to neuroinflammation [2]. Depending on the duration and intensity of neuroinflammation, both positive and negative outcomes have been described. Thus, transiently low and medium inflammation leads to enhanced plasticity, tissue repair and neuroprotection, while chronic low- and high-level inflammation seems to be associated with reduced plasticity, neuronal damage and cognitive impairment in neurodegenerative diseases and aging [3].
One of the contributing inflammation mechanisms is related to Toll-like receptors (TLRs) signaling [4]. TLRs are transmembrane proteins expressed by both immune and non-immune cells. Intracellular TLRs mainly recognize nucleic acids derived from bacteria and viruses, while cell surface TLRs interact with microbial membrane components as well as with damage-associated molecular patterns (DAMPs) [5]. In the AD brain, besides pathogens, abnormally folded proteins and protein aggregates, such as TAU and amyloid beta, have been shown to activate TLRs [6], and to trigger local inflammatory responses that affect synaptic plasticity, microglial activity and TAU phosphorylation [7]. In this context, DAMPs have been shown to be associated with neuronal dysfunction, leading to neuronal death in AD. For instance, signaling events related to DAMPs-induced unfolded protein response may regulate the expression of different AD-related proteins and early processes of β-amyloid precursor protein (APP) maturation. Hence, some DAMPs, including neuronal stress-induced HSP72 and TLR2, 4 and 9, have been reported to be involved in AD-associated neuroinflammation [8].
Except for TLR3, the myeloid differentiation factor-88 adaptor protein (MYD88) mediates the intra-cellular signaling of all other TLRs [9]. MYD88 binds to the cytoplasmic region of TLRs and activates the interleukin-1 receptor-associated kinase (IRAK) family, leading to a variety of functional outputs, including the activation of the nuclear factor-kappa B (NFκB) [10], a key mediator of inflammatory responses that regulates the survival, activation and differentiation of innate immune cells and the inflammatory Th1 and Th17 cells [11]. Moreover, it has been demonstrated that TLR/MYD88 signaling plays an important role in sustaining the chronic low-grade sterile inflammation associated with AD [12].
Post-mortem brain studies in AD patients and animal models have evidenced high levels of MYD88 in the brain, and it has been shown that experimental MYD88 deficiency ameliorates both β-amyloidosis and cognitive functions [13,14,15]. The research on MYD88 has remained confined to the AD brain, and no study has assessed so far its levels in blood leukocytes. Therefore, the aim of the present study was to evaluate the blood mRNA levels of MYD88 in a mouse model of AD, and to evaluate the putative effect of AD-specific medication on gene expression.
Methods
Transgenic mouse model of AD
Double transgenic APP/TAU (AT) mice were used as animal model of AD. These mice express the human genes APP and TAU under the control of the mouse Thy1 gene promoter. APP/TAU mice were obtained by crossing for more than eight generations mice presenting the human APP-V717I mutation with TAUP301L transgenic mice, both having a C57/BL6j background [16]. The characteristics of the APPV717I and TAUP301L transgenic mice have been previously described [17]. The double transgenic APP/TAU (AT) mouse model shows a combined amyloid and TAU-pathology, which mimics the pathology of AD patients, including diffuse and senile plaques, vascular amyloid and neurofibrillary tangles in brain [18]. Amyloid accumulation begins at an early age, intracellularly, decreases with age and is progressively replaced by extracellular amyloid deposits, first in the form of diffuse plaques (10–12 months), followed by senile plaques (12–15 months) and deposits of amyloid at the vascular level (15–18 months). The formation of Tau protein fibrils, especially in the anterior brain and hippocampus, occurs around the age of 13 months [18]. Twenty-eight AT mice and twenty-two control WT mice, all on C57/BL6j background, with a mean age of 55.26 ± 6.44 weeks, were investigated. The transgenic AT and WT mice were homogeneous for age and sex (Table 1). Mice were group-housed in simple cages under standard conditions (normal 12 h light/dark cycle, constant temperature and humidity), with ad libitum access to food and water. The study was conducted according to the guidelines of the European Directive 2010/63/EU and approved by the Ethics Committee of “Victor Babes” National Institute of Pathology, Bucharest, Romania, authorization No. 39/11.04.2017, and by the Romanian National Authority for Veterinary Research, authorization No. 385/09.02.2018.
Rivastigmine treatment
Five transgenic AT mice and five control WT mice were treated with Rivastigmine (Sandoz, 2 mg/mL oral solution) by daily oral gavage (0.75 mg/Kg) for 50 days, with intermediary blood collection at 20 days of treatment.
Blood collection
Blood was collected by retro-orbital sinus sampling in PAXgene RNA stabilizer solution (Qiagen). The volume of the collected blood was between 140 and 180 µl, according to the body weight of the mice. Blood was collected before (T0), during (T1, 20 days) and at the end (T2, 50 days) of Rivastigmine treatment.
Gene expression analysis
RNA isolation was performed using the modified PAXgene method [19], and was quantified using a NanoDrop 2000 spectrophotometer (Thermo Scientific). Reverse transcription of 500 ng total RNA was performed using the High-Capacity cDNA Reverse Transcription Kit (Thermo Scientific) according to the manufacturer’s protocol. The expression level of MYD88 was assessed by qPCR on 7500 Fast Real-Time PCR System (Applied Biosystems), using the primers described in Table 2. MYD88 levels were normalized on the geometric mean of two reference genes, GAPDH and TBP (Table 2). The stability of the reference genes was assessed with the RefFinder algorithm (http://leonxie.esy.es/RefFinder/) [20]. The gene expression levels of MYD88 were presented as 2−∆CT values and are available in the Additional file 1: https://doi.org/10.7910/DVN/XRQWSE.
Statistical analysis
Whenever possible, results were expressed as mean ± standard error of the mean (SEM) or standard deviation (SD). The Statistical Package for Social Science (SPSS version 17.0) was used for data analysis. Categorical variables were compared with the chi-square test, and continuous variables with the Student’s t-test (age) or the Mann Whitney test (gene expression levels between two independent groups). Rivastigmine-treated and non-treated mice were compared regarding the effect of medication on MYD88 transcript levels at T1 and T2 by Repeated Measures ANOVA with the Bonferroni Corrected t-tests.
Results
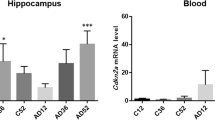
The expression analysis on MYD88 in whole blood showed that the gene was significantly upregulated in AT mice as compared to WT controls (FC = 2.04, p < 0.001) (Fig. 1). The two groups were homogenous for age and sex. However, since AD transgenic female mice have been shown to be more susceptible to amyloid beta plaques and tangles and also TAU pathology compared to the males [21], an additional analysis has been performed. Comparing the MYD88 blood levels between the AT transgenic males (N = 18) vs. females (N = 10) no significant differences were observed (FC = 1.01, p = 1).To investigate a potential dependence of the registered gene over-expression on age, mice in each group were divided in three age categories, as follows: young-middle aged (41–51 weeks), middle aged (52–54 weeks) and old-middle aged (57–74 weeks) mice, and MYD88 levels were analyzed accordingly. Results evidenced a significant MYD88 upregulation in the AT group as compared to WT mice in each of the considered age categories (Fig. 2). Within the AT mice group, an increase of MYD88 levels was observed especially in old-middle aged mice when compared to young-middle aged (p = 0.030) (Fig. 2). Since such an upregulation was not detected in the control WT group (Fig. 2), results suggested that MYD88 overexpression was not age-dependent, but the levels of transcript are apparently increasing in time due to disease progression.

The mRNA levels of MYD88±SEM in the blood of AT (N = 28) and WT (N = 22) mice (FC = 2.04, p < 0.001). Comparison between mice groups was performed with the Mann Whitney test, and differences were considered significant for p < 0.05

MYD88 mRNA levels ±SEM in the blood of WT (N = 22) and AT (N = 28) mice in different age categories. Comparison between groups was performed using the Mann Whitney test, and differences were considered significant for p < 0.05
Considering that AD patients are generally treated with acetylcholinesterase inhibitors, such as Rivastigmine, we investigated if Rivastigmine treatment could influence the transcript levels of MYD88 in the examined mice groups. Five AT and five WT mice were treated by daily oral gavage with Rivastigmine (0.75 mg/kg) for 50 days, and blood MYD88 mRNA levels were measured at baseline (T0), at 20 (T1) and 50 days (T2) of treatment. Results showed that Rivastigmine did not significantly change MYD88 transcript levels in the WT group, after neither 20 nor 50 days of therapy (Fig. 3). An increase of MYD88 mRNA levels was registered in the AT group only at 50 days of therapy (Fig. 3). This increase was most probably related to the disease evolution, and not to therapy (Fig. 3), considering the same ascending trend without (Fig. 2) or with Rivastigmine therapy.

MYD88 mRNA blood levels at different time points before and during Rivastigmine treatment: T0-before treatment, T1-at 20 days, T2-at 50 days after treatment initiation. The mean±SD of mice age at the investigated time points during therapy is mentioned. Bars represent the mean of MYD88 levels±SEM. Comparison among T0, T1 and T2 was performed using Repeated Measures ANOVA with the Bonferroni Corrected t-tests, and differences were considered significant for p < 0.05. RIV Rivastigmine
Discussion
MYD88, encoding for a key signal transductor in sterile inflammation, was found significantly upregulated in AD double transgenic mice, partially mimicking human AD, as compared to WT control mice. The increased of MYD88 mRNA levels was not age-dependent, but apparently the transcript levels increased during disease progression. To our knowledge, this is the first study investigating blood mRNA levels of MYD88 in an AD mice model. Moreover, we did not find so far human studies focused on blood MYD88 levels in AD. The increased blood levels of MYD88, evidenced in the present study, indicate that blood leukocytes from AD transgenic mice, might have an increased susceptibility to respond to TLR ligands, either Pathogen-Associated Molecular Patterns (PAMPs) or Damage-Associated Molecular Patterns (DAMPs) [22, 23]. This could increase the immune competence to fight infection and to repair tissue damages, but may also perpetuate through a vicious cycle systemic sterile inflammation when the coordinated leukocyte effort cannot clear the immunostimulatory cues [24, 25]. The pathologic significance of the reported increase of MYD88 transcript levels in blood was not investigated in the present study. Moreover, it is not clear, if the observed increase in gene expression is translated into higher protein levels of MYD88, or if such an overexpression has functional significance for immune homeostasis.
Preclinical and clinical evidence stated that MYD88 levels are increased in the AD brain. Immunofluorescence studies on 5XFAD mice showed higher MYD88 levels in the hippocampus and cortex of these transgenic mice, resulting in increased protein levels [13]. The authors found similar results also in humans, as MYD88 was increased in post mortem brains from AD patients as compared to MCI or to non-demented controls, and MYD88 protein levels positively correlated with the Braak staging. Another study, performed on the APP/PS1 mouse model, showed that neural stem cell transplantation decreased neuroinflammation and induced cognitive improvement, accompanied by a decreased expression of the MYD88 protein [26]. Moreover, the deletion of one MYD88 allele in APP/PS1 transgenic mice was associated with a decrease of cerebral Aβ load and with an improvement of cognition [15]. Additionally, MYD88 levels were found increased also in the hippocampus of Balb/c mice where AD-like dementia was induced through the administration of Aβ1-42 oligomers [27]. In this study, MYD88 levels were down-regulated by minocycline treatment that ameliorated cognitive impairment, possibly through its anti-inflammatory action.
Considering the data evidencing increased levels of MYD88 in the AD brain, it is possible that neuronal damage in AD and the consequent release of DAMPs could have an echo in the periphery and prime blood leukocytes [28] by increasing the transcription of MYD88, as reported in the present study. Alternatively, circulating inflammatory factors or activated/primed inflammatory blood cells arising from a peripheral or distant inflammation might be recruited in the AD brain and increase locally the inflammatory microenvironment [29].
Although, the mechanisms underlying the brain-blood connection relative to MYD88 is not known, this adaptor protein might represent a valuable therapeutic target [24, 30,31,32], albeit being hypothetically beneficial in AD, raises concerns due to its critical role in anti-infectious defence. In this context, we have investigated if the AD-specific treatment with Rivastigmine, an acetylcholinesterase inhibitor, having also anti-inflammatory properties [33], is impacting on MYD88 transcript levels in the blood of the investigated AT mice. Albeit its known anti-inflammatory action, Rivastigmine treatment could not decrease the mRNA levels of MYD88 neither in AT nor in WT mice, probably because an unknown inflammatory stimulus, not affected by Rivastigmine, is decisively dictating gene over-expression.
Conclusions
In conclusion, the study emphasized elevated transcript levels of the MYD88 gene in the blood of double transgenic AT mice used as AD animal model. MYD88 expression increased during disease evolution, and was not affected by Rivastigmine therapy. Accordingly, our results point towards MYD88 as a promising blood biomarker to monitor AD progression, which has to be further validate in human studies.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional information files. The mRNA data are also available in the Harvard Dataverse repository https://doi.org/10.7910/DVN/XRQWSE.
Abbreviations
- AD:
-
Alzheimer’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- APP:
-
β-Amyloid precursor protein
- AT:
-
APP/TAU
- CNS:
-
Central nervous system
- DAMPs:
-
Damage-associated molecular patterns
- IRAK:
-
Interleukin-1 receptor-associated kinase
- MYD88:
-
Myeloid differentiation factor-88 adaptor protein
- NFB:
-
Nuclear factor-kappa B
- PAMPs:
-
Pathogen-Associated Molecular Patterns
- PD:
-
Parkinson disease
- TLR:
-
Toll-like receptor
- WT:
-
Wild type
References
Banjara M, Ghosh C. Sterile neuroinflammation and strategies for therapeutic intervention. Int J Inflam. 2017;2017:8385961.
Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17:157–72.
DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem. 2016;139 Suppl:136–53.
M. Zolezzi J, Bastías-Candia S, C. Inestrosa N. Toll-Like Receptors (TLRs) in Neurodegeneration: Integrative Approach to TLR Cascades in Alzheimer’s and Parkinson’s Diseases. In: Toll-like Receptors. IntechOpen; 2020.
Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461.
Frederiksen HR, Haukedal H, Freude K. Cell Type Specific Expression of Toll-Like Receptors in Human Brains and Implications in Alzheimer’s Disease. Biomed Res Int. 2019;2019:7420189.
Momtazmanesh S, Perry G, Rezaei N. Toll-like receptors in Alzheimer’s disease. J Neuroimmunol. 2020;348:577362.
Land WG. The Role of Damage-Associated Molecular Patterns (DAMPs) in Human Diseases: Part II: DAMPs as diagnostics, prognostics and therapeutics in clinical medicine. Sultan Qaboos Univ Med J. 2015;15:e157-70.
Brown J, Wang H, Hajishengallis GN, Martin M. TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J Dent Res. 2011;90:417–27.
Deguine J, Barton GM. MyD88: a central player in innate immune signaling. F1000Prime Rep. 2014;6:97.
Liu T, Zhang L, Joo D, Sun S-C. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023. https://doi.org/10.1038/sigtrans.2017.23.
Xiang W, Chao Z-Y, Feng D-Y. Role of Toll-like receptor/MYD88 signaling in neurodegenerative diseases. Rev Neurosci. 2015;26:407–14.
Rangasamy SB, Jana M, Roy A, Corbett GT, Kundu M, Chandra S, et al. Selective disruption of TLR2-MyD88 interaction inhibits inflammation and attenuates Alzheimer’s pathology. J Clin Invest. 2018;128:4297–312.
Lim J-E, Kou J, Song M, Pattanayak A, Jin J, Lalonde R, et al. MyD88 deficiency ameliorates β-amyloidosis in an animal model of Alzheimer’s disease. Am J Pathol. 2011;179:1095–103.
Quan W, Luo Q, Hao W, Tomic I, Furihata T, Schulz-Schäffer W, et al. Haploinsufficiency of microglial MyD88 ameliorates Alzheimer’s pathology and vascular disorders in APP/PS1-transgenic mice. Glia. 2021;69:1987–2005.
Rojo AI, Pajares M, Rada P, Nuñez A, Nevado-Holgado AJ, Killik R, et al. NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol. 2017;13:444–51. https://doi.org/10.1016/j.redox.2017.07.006.
Samaey C, Schreurs A, Stroobants S, Balschun D. Early Cognitive and Behavioral Deficits in Mouse Models for Tauopathy and Alzheimer’s Disease. Front Aging Neurosci. 2019;11:335. https://doi.org/10.3389/fnagi.2019.00335.
Terwel D, Muyllaert D, Dewachter I, Borghgraef P, Croes S, Devijver H, et al. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am J Pathol. 2008;172:786–98. https://doi.org/10.2353/ajpath.2008.070904.
Krawiec JA, Chen H, Alom-Ruiz S, Jaye M. Modified PAXgene method allows for isolation of high-integrity total RNA from microlitre volumes of mouse whole blood. Lab Anim. 2009;43:394–8.
Xie F, Xiao P, Chen D, Xu L, Zhang B. miRDeepFinder: a miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol Biol. 2012. https://doi.org/10.1007/s11103-012-9885-2.
Jankowsky JL, Zheng H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol Neurodegener. 2017;12:89. https://doi.org/10.1186/s13024-017-0231-7.
Evavold CL, Kagan JC, Inflammasomes. Threat-assessment organelles of the innate immune system. Immunity. 2019;51:609–24.
Patel S. Danger-Associated Molecular Patterns (DAMPs): the derivatives and triggers of inflammation. Curr Allergy Asthma Rep. 2018;18:63.
Saikh KU. MyD88 and beyond: a perspective on MyD88-targeted therapeutic approach for modulation of host immunity. Immunol Res. 2021;69:117–28.
Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathol. 2020;15:493–518.
Zhang Q, Wu H-H, Wang Y, Gu G-J, Zhang W, Xia R. Neural stem cell transplantation decreases neuroinflammation in a transgenic mouse model of Alzheimer’s disease. J Neurochem. 2016;136:815–25.
Garcez ML, Mina F, Bellettini-Santos T, da Luz AP, Schiavo GL, Macieski JMC, et al. The Involvement of NLRP3 on the effects of minocycline in an AD-like pathology induced by β-amyloid oligomers administered to mice. Mol Neurobiol. 2019;56:2606–17.
Liesz A, Dalpke A, Mracsko E, Antoine DJ, Roth S, Zhou W, et al. DAMP signaling is a key pathway inducing immune modulation after brain injury. J Neurosci. 2015;35:583–98.
D’Mello C, Le T, Swain MG. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci. 2009;29:2089–102.
Qiang W, Cai W, Yang Q, Yang L, Dai Y, Zhao Z, et al. Artemisinin B improves learning and memory impairment in ad dementia mice by suppressing neuroinflammation. Neuroscience. 2018;395:1–12.
Zhou J, Deng Y, Li F, Yin C, Shi J, Gong Q. Icariside II attenuates lipopolysaccharide-induced neuroinflammation through inhibiting TLR4/MyD88/NF-κB pathway in rats. Biomed Pharmacother. 2019;111:315–24.
Guo M-F, Zhang H-Y, Li Y-H, Gu Q-F, Wei W-Y, Wang Y-Y, et al. Fasudil inhibits the activation of microglia and astrocytes of transgenic Alzheimer’s disease mice via the downregulation of TLR4/Myd88/NF-κB pathway. J Neuroimmunol. 2020;346:577284.
Goschorska M, Baranowska-Bosiacka I, Gutowska I, Tarnowski M, Piotrowska K, Metryka E, et al. Effect of acetylcholinesterase inhibitors donepezil and rivastigmine on the activity and expression of cyclooxygenases in a model of the inflammatory action of fluoride on macrophages obtained from THP-1 monocytes. Toxicology. 2018;406–407:9–20.
Funding
Research and publication of the present study were funded by Competitiveness Operational Programme 2014–2020 project P37_732 (contract no. 29/2016), Priority Axis 1, Action 1.1.4, cofinanced by the European Funds for Regional Development and Romanian Government funds. The contents of this publication do not necessarily reflect the official position of the European Union or Romanian Government.
Author information
Authors and Affiliations
Contributions
CAC designed and coordinated the study, CAC and EM performed the statistical analysis, and wrote the first draft of the manuscript. MD and EMD performed the laboratory experiments, MD contributed to data processing, and writing. GM contributed in data interpretation, writing and critical reading, and revision. All authors contributed to the article and agreed to the published version of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experiments involving mice were carried out in accordance with the guidelines of the European Directive 2010/63/EU and approved by the Ethics Committee of “Victor Babes” National Institute of Pathology, Bucharest, Romania, authorization No. 39/11.04.2017, and by the Romanian National Authority for Veterinary Research, authorization No. 385/09.02.2018. The study was carried out in compliance with the ARRIVE guidelines.
Consent for publication
not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
All data generated or analyzed during this study are included in this published article and its additional information files. The mRNA data are also available in the Harvard Dataverse repository https://doi.org/10.7910/DVN/XRQWSE.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cucos, C.A., Dobre, M., Dragnea, E.M. et al. Increased MYD88 blood transcript in a mouse model of Alzheimer’s disease. BMC Neurosci 23, 13 (2022). https://doi.org/10.1186/s12868-022-00699-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12868-022-00699-8