
Abstract
Background
The aim of this study was to investigate whether AMN082 exerts its neuroprotective effect by attenuating glutamate receptor-associated neuronal apoptosis and improving functional outcomes after traumatic brain injury (TBI).
Methods
Anesthetized male Sprague–Dawley rats were divided into the sham-operated, TBI + vehicle, and TBI + AMN082 groups. AMN082 (10 mg/kg) was intraperitoneally injected 0, 24, or 48 h after TBI. In the 120 min after TBI, heart rate, mean arterial pressure, intracranial pressure (ICP), and cerebral perfusion pressure (CPP) were continuously measured. Motor function, the infarct volume, neuronal nitrosative stress-associated apoptosis, and N-methyl-d-aspartate receptor 2A (NR2A) and NR2B expression in the pericontusional cortex were measured on the 3rd day after TBI.
Results
The results showed that the AMN082-treated group had a lower ICP and higher CPP after TBI. TBI-induced motor deficits, the increase in infarct volume, neuronal apoptosis, and 3-nitrotyrosine and inducible nitric oxide synthase expression in the pericontusional cortex were significantly improved by AMN082 therapy. Simultaneously, AMN082 increased NR2A and NR2B expression in neuronal cells.
Conclusions
We concluded that intraperitoneal injection of AMN082 for 3 days may ameliorate TBI by attenuating glutamate receptor-associated nitrosative stress and neuronal apoptosis in the pericontusional cortex. We suggest that AMN082 administration in the acute stage may be a promising strategy for TBI.
Similar content being viewed by others

Introduction
Excessive glutamate excitotoxicity is well known to be a leading cause of neurological complications after traumatic brain injury (TBI) [1]. Metabotropic receptors (mGluRs) [2] and ionotropic receptors are two major glutamate receptors in the central nervous system (CNS) [3, 4]. According to molecular cloning data, there are eight different metabotropic receptor subtypes, i.e., mGluR1 to mGluR8, with different molecular structures and pharmacological properties [2]. Among ionotropic receptors, N-methyl-d-aspartate receptor 2A (NR2A) and 2B (NR2B) are two major NMDA receptors [3, 4]. Triggering of astroglial and neuronal glutamate excitotoxicity generates inducible nitric oxide synthase (iNOS) and induces reactive nitrosative stress (RNS), eventually contributing to cell apoptosis [5, 6]. Therefore, inhibition of glutamate-related RNS processes may be a therapeutic strategy for TBI.
Although there is little data in the literature, some authors have reported changes in the expression of mGluRs in TBI. Gong et al. showed that at 7 days after TBI, mGluR2/3 and mGluR5 expression was reduced in the ipsilateral hippocampus and cortex [7]. Fei et al. demonstrated a significant rise in the expression of mGluR1 and mGluR5 (peaking at 24 h) and mGluR4, mGluR7 and mGluR8 (peaking at 6 h) and a decrease in the expression of mGluR2 and mGluR3 (peaking at 24 h) after diffuse brain injury [8, 9]. Therefore, modification of the expression of mGluR may provide neuroprotection after brain injury.
AMN082 (N,N′-dibenzhydrylethane-1,2-diamine dihydrochloride) is a blood–brain barrier-permeable and highly selective mGLu7 receptor allosteric agonist with clear pharmacokinetic activities [10]. Previously, the neuroprotective effects of AMN082 against the neurotoxic and proapoptotic effects of sevoflurane were demonstrated in both in vitro and in vivo models [11]. Moreover, AMN082 has been found to exert neuroprotective effects against oxygen–glucose deprivation- and kainate-evoked neuronal cell damage in in vitro models [12]. The neuroprotective and glioprotective effects of AMN082 have also been observed in models of apoptotic (induced by staurosporine and doxorubicin) and apoptotic-necrotic [induced MPP (+)] damage [13,14,15,16].
Furthermore, AMN082 has been shown to affect the CNS to exert anti-anxiety, antidepressive effects [17,18,19] and antiparkinsonian-like effects [20], and it is used in the treatment of cocaine or opioid addiction [21]. Its mechanisms include regulating glutamate release at the synapse [22], binding to the serotonin transporter [10], activating prosurvival MAPK/ERK 1/2 and PI3-K/Akt pathways [15], and stimulating mammalian target of rapamycin (mTOR) activation [18].
However, to date, AMN082 has not been applied for the treatment of TBI. The effects of AMN082 on neurons and glia as well as TBI-induced nitrosative stress-associated neuronal apoptosis have not been well investigated.
In the current study, we hypothesized that AMN082 has therapeutic effects against TBI because it is a selective mGlu7 receptor that affects glutamate release. We aimed to elucidate the beneficial effects of AMN082 against TBI-induced nitrosative stress-related apoptosis. We hope our results provide new evidence that AMN082 may play a role in clinical treatment in the field of neurotrauma in the future.
Methods
Experimental design
The overall experimental protocol is shown in Fig. 1. First, we continuously measured physiological changes, including heart rate (HR), mean arterial pressure (MAP), intracranial pressure (ICP) and cerebral perfusion pressure (CPP), for 120 min after TBI. Then, we tested whether the changes induced by TBI were associated with nitrosative stress-related neuronal apoptosis and cerebral infarction on the ipsilateral side of the cortex using immunofluorescence and TTC staining. Motor function was assessed by the inclined plane test on the 3rd day after TBI. At the same time point, the effects of AMN082 on TBI-related parameters were also evaluated. All methods were carried out in accordance with relevant guidelines and regulations.

Summary of the experimental protocol
Animals
Ten-week-old adult male Sprague–Dawley rats were obtained from a commercial source (BioLASCO Taiwan Co., Ltd.) and used in the experiments. The animals were housed under a 12/12-h light/dark cycle and provided free access to food and water. The Chi Mei Medical Center’s Animal Care and Use Committee approved all of the experimental procedures, which conformed to the NIH guidelines, including minimization of discomfort to the animals during surgery and during the recovery period (approval no. 108080602). At the end of the experiments, the rats were euthanized by an overdose of urethane (intraperitoneal injections of 2 ml of 500 mg/ml urethane solution) until deep unconscious condition determined by the absence of visible breathing. The study was carried out in compliance with the ARRIVE guidelines.
Traumatic brain injury
The animals were anesthetized with a mixture of ketamine (44 mg/kg, i.m.; Nankuang Pharmaceutical, Taiwan), atropine (0.063 mg/kg, i.m.; Sintong Chemical Ind. Co., Taiwan), and xylazine (6.77 mg/kg, i.m.; Bayer, Germany). On a stereotaxic frame (Kopf 1406; Grass Instrument, Quincy, MA), 2-mm-radius craniectomy was performed 3 mm from the sagittal suture and 4 mm from bregma above the right parietal cortex. After implantation of the injury cannula, a fluid percussion device (VCU Biomedical Engineering, Richmond, VA, USA) was connected to the craniectomy site via Luer-lock fitting, and the brain was subjected to percussion injury at 2.2 atm for 25 ms as previously described by Mclntosh et al. [23]. The selection criteria for successful TBI in the rats were transient hypertensive response, apnea, and seizure observed immediately following fluid percussion injury.
Surgery and physiological parameter monitoring
The right femoral artery of each rat was cannulated with polyethylene tubing (PE50) for blood pressure monitoring. The mean arterial pressure (MAP) and heart rate (HR) were monitored continuously after TBI with a pressure transducer. An intracranial pressure (ICP) microsensor (Codman and Shurtlef, Inc., Rayman, MA, USA) was placed in the parenchyma of the left frontal lobe of each rat. The ICP was monitored continuously, and ICP and cerebral perfusion pressure (CPP) values were recorded at 5-min intervals for 120 min after TBI. The CPP is the MAP-ICP [24]. Colon temperatures were measured with an analog electronic thermometer (model 43 TE; YSI, Inc, Yellow Springs, OH, USA) and temperature probe (series 400; YSI, Inc).
Treatment intervention
Using a random number table, the rats were randomly assigned numbers and divided into three groups: the sham operated group (n = 6); the vehicle (dimethylsulfoxide (DMSO), 4%, 1 ml/kg, intraperitoneal; K42088831, Merck, Darmstadt, Germany)-treated TBI control group; and AMN082 (10 mg/kg, dissolved in DMSO, intraperitoneal; U.S. Pharmacopeia)-treated TBI group (n = 6). The vehicle-treated TBI group received an equal volume of vehicle via intraperitoneal injection. Vehicle or AMN082 was administered for three consecutive days after TBI. The first injection occurred immediately after TBI, the second injection was administered 24 h later, and the third injection was administered 48 h later. All tests were performed by investigators and assessors blinded to the experimental groups, which were revealed only at the end of the analyses. Table 1 provides detailed information about animal grouping and principal component analysis.
Motor function test
An inclined plane was used to measure limb strength in the motor function test [25]. This test assesses the motor function of rats after neural injury by evaluating a rat’s ability to prevent itself from falling and the endurance strength of the upper and lower limbs on an inclined plane. The animals were initially placed on a 20 × 20-cm inclined plane with a ribbed surface and a 30° angle. To determine the maximal angle at which an animal could remain on the inclined plane without falling, the angle was increased in 1° increments. Motor deficits were assessed by determining the mean maximal angles of the left upper and lower limbs on the 3rd day after TBI.
Cerebral infarction assay
Triphenyltetrazolium chloride (TTC) staining was performed as described elsewhere [26]. Briefly, brain tissues were removed, immersed in cold saline for 5 min, and sliced into 1-mm-thick sections. The brain slices were incubated in 2% TTC dissolved in phosphate-buffered saline for 30 min at 37 °C and then transferred to 10% formaldehyde solution for fixation. The volume of infarction, as revealed by negative TTC staining, which indicated dehydrogenase-deficient tissue, was measured in each slice and summed using computerized planimetry (PC-based Image Tools software). The volume of infarction was calculated as 1 mm (the thickness of the slice) × (the sum of the infarct area in all brain slices [mm2]) [27].
Immunofluorescence assay
Immunofluorescence was performed on the 3rd day after TBI [28]. Adjacent 6-μm sections corresponding to the region 0.20–0.70 mm anterior to bregma were incubated in 2 mol/L HCl for 30 min, rinsed in 0.1 mol/L boric acid (pH 8.5) for 3 min at room temperature, and then incubated with primary antibodies in PBS containing 0.5% normal bovine serum at 4 °C overnight. After being washed in PBS, the sections were incubated with secondary antibodies for 1 h at room temperature. The number of positive cells in the pericontusional cortex was calculated using computerized planimetry (400 × magnification, Image-Pro Plus Media Cybernetics, Inc. Washington Street, Rockville, USA), and the number of positive cells per mm2 of the region of interest is presented. Details about the antibodies used are provided in Table 2.
Statistical analysis
According to analysis of variance (ANOVA) with a type I error of 0.05 and power calculation for the smallest effect size at 0.2, at least 6 rats per group were needed. This proposed sample size was sufficient to detect a significant difference by ANOVA at the 0.05 level with a power of 80%. All of the data in this study were analyzed using SigmaPlot, version 10.0, for Windows (Systat Software, San Jose, CA, USA). The results of the experiments are expressed as the means ± standard deviations of the means. For each time point, the mean difference between the three groups was analyzed using ANOVA followed by Scheffe’s post hoc test was used to analyze the significance of the differences between the three groups. p-values < 0.05 were considered statistically significant.
Results
Basic data for the experimental rats
A total of 54 10-week-old male rats weighing 413 ± 8.7 g were used in the experiments. The fluid percussion force was 2.20 ± 0.01 atm. The colon temperature was maintained at ~ 36–37 °C with a lamp during the procedure and for up to 120 min after injury. No animals died over the course of the experiments. Table 2 provides detailed information about animal grouping and principal component analysis.
Trend of a lower ICP and higher CPP in the AMN082-treated group compared with the TBI + vehicle group in the initial 120 min after TBI
The ICP was higher in the TBI group than in the sham-operated control group from 0 to 120 min after the start of FPI. In contrast, the CPP was significantly lower in the TBI group than in the sham-operated control group (*p < 0.05; n = 6 in the sham group and n = 6 in the TBI group; Table 3).
Treatment with AMN082 a TBI-induced motor impairment on the 3rd day after TBI
The maximal grip angle of the TBI group was significantly lower than that of the sham controls on the 3rd day after TBI (45.3° ± 0.72° versus 53.9 ± 0.37°, ***p < 0.001). TBI-induced motor impairment was significantly ameliorated by AMN082 treatment (the TBI group versus the AMN082 group, 45.3° ± 0.72° versus 47.5 ± 0.34, $p < 0.05; n = 6 in each group; Fig. 2).

Effects of AMN082 on TBI-induced motor deficits on the 3rd day after TBI. ***p < 0.001, the sham group compared with the TBI group; ###p < 0.05, the sham group compared with the AMN082-treated TBI + group; $p < 0.005, the TBI group compared with the AMN082-treated TBI group; n = 6 in each group
Treatment with AMN082 significantly decreased the TBI-induced cerebral infarct volume on the 3rd day after TBI
Compared with the sham group, the TBI group showed a significant increase in infarct volume (134.4 ± 3.93 mm3 versus 0 ± 0 mm3, ***p < 0.001). However, compared with the TBI group, the AMN082-treated TBI group exhibited a significant reduction in the infarct volume (66.9 ± 19.62 mm3 versus 134.4 ± 3.93 mm3, #p < 0.05; n = 6 in each group; Fig. 3).

Effects of AMN082 on the TBI-induced infarct volumes in the pericontusional cortex on the 3rd day after TBI. ***p < 0.001, the sham group compared with the TBI group; #p < 0.05, the sham group compared with the AMN082-treated TBI + group; n = 6 in each group
Treatment with AMN082 significantly attenuated neuronal apoptosis in the pericontusional cortex on the 3rd day after TBI
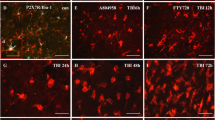
Caspase-3 and TUNEL staining revealed that the number of apoptotic neuronal cells (in the Neu-N and Caspase-3 staining and Neu-N and TUNEL staining assays) in the pericontusional cortex was significantly increased in the TBI group compared with the sham group on the 3rd day after TBI (***p < 0.001). The number of positive apoptotic cells in rats subjected to TBI was significantly reduced after AMN082 treatment (#p < 0.05, ##p < 0.01; n = 6 in each group; Fig. 4a, b).

Effects of AMN082 on TBI-induced neuronal apoptosis in the pericontusional cortex on the 3rd day after TBI. Expression of the markers Neu-N and Caspase-3. ***p < 0.001 compared with the sham group; #p < 0.05 compared with the TBI group; n = 6 in each group (a). Expression of the marker Neu-N and TUNEL staining. ***p < 0.001 compared with the sham group; ##p < 0.01 compared with the TBI group; n = 6 in each group (b)
Treatment with AMN082 significantly alleviated neuronal nitrosative stress in the pericontusional cortex on the 3rd day after TBI
Compared with that in the sham controls (62.1 ± 19.87), the number of n-NOS-Neu-N-positive cells in the pericontusional cortex in TBI rats (37.6 ± 4.99; Fig. 5a) was nonsignificantly decreased. 3-NT and Neu-N staining revealed that the number of 3-NT-positive neuronal cells in the pericontusional cortex in vehicle-treated rats was significantly increased compared with that in the sham controls (56.0 ± 4.35 versus 0 ± 0; Fig. 5b). The TBI-induced decrease in the number of n-NOS-positive neurons and increase in the number of 3-NT-positive cells were significantly ameliorated by AMN082 therapy.

Effects of AMN082 on TBI-induced neuronal nitrosative stress in the pericontusional cortex on the 3rd day after TBI. Expression of the markers Neu-N and n-NOS. #p < 0.05 compared with the TBI group; n = 6 in each group (a). Expression of the markers Neu-N and 3-NT. ***p < 0.001 compared with the sham group; ##p < 0.01 compared with the TBI group; n = 6 in each group (b)
Treatment with AMN082 significantly alleviated glial nitrosative stress in the pericontusional cortex on the 3rd day after TBI
TBI-induced iNOS expression in activated microglia (29.6 ± 3.92, ***p < 0.001; Fig. 6a) and astrocytes (28.2 ± 3.48, ***p < 0.001; Fig. 6b) was significantly lower in the AMN082-treated rats than in the control-treated TBI rats (17.9 ± 3.14 versus 19.1 ± 2.06, #p < 0.05; n = 6 in each group).

Effects of AMN082 on TBI-induced nitrosative stress in glia in the pericontusional cortex on the 3rd day after TBI. Expression of the markers OX-42-N and iNOS. ***p < 0.001 compared with the sham group, #p < 0.05 compared with the TBI group; n = 6 in each group (a). Expression of the markers GFAP and iNOS. ***p < 0.001 compared with the sham group; #p < 0.05 compared with the TBI group; n = 6 in each group (b)
AMN082-treated rats had a higher NR2A/NR2B ratio in the pericontusional cortex than control-treated TBI rats
Compared with that in the sham control group (0 ± 0), the number of NR2A- and Neu-N-positive cells in the pericontusional cortex in TBI rats (27.1 ± 2.74, ***p < 0.001; Fig. 7a) was significantly increased. The AMN082-treated group exhibited a significantly increased number of neuronal NR2A-positive cells after TBI (50.0 ± 4.18, ##p < 0.01; n = 6 in each group; Fig. 7a).

Effects of AMN082 on TBI-induced NR2A and NR2B expression in the pericontusional cortex on the 3rd day after TBI. Expression of the markers NeuN and NR2A. ***p < 0.001 compared with the sham group; ##p < 0.01 compared with the TBI group; n = 6 in each group (a). Expression of the markers NeuN and NR2B. ***p < 0.001 compared with the sham group; #p < 0.05 compared with the TBI group; n = 6 in each group (b). Compared with the vehicle-treated TBI group, the AMN082-treated TBI group had a higher NR2A/NR2B ratio (1.09 ± 0.08 versus 0.89 ± 0.16) (c)
Compared with that in the TBI group, the number of NR2B- and Neu-N-positive cells (33.6 ± 4.76; n = 6; Fig. 7b) was significantly lower than that in the sham group (101.1 ± 4.03, ***p < 0.001; n = 6) and AMN082-treated group (45.7 ± 2.47, #p < 0.05; n = 6).
We further measured the NR2A/NR2B ratio in the vehicle-treated TBI and AMN082-treated TBI groups, and we found that the AMN082-treated TBI group had a higher ratio than the vehicle-treated TBI group (1.09 ± 0.08 versus 0.89 ± 0.16; Fig. 7c).
Discussion
Summary of the current study
According to our review of the literature, this study is the first to demonstrate the effects of AMN082 on TBI. The results of this study add new information to the field of traumatology. We found that treatment with AMN082 improved neuropathology and functional outcomes after TBI. At the cellular level, AMN082 decreased iNOS and 3-NT expression, decreased neuronal apoptosis, and increased NR2A/2B glutamic receptor expression in astroglia and neuronal cells. Regarding functional outcomes, the AMN082-treated TBI group exhibited a very significant improvement in motor function. Therefore, we suggest that AMN082 may be a promising treatment agent for the treatment of TBI-induced nitrosative stress-associated neuronal apoptosis, which leads to functional impairment.
Dosage of AMN082 and time course of AMN082 treatment
Several dosages of AMN082 and treatment durations are used in the neurobehavior field. AMN082 (1 mg/kg, I.P.) exerts significant antidepressant-like effects in the tail suspension test [29]. AMN082 (1 and 3 mg/kg, I.P.) decreases haloperidol (0.25 mg/kg)-induced parkinsonian-like effects [20]. AMN082 at doses of 2.5 and 5 mg/kg (I.P.) also decreases ethanol- and morphine withdrawal-induced anxiety-like behavior in the elevated plus-maze test [19]. AMN082 at a dose of 10 mg/kg (I.P.) suppresses the locomotor effect of both cocaine- and morphine-induced hyperactivity [21] When AMN082 is injected I.P. at a concentration of 10 ml/kg, activation of the mGlu7 receptor elicits antidepressant-like effects [30]. In the current study, we administered 10 mg/kg AMN082 for three consecutive days after TBI. These dose and time point of 3 days after TBI were selected because lateral fluid percussion causes motor dysfunction from 3 days to 1 year after TBI [31], and considerable evidence suggests that the brain edema volume peaks 2–3 days after TBI, which is usually when the ICP also reaches a maximum [32]. Our results support the idea that AMN082 at this dose may have therapeutic effects against TBI.
Early effects of AMN082 on physiological parameters during the initial 120 min after TBI
In the current study, there was a trend for the AMN082 (10 mg/kg, I.P.)-treated group to have a lower ICP and higher CPP than the TBI group, specifically at approximately 0 min of the initial 120-min period after TBI. We speculate that these early beneficial effects of AMN082 on ICP and CPP may lead to attenuation of secondary injury and improvements in outcomes on the 3rd day after TBI. Sukoff Rizzo et al. demonstrated that following a single injection of 10 mg/kg AMN082 (I.P.), the drug crossed the blood–brain barrier and reached peak concentrations in the brain 30 min posttreatment [10]. However, we did not investigate the relationship between the AMN082 concentration in the brain and changes in physiological parameters after TBI. Therefore, this relationship warrants further investigation.
Possible mechanisms of action of AMN082 against TBI-relate neuropathological changes and functional outcomes
Inducible NOS produces large amounts of nitric oxide (NO) and then causes the generation of 3-NT, leading to cell apoptosis [6, 33]. The occurrence of such nitrosative stress-associated neuronal apoptosis in in vivo and in vitro TBI models is consistent with peroxynitrite-mediated inhibition of the effects of caspase [33]; brain cooling [6] or memantine [5] can attenuate brain nitrosative damage, decrease the infarct volume in the cortex and improve functional outcomes after TBI, and coculture with 3NT induces motor neuron apoptosis [34]. In the current study, we found that the expression of iNOS was increased in microglia and astrocytes, neuronal apoptosis was increased in the TBI group, and these parameters were attenuated in the AMN082-treated TBI group (Table 2). Therefore, we speculate that the neuroprotective effects of AMN082 may be related to its attenuation of the nitrosative stress-associated neuronal apoptotic pathway, which leads to functional impairment after TBI.
The glutamate receptor subunit NR2A is involved in neuroprotection, and NR2B triggers destructive pathways. Both NR2A and NR2B are mainly located in neurons [4]. To date, the expression of NR2A and NR2B following AMN082 treatment after TBI has not been investigated. In the current study, we found that the expression of both NR2A and NR2B was significantly increased in the AMN082-treated TBI group and that the AMN082-treated TBI group had a higher NR2A/NR2B ratio than the vehicle-treated TBI group (Table 2). Therefore, we speculate that AMN082 may affect NR2A and NR2B expression, although AMN082 is a highly selective mGlu7 receptor agonist. However, the detailed mechanisms need to be investigated in the future.
Neuronal nitric oxide synthase (nNOS) is the predominant source of NO in neurons and has dual biological activities [35]. Its activity is related to NMDA receptor-mediated excitotoxicity [36]. In the current study (Table 2), we found that AMN082 increased nNOS expression while reducing both neuronal 3-NT expression and apoptosis and increasing the NR2A/NR2B ratio, which may be an indicator of neuroprotection [5]. Under such conditions, nNOS may play a role in neuroprotection, and nNOS may be activated by AMN082 administration.
Therefore, we infer that there are several possible mechanisms of action that may be involved. First, AMN082, a highly selective mGlu7 receptor agonist, inhibits presynaptic glutamate release from rat cerebrocortical nerve terminals [5]. Therefore, it appears to inhibit nitrosative stress development and exert neuroprotective effects by reducing glutamate release into the postsynaptic space or extrasynaptic space. Second, AMN082, through postsynaptic mGlu7 receptors, can modulate NMDA receptor activity [17, 37] or directly bind to NMDA receptors such as NR2A and NR2B-containing receptors, resulting in neuroprotection [5, 10]. Third, AMN082 binds to the mGlu7 receptor in astrocytes and microglia [23] and attenuates TBI-induced iNOS expression in activated microglia and astroglia in the pericontusional cortex. These possible mechanisms may result in reductions in neuronal 3NT expression and apoptosis and ultimately improve functional outcomes after TBI. Although there are various possibilities, the results show that AMN082 is an effective and practical agent for TBI treatment, particularly in that it exerts neuroprotective effects by attenuating nitrosative stress-associated apoptosis in the brain, which is a serious form of secondary injury after TBI.
Limitations of the current study
Several drawbacks of the current study should be mentioned. First, we only investigated the NMDA receptor subunits NR2A and NR2B but did not assess the expression of kainite or α-amino–3-hydroxy-5 methyl-4-isoxazole propionic acid (AMPA) receptors, which may influence nitrosative stress and apoptosis after TBI. Second, we only provided proof of associations between the parameters at a specific time point, i.e., 3 days, after TBI. The long-term effect of AMN082 on TBI needs to be clarified in the future. Third, we only assessed the effects of AMN082 in male rats; it should be determined whether these results can be generalized to female rats or other species and whether they are applicable to clinical practice in the future. Fourth, we only used a single functional test, the inclined plane test, to evaluate the motor functions of rats after neural damage. However, our results showed a small absolute difference in angle between the TBI + vehicle and TBI + AMN082 groups. Whether these results indicate a clinically meaningful difference in humans needs further confirmation. Additionally, multiple tests of motor function should be used in the future. Finally, this study included 3 groups: the sham-operated group, vehicle-treated TBI group and AMN082-treated TBI group. Because AMN082 has not been applied in TBI, these novel data regarding the effects of AMN082 on neurons and glia as well as TBI-induced nitrosative stress-associated neuronal apoptosis and neuronal NR2A/2B expression may be useful for critical care physicians in the clinic. However, we could consider adding another group subjected to TBI and treated with an AMN082 antagonist group, such as 6-(4-methoxyphenyl)-5-methyl-3-pyridinyl-4-isoxazolo [4,5-c] pyridin-4(5H)-one (MMPIP) [38], which is a selective mGluR7 antagonist; this addition would make the results stronger and more convincing. Therefore, many more studies are required in the future.
Conclusion
Our results demonstrate that intraperitoneal administration of AMN082 for 3 days after TBI may ameliorate TBI insults by affecting glutamate receptor-related nitrosative stress and NR2A/2B expression and decreasing neuronal apoptosis in the injured cortex. These mechanisms might explain the observed functional recovery. We also speculate that AMN082 may be a promising agent for the treatment of TBI in the acute stage.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Abbreviations
- TBI:
-
Traumatic brain injury
- SD:
-
Sprague–Dawley
- TTC:
-
Triphenyltetrazolium chloride
- TUNEL:
-
Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling
- GFAP:
-
Glial fibrillary acidic protein
- DAPA:
-
4′, 6-Diamidino-2-phenylindole
- HR:
-
Heart rate
- MAP:
-
Mean arterial pressure
- ICP:
-
Intracranial pressure
- CPP:
-
Cerebral perfusion pressure
- NR2A:
-
N-Methyl-d-aspartate receptor 2A
- NR2B:
-
N-Methyl-d-aspartate receptor 2B
- mGlu:
-
Metabotropic receptor
- iNOS:
-
Inducible nitric oxide synthase
References
Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330(9):613–22.
Ferraguti F, Shigemoto R. Metabotropic glutamate receptors. Cell Tissue Res. 2006;326(2):483–504.
Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51(1):7–61.
Brassai A, Suvanjeiev RG, Bán EG, Lakatos M. Role of synaptic and nonsynaptic glutamate receptors in ischaemia induced neurotoxicity. Brain Res Bull. 2015;112:1–6.
Wang CC, Wee HY, Hu CY, Chio CC, Kuo JR. The effects of memantine on glutamic receptor-associated nitrosative stress in a traumatic brain injury rat model. World Neurosurg. 2018;112:e719–31.
Kuo JR, Lo CJ, Chang CP, Lin MT, Chio CC. Attenuation of brain nitrostative and oxidative damage by brain cooling during experimental traumatic brain injury. J Biomed Biotechnol. 2011;2011:145214.
Gong QZ, Phillips LL, Lyeth BG. Metabotropic glutamate receptor protein alterations after traumatic brain injury in rats. J Neurotrauma. 1999;16(10):893–902.
Fei Z, Zhang X, Jiang XF, Huang WD, Bai HM. Altered expression patterns of metabotropic glutamate receptors in diffuse brain injury. Neurosci Lett. 2005;380:280–3.
Fei Z, Zhang X, Bai HM, Jiang XF, Li X, Zhang W, Hu W. Posttraumatic secondary brain insults exacerbates neuronal injury by altering metabotropic glutamate receptors. BMC Neurosci. 2007;8:96.
Rizzo S, et al. The metabotropic glutamate receptor 7 allosteric modulator AMN082: a monoaminergic agent in disguise? J Pharmacol Exp Ther. 2011;338(1):345–52.
Wang WY, Wang H, Luo Y, Jia LJ, Zhao JN, Zhang HH, Ma ZW, Xue QS, Yu BW. The effects of metabotropic glutamate receptor 7 allosteric agonist N,N′-dibenzhydrylethane-1,2-diamine dihydrochloride on developmental sevoflurane neurotoxicity: role of extracellular signal-regulated kinase 1 and 2 mitogen-activated protein kinase signaling pathway. Neuroscience. 2012;205:167–77.
Domin H, Jantas D, Śmiałowska M. Neuroprotective effects of the allosteric agonist of metabotropic glutamate receptor 7 AMN082 on oxygen-glucose deprivation- and kainate-induced neuronal cell death. Neurochem Int. 2015;88:110–23.
Jantas D, Greda A, Golda S, Korostynski M, Grygier B, Roman A, Pilc A, Lason W. Neuroprotective effects of metabotropic glutamate receptor group II and III activators against MPP(+)-induced cell death in human neuroblastoma SH-SY5Y cells: the impact of cell differentiation state. Neuropharmacology. 2014;83:36–53.
Jantas D, Greda A, Leskiewicz M, Grygier B, Pilc A, Lason W. Neuroprotective effects of mGluR II and III activators against staurosporine- and doxorubicin-induced cellular injury in SH-SY5Y cells: new evidence for a mechanism involving inhibition of AIF translocation. Neurochem Int. 2015;88:124–37.
Jantas D, Gręda A, Gołda S, Korostyński M, Lasoń W. The neuroprotective effects of orthosteric agonists of group II and III mGluRs in primary neuronal cell cultures are dependent on developmental stage. Neuropharmacology. 2016;111:195–211.
Jantas D, Lech T, Gołda S, Pilc A, Lasoń W. New evidences for a role of mGluR7 in astrocyte survival: possible implications for neuroprotection. Neuropharmacology. 2018;141:223–37.
Bradley SR, Uslaner JM, Flick RB, Lee A, Groover KM, Hutson PH. The mGluR7 allosteric agonist AMN082 produces antidepressant-like effects by modulating glutamatergic signaling. Pharmacol Biochem Behav. 2012;101(1):35–40.
Pałucha-Poniewiera A, Szewczyk B, Pilc A. Activation of the mTOR signaling pathway in the antidepressant-like activity of the mGlu5 antagonist MTEP and the mGlu7 agonist AMN082 in the FST in rats. Neuropharmacology. 2014;82:59–68.
Kotlinska JH, et al. Impact of the metabotropic glutamate receptor7 (mGlu7) allosteric agonist, AMN082, on fear learning and memory and anxiety-like behavior. Eur J Pharmacol. 2019;858:172512.
Konieczny J, Lenda T. Contribution of the mGluR7 receptor to antiparkinsonian-like effects in rats: a behavioral study with the selective agonist AMN082. Pharmacol Rep. 2013;65(5):1194–203.
Jenda M, Gawel K, Marszalek M, Komsta L, Kotlinska JH. AMN082, a metabotropic glutamate receptor 7 allosteric agonist, attenuates locomotor sensitization and cross-sensitization induced by cocaine and morphine in mice. Prog Neuropsychopharmacol Biol Psychiatry. 2015;57:166–75.
Wang CC, Kuo JR, Huang SK, Wang SJ. Metabotropic glutamate 7 receptor agonist AMN082 inhibits glutamate release in rat cerebral cortex nerve terminal. Eur J Pharmacol. 2019;823:11–8.
McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Soares H, Faden AL. Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience. 1989;28(1):233–44.
The Brain Trauma Foundation. The American Association of Neurological Surgeons. The Joint Section on Neurotrauma and Critical Care. Cerebral perfusion pressure thresholds. J Neurotrauma. 2007;24(Suppl 1):S59–S64.
Hallam TM, Floyd CL, Folkerts MM, Lee LL, Gong QZ, Lyeth BG, Muizelaar JP, Berman RF. Comparison of behavioral deficits and acute neuronal degeneration in rat lateral fluid percussion and weight-drop brain injury models. J Neurotrauma. 2004;21(5):521–39.
Kuo JR, Lo CJ, Chio CC, Chang CP, Lin MT. Resuscitation from experimental traumatic brain injury by agmatine therapy. Resuscitation. 2007;75(3):506–14.
Wang Y, Lin SZ, Chiou AL, Williams LR, Hoffer BJ. Glial cell line-derived neurotrophic factor protects against ischemia induced injury in the cerebral cortex. J Neurosci. 1997;17(11):4341–8.
Chong AJ, Lim SW, Lee YL, Chio CC, Chang CH, Kuo JR, Wang CC. The neuroprotective effects of simvastatin on high cholesterol following traumatic brain injury in rats. World Neurosurg. 2019;132:e99–108.
Podkowa K, Pilc A, Podkowa A, Sałat K, Marciniak M, Pałucha-Poniewiera A. The potential antidepressant action and adverse effects profile of scopolamine co-administered with the mGlu7 receptor allosteric agonist AMN082 in mice. Neuropharmacology. 2018;141:214–22.
Palucha A, Klak K, Branski P, van der Putten H, Flor PJ, Pilc A. Activation of the mGlu7 receptor elicits antidepressant-like effects in mice. Psychopharmacology. 2007;194(4):555–62.
Pierce JE, Smith DH, Trojanowski JQ, McIntosh TK. Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience. 1998;87(2):359–69.
Marmarou A. Pathophysiology of traumatic brain edema: current concepts. Acta Neurochir Suppl. 2003;86:7–10.
Lau A, Arundine M, Sun HS, Jones M, Tymianski M. Inhibition of caspase-mediated apoptosis by peroxynitrite in traumatic brain injury. J Neurosci. 2006;26(45):11540–53.
Peluffo H, Shacka JJ, Ricart K, Bisig CG, Martìnez-Palma L, Pritsch O, Kamaid A, Eiserich JP, Crow JP, Barbeito L, Estèvez AG. Induction of motor neuron apoptosis by free 3-nitro-l-tyrosine. J Neurochem. 2004;89(3):602–12.
Zhou L, Zhu DY. Neuronal nitric oxide synthase: structure, subcellular localization, regulation, and clinical implications. Nitric Oxide. 2009;20(4):223–30.
Eliasson MJ, Huang Z, Ferrante RJ, Sasamata M, Molliver ME, Snyder SH, Moskowitz MA. Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. J Neurosci. 1999;19(14):5910–8.
Gu Z, Liu W, We J, Yan Z. Regulation of N-methyl-d-aspartic acid (NMDA) receptors by metabotropic glutamate receptor 7. J Biol Chem. 2012;287(13):10265–75.
Palazzo E, Romano R, Luongo L, Boccella S, De Gregorio D, Giordano ME, Rossi F, Marabese I, Scafuro MA, de Novellis V, Maione S. MMPIP, an mGluR7-selective negative allosteric modulator, alleviates pain and normalizes affective and cognitive behavior in neuropathic mice. Pain. 2015;156(6):1060–73.
Acknowledgements
The authors thank Chiao-Ya Hu, who participated in this study.
Funding
This study was supported by Grants from Chi-Mei Medical Center, CMFHR10755.
Author information
Authors and Affiliations
Contributions
CCL, CCW and JRK conceived and designed the experiments. CCL and TT Eric N performed the experiments. CCW, TT Eric N and JRK analyzed the data. CCC contributed reagents/materials/analysis tools. CCL, YLL and CCW wrote the paper. All authors reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Chi Mei Medical Center’s Animal Care and Use Committee (approval no. 107061903). This work was supported by Grant No. CMFHR10755.
Consent for publication
Not applicable.
Competing interests
The authors report no biomedical financial interests or potential conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lu, CC., Nyam, TT.E., Kuo, JR. et al. The neuroprotective effects of AMN082 on neuronal apoptosis in rats after traumatic brain injury. BMC Neurosci 22, 44 (2021). https://doi.org/10.1186/s12868-021-00649-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12868-021-00649-w