
Abstract
Background
Systemic infections can influence the course of multiple sclerosis (MS), especially by driving recurrent acute episodes. The question whether the infection enhances tissue damage is of great clinical importance and cannot easily be assessed in clinical trials. Here, we investigated the effects of a systemic infection with Escherichia coli, a Gram-negative bacterium frequently causing urinary tract infections, on the clinical course as well as on neurodegeneration in experimental autoimmune encephalomyelitis (EAE), an animal model of MS.
Methods
Rats were immunized with myelin oligodendrocyte glycoprotein (MOG1–125) and challenged intraperitoneally with live E. coli K1 in the preclinical or in the clinical phase of the disease. To ensure the survival of animals, antibiotic treatment with ceftriaxone was initiated 36 h after the infection and continued for 3 consecutive days.
Results
Systemic infection with E. coli did not influence the onset of clinical EAE symptoms or disease severity. Analysis of the optic nerve and retinal ganglion cells revealed no significant changes in the extent of inflammatory infiltrates, demyelination and neurodegeneration after E. coli infection.
Conclusions
We could not confirm the detrimental effect of lipopolysaccharide-induced systemic inflammation, a model frequently used to mimic the bacterial infection, previously observed in animal models of MS. Our results indicate that the effect of an acute E. coli infection on the course of MS is less pronounced than suspected and underline the need for adequate models to test the role of systemic infections in the pathogenesis of MS.
Similar content being viewed by others

Background
Multiple sclerosis (MS) is the most frequent inflammatory disease of the central nervous system that often manifests with acute optic neuritis. The cause of MS is not known, but several factors have been shown to be associated with the risk of developing this disease including vitamins [1], smoking [2], genetic factors [3, 4], and infections [5]. A link between the first clinical attack or relapses in MS and infections has been proposed on the basis of epidemiological studies [6]. There is a growing body of evidence that the onset and progression of MS is influenced by systemic bacterial infections. In MS patients, resolution of neurological symptoms that occurred during an infection is often incomplete [7, 8]. Whether this phenomenon is a consequence of truly aggravated neurodegeneration during the systemic infection is a question of great clinical importance.
In the majority of experimental studies in this area, lipopolysaccharide (LPS), a cell wall component of Gram-negative bacteria with strong immunostimulatory properties, has been used to induce systemic inflammation. Systemic treatment of animals with LPS has been shown to promote relapses [9] and to enhance neurodegeneration in experimental models of MS [10]. Although frequently used for this purpose in animal models, LPS-induced inflammation does not accurately mimic a systemic infection with viable bacteria [11, 12]. Only few studies in humans or animal models demonstrated an exacerbating effect of an acute infection with Gram-negative bacteria on the disease course of MS and EAE [13–15]. Here, the upper respiratory tract pathogen Clamydia pneumoniae is the most thoroughly investigated candidate. However, a large number of MS patients suffers from urinary bladder dysfunction, which in turn leads to urinary tract infections (UTIs) [16] and results in significant morbidity and impairment in quality of life. Escherichia coli (E. coli) is a Gram-negative bacterium and the most common cause of UTI in MS patients. In the present study, we investigated the impact of a systemic E. coli infection during the preclinical and clinical phase of experimental autoimmune encephalomyelitis (EAE), an animal model of MS induced by immunization with myelin oligodendrocyte glycoprotein (MOG) in female Brown Norway rats, which shows a progressive disease course without any definite form of remission. We have previously demonstrated that this model strongly reflects the neurodegenerative aspects of MS. 12–14 days after immunization with MOG, more than 90% of affected animals develop acute optic neuritis, which leads to acute axonal degeneration of the optic nerve and consecutive apoptosis of retinal ganglion cells (RGCs) [17–19]. In contrast, the amount of spinal cord lesions shows certain variability in this model [20–22].
Methods
Rats
Female Brown Norway rats, 8–10 weeks of age, were used in all experiments. Animals were obtained from Charles River (Sulzfeld; Germany) and kept under environmentally controlled and pathogen-free conditions. All experiments involving animal use were performed in accordance with the relevant laws and institutional guidelines. All animal experiments were approved by the Animal Care Committee of the University Hospital of Göttingen, Germany, and by the Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit (LAVES), Braunschweig, Lower Saxony; Germany.
Retrograde labelling of RGCs
For retrograde labelling of retinal ganglion cells, rats were anesthetized by intraperitoneal injection of ketamine (Ketanest 10; 0.95 ml/kg; Atarost, Twistringen; Germany) and xylazine 2% (0.25 ml/kg; Albrecht, Aulendorf; Germany) and positioned in a stereotaxic frame. The skin was incised mediosagittaly, and holes were drilled into the skull above each superior colliculus (6.8 mm dorsal and 2 mm lateral from bregma). 2 µl of the fluorescent dye Fluoro-Gold (5% in distilled water; Fluorochrome, Englewood, CO, USA) were injected into both superior colliculi using a 10 µl Hamilton syringe.
Induction of EAE and infection with E. coli
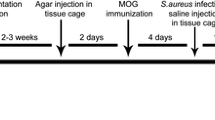
Rats were anesthetized by inhalation anaesthesia with isoflurane and injected subcutaneously at the base of the tail with a total volume of 200 µl inoculum, containing 50 µg recombinant rat MOGIgd (Kindly provided by C. Stadelmann, Department of Neuropathology, Göttingen; Germany) in saline emulsified (1:1) with complete Freund’s adjuvant (CFA; Sigma-Aldrich, St. Louis, MO, USA) containing 200 µg heat-inactivated Mycobacterium tuberculosis (strain H 37 RA; Difco Laboratories, Detroit, MI, USA). Immunized animals were randomly assigned into four different groups. The animals were infected intraperitoneally with 106 colony-forming units (CFU) E. coli K1 (originally isolated from the cerebrospinal fluid of a child with meningitis; gift G. Zysk, Düsseldorf; Germany) in 400 µl 0.9% saline (B. Braun, Melsungen; Germany) either on day 7 post immunization (“early infection” group; Figure 1a) or on day one of clinical manifestation of EAE (“late infection” group; Figure 1b). Control rats received an intraperitoneal injection of an equal volume of saline.

Experimental design. Fluoro-Gold (FG) injection was performed 2 weeks prior to MOG immunization. a In the early infection group, intraperitoneal injection with E. coli or saline was performed on day (D) seven post immunization followed by antibiotic treatment (AB) for 3 consecutive days. Day 12–17 was the usual time for clinical onset of the disease. Animals were followed until day 8 of EAE or day 21 post immunization in case of clinically healthy animals. b In the late infection group intraperitoneal injection with E. coli or saline was performed in animals on the day of clinical manifestation of EAE. Animals were followed until day 8 of EAE. Tissue was harvested from the optic nerve for histopathological analysis, retinas were flat-mounted for quantification of retinal ganglion cells.
The infection time point 7 days after immunization was chosen to elucidate whether an infection during the asymptomatic phase could modulate the time point of disease onset in MOG-EAE. The number of animals in this experiment used for analysis was 28 (n = 15, “early infection”; n = 13, control group) excluding the animals which died after E. coli infection or were sacrificed for ethical reasons. To mimic the clinical situation in MS patients, animals were infected on day one of EAE (“late infection” group). Here, we addressed the question whether the infection aggravates the clinical symptoms of EAE when it occurs in the clinical phase of the disease. The number of animals included in this experiment used for analysis was 29 (n = 15, “late infection”; n = 14, control group). Data of animals that died as a consequence of the E. coli infection were not included in the analysis.
To ensure the survival of E. coli-infected animals, antibiotic treatment was started 36 h after the infection in both groups. All animals received 100 mg/kg ceftriaxone (Hikma pharma; Klein-Winternheim; Germany) twice daily for 3 consecutive days. To confirm the systemic infection in the “late infection” group, blood samples were collected from the tail vein under inhalation anaesthesia with isoflurane 24 h after the infection (n = 8 in each group). Bacterial concentrations in blood were determined by quantitative plating of blood samples on blood agar plates followed by incubation for 24 h at 37°C. The bacterial detection limit was 100 colony forming units (CFU)/ml. Animals were followed until day 8 after the clinical manifestation of the disease (EAE d8) in the “early infection” and the “late infection” experiment or until day 21 post immunization in case of clinically healthy animals in the “early infection” experiment. The rationale for the study end point at day 8 of EAE lies in our previous studies in this animal model showing a gradual neurodegeneration with significant reduction of RGCs until day 8 of EAE. No further substantial changes neither in RGCs density nor in the histopathological changes of the optic nerve were observed beyond this time point when compared to day 21 of EAE [17, 18].
Clinical evaluation
Rats were scored for clinical signs of EAE and weighed daily. Clinical signs were scored as follows: grade 0, no symptoms; grade 0.5, distal paresis of the tail; grade 1, complete tail paralysis; grade 1.5, paresis of the tail and mild hind limb paresis; grade 2.0, unilateral severe hind limb paresis; grade 2.5, bilateral severe hind limb paresis; grade 3.0, complete bilateral hind limb paralysis; grade 3.5, complete bilateral hind limb paralysis and paresis of one front limb; grade 4, complete paralysis (tetraplegia), moribund state, or death.
Histopathology and immunohistochemistry
Histopathological evaluations were performed on paraformaldehyde-fixed, paraffin-embedded 2 µm-thick slices of the optic nerves (ONs). Luxol Fast Blue staining was performed to assess demyelination. Photographs of vertical cross sections were taken using an AxioCam MR Microscopy camera (Zeiss, Göttingen; Germany). The images were processed using Zeiss AxioVision 4.2 software to evaluate the demyelinated area as a percentage of the whole optic nerve cross-section. Immunohistochemistry was performed on paraffin-embedded ON cross-sections representing three different levels of an optic nerve. ED1 macrophages/activated microglia (MCA341R diluted 1:500; Serotec, Oxford; UK) were detected in cross-sections using avidin–biotin detection. Spleen sections served as positive control for ED1 staining. ED1-positive cells were evaluated using the following score: 0, no labelled cells; 1, a few ED1-positive cells (number countable, infiltration of less than 10% of the ON cross-section area); 2, infiltration of less than 10% of the ON cross-section area with ED1 positive cells in at least one of the three cross-sections of an ON, number of ED1-positive cells not countable; 3, infiltration of 10–50% of the ON cross-section area with labelled ED1-positive cells in at least in one of three cross-sections of an ON; 4, infiltration of 50–80% area of the ON cross-section area in at least one of the three cross-sections of an ON; 5, infiltration of more than 80% of the ON cross-section area in at least one of the three cross-sections of an ON. Bielschowsky’s silver impregnation was performed on 2 µm-thick cross-section slices of the ON to access the axonal density on the ON cross-sections. The density of axons in each ON was measured semi-quantitatively in at least nine standardized microscopic fields for each ON by using a 25 points grid system. Mean axon density was calculated for each ON.
Quantification of RGC density
At day 8 of MOG-EAE, retinas were dissected, flat-mounted on glass slides, and examined by fluorescence microscopy (Zeiss Axioplan 2) using a DAPI filter (315/395 nm). RGC densities were determined by counting labelled cells in three areas per retinal quadrant at three different eccentricities of 1/6, 3/6, and 5/6 of the retinal radius. Cell counts were performed by two independent investigators following a blinded protocol.
Statistics
Data presented here are the cumulative results of three independent experiments and presented as mean ± SEM. Student’s t test (parametric analysis) was used to compare RGC densities, demyelination and axonal densities within the ONs between two groups. For the comparison of bacterial concentrations, ED1 scores, and mean clinical scores of EAE between two groups (nonparametric analysis), the Mann–Whitney U-test was used. A P value (P) ≤0.05 was considered statistically significant.
Results
Intraperitoneal injection of E. coli leads to systemic infection with weight loss or bacteraemia
E. coli infection was either performed on day 7 post immunization (“early infection”) or on day 1 of clinical EAE (“late infection”). In the “early infection” experiment, animals infected with E. coli showed a significant weight loss after the infection whereas animals in the control group did not lose weight (P < 0.001; Figure 2a). Infected animals recovered their weight after the antibiotic treatment had been started. In contrast, in the “late infection” experiment, animals infected with E. coli did not show any significant difference in weight loss compared to the control group (P = 0.69; Figure 2b). This is probably due to the fact that during the clinical disease course of EAE both infected and control groups lost their weight rapidly. To confirm the systemic infection, E. coli concentrations were determined in blood samples collected 24 h after the infection. Animals infected with E. coli showed high bacterial counts in blood (Log10 3.64 CFU/ml ± 3.32), whereas no bacteria were detected in blood of control animals, which received saline instead of the bacterial inoculum. The overall animal mortality after E. coli infection was around 20%.

Comparison of weight loss during the course of the experiments; a represent the weight curve in the animals infected on day 7 post immunization along with their control (early infection group). b represent the weight curve of the group of animals, when infection was performed on the day of clinical onset of the disease (late infection group) with their respective control.
E. coli infection has no impact on onset and severity of clinical symptoms in MOG-EAE
In the “early infection” experiment, 7 out of 15 E. coli infected animals showed clinical disease symptoms related to the spinal cord lesions, which did not differ much from the control, where 7 out of 13 animals developed clinical score of EAE. In contrast, the incidence of optic neuritis in “early infection” experiment was 87% in animals infected with E. coli and 92% in control group. The mean clinical score of infected animals and control animals, at the end of the experiments was not significantly different [0.9 ± 0.35 (n = 15) vs. 1.04 ± 0.39 (n = 13); P = 0.8; Figure 3a]. Also the mean time point of clinical onset of EAE was similar in both groups (14.17 ± 0.48 days post immunization in the infection group and 13.50 ± 0.62 days post immunization in the control group; P = 0.41).

Comparison of clinical course of the disease: the mean clinical score represents the neurological deficit based on spinal cord lesion. a represent the clinical course of the disease in the animals infected on day 7 post immunization (early infection group). b represent the clinical course of the disease when infection was performed on the day of clinical onset of the disease (late infection group). Data are shown as mean ± SEM of the daily score.
In the “late infection” experiment, infection was induced only in animals, which showed clinical sign of EAE. The incidence of optic neuritis was 100% in both the animal groups. In this experiment, we observed that E. coli infection performed on day 1 of clinical onset of the disease also did not substantially modulate the course of EAE. Here, the mean clinical score at the end of the experiment was not significantly different between the group infected with E. coli and the respective control group [2.47 ± 0.22 (n = 15) vs. 2.71 ± 0.19 (n = 14); P = 0.51; Figure 3b].
E. coli infection does not influence demyelination, inflammatory infiltration and axonal density of the optic nerve
In optic nerves evaluated histopathologically at the end of the experiment, the extent of demyelination was similar in animals infected with E. coli on day 7 post immunization and the respective control animals [62.90 ± 9.43% (n = 15) vs. 79.18 ± 8.06% (n = 13); P = 0.20; Figure 4a, b, g]. Analysis of inflammatory infiltrates by ED1 staining also revealed no significant difference between the infection group and the control group in the “early infection” experiment [2.84 ± 0.39 (n = 15) vs. 3.45 ± 0.3 (n = 13) in the control group; P = 0.50; Figure 4c, d, h]. Bielschowsky silver impregnation was performed to assess the axonal density of the ON. The axonal loss in infected animals and control animals did not differ significantly [66 ± 7.64% (n = 15) vs. 82.19 ± 5.69% (n = 13); P = 0.10; Figure 4e, f, i].

Optical nerve (ON) histopathology for the early infection group. a, b Representative Luxol Fast Blue-stained cross sections of ONs of an animal infected with E. coli (a) and an animal from the respective control group (b). Representative examples for the number of infiltrated ED1 macrophages/activated microglia detected in the ON of an E. coli infected animal (c) and an animal from the non-infected control group (d). e, f Bielschowsky’s silver impregnation of an ON cross-section revealed no differences in axonal densities in a MOG immunized animal infected on day 7 post immunization (e) compared to a non-infected respective control animal (f). j Represents the quantitative data for numbers of surviving retinal ganglion cells (RGCs) of the animals infected with E. coli and of the animals without infection. g, h and i represent the quantitative data for demyelination, inflammatory infiltrates and for axonal counts in both the E. coli infected and control group (n = 15 infected; n = 13 control). Bar length for Luxol Fast Blue, ED1, 100 µm, and for Bielschowsky’s silver impregnation = 20 µm.
In the “late infection” experiment, we also did not find any effect of E. coli infection on inflammatory infiltration [3.94 ± 0.24 (n = 15) in the infection group vs. 3.83 ± 0.26 (n = 14) in the control group; P = 0.74; Figure 5b] or demyelination [77.06 ± 5.77% (n = 15) in the infection group vs. 72.87 ± 6.46% (n = 14) in the control group; P = 0.63; Figure 5a] of the optic nerve at the end of the experiment. Moreover, we did not find any difference in axonal density. The extent of axonal loss was 71.84 ± 5.03% (n = 15) in the infection group and 70.07 ± 5.39% (n = 14) in the control group (P = 0.81; Figure 5c).

Optic nerve (ON) histopathology in the late infection group. a, b, and c represents the quantitative data for demyelination, inflammatory infiltrates (ED1 score), and axonal counts in the optic nerve from the animals infected on the day of clinical manifestation of EAE and the animals from respective control group. d Represents the quantitative data for numbers of surviving retinal ganglion cells (RGCs) of the animals infected with E. coli on day 1of EAE and of the animals without infection.
E. coli infection does not affect neuronal viability
To evaluate the impact of E. coli infection on neuronal damage in MOG-EAE, numbers of surviving RGCs were counted on Fluoro-Gold-labeled retinal flat mounts. In accordance with the histopathological changes in the ON, analysis of RGC density did not reveal any significant differences between infected animals and the respective control animals both in the “early infection” experiment [1,411 ± 88 vs. 1,248 ± 69/mm2; P = 0.15; Figure 4j, (n = 25 each)] and the “late infection” experiment [967 ± 33 vs. 1,116 ± 78/mm2; P = 0.08; Figure 5d, (n = 16 each)]. For RGC counts ‘‘n’’ indicate the number of eyes used for quantification. RGC counts in both experiments were significantly lower than those of healthy control animals in our previous studies (2,567 ± 89; mean ± SEM) [23], indicating a loss of RGCs in infected and non-infected animals.
Discussion and conclusions
There is a growing body of evidence that the onset and progression of MS is influenced by systemic infections caused by bacteria or viruses. Bacterial infections can aggravate neurological symptoms, which remain even after the infection is resolved, while some appears to be protective [24]. A better understanding of the mechanisms underlying these phenomena would be an essential step for a rational treatment of infections in MS patients. In spite of the rising interest in the field linking infection and MS, only few studies examined the impact of real systemic infections with viable bacteria in animal models of MS. In the present study, we investigated whether systemic infection with E. coli, a Gram-negative bacterium frequently causing urinary tract infection, has a disease-modulating effect on EAE.
Clinical as well as experimental data indicate the preclinical phase of MS to be an “at risk” period with the greatest susceptibility to infections [25, 26]. Systemic infection with Streptococcus pneumoniae 7 days after immunization with MOG aggravated EAE symptoms in mice, whereas the severity of EAE symptoms was unaltered in mice infected 2 days before or 21 days after MOG-immunization [27]. For this reason, in our study, animals were infected at day 7 post immunization corresponding to the induction phase of EAE. Intraperitoneal injection of E. coli induced weight loss in EAE animals indicating severe systemic infection and sickness behavior in animals. However, systemic E. coli infection during the latent phase of EAE did not influence the time point of onset of clinical symptoms or the disease severity of EAE in this model. To mimic the situation in MS patients at the beginning of a clinical event, animals were infected on the first day of onset of EAE symptoms. Despite the presence of high bacterial counts in the systemic circulation of infected animals, the severity of EAE symptoms was not influenced by E. coli infection.
It is poorly understood whether systemic infections lead to tissue damage in the CNS with increased neurodegeneration, a major histopathological correlate of disability in MS patients. The results of magnetic resonance imaging (MRI) studies on the influence of extra cerebral infections on the number of contrast-enhancing MS lesions as a marker of disease activity are inconsistent [28, 29]. Moreover, reliable markers for neurodegeneration in MS patients do not exist. For this reason, much of the knowledge about the interaction of systemic infection and autoimmunity in MS is derived from studies in EAE models. Previous histopathological analyses revealed that a single dose of LPS increased the inflammatory response and demyelination in EAE lesions [30]. In addition, it led to a switch in the macrophage/microglia phenotype towards proinflammation increasing axonal damage in an EAE model [10]. In contrast, our histopathological analysis performed on cross-sections of the optic nerve did not reveal any significant increase in inflammatory infiltrates in the animals infected with E. coli either on day 7 post immunization or on day 1 of EAE. In our study, we also did not observe any significant differences in other histopathological hallmarks such as demyelination and axonal damage between animals infected with E. coli and the respective control animals. In accordance with the histopathology of the optic nerve, the survival of RGCs was also not significantly influenced by the E. coli infection.
Given the disease-modulating effect of systemic LPS in EAE [9, 10] our data are surprising. In accordance with our observations, the disease-aggravating effect of LPS observed in other neurodegenerative disease models [31, 32] could also not be confirmed in mouse models of Alzheimer’s and Parkinson’s disease when real systemic infections were induced by viable S. pneumoniae [31, 33] or E. coli (Schütze et al., unpublished observation).
There are several hypotheses that may explain the discrepancy between the effect of LPS-induced systemic inflammation and systemic E. coli infection on clinical symptoms and neurodegeneration in EAE. The LPS doses applied in the studies mentioned above were very high in comparison to plasma endotoxin levels measured in patients with sepsis [34]. Consequently, the levels of the released pro-inflammatory cytokines (e.g. TNF-alpha, IL-1) in sepsis patients are much lower than those typically found after endotoxin infusion [35–37]. In-vitro data suggest that microglial activation and microglia-mediated neurodegeneration depend on the dose of the inflammatory stimulus [38, 39]. We assume that stimulation of the innate immune response in the CNS induced by the systemic E. coli infection in our study was less severe than that achieved by high doses of LPS and did not reach a sufficient level to induce neurodegeneration.
Treatment with the β-lactam antibiotic ceftriaxone was initiated 36 h after the infection in our experiments which stops bacterial application and terminates the effect of infection. Ceftriaxone has a bacteriolytic mode of action, thereby causing the rapid release of high amounts of bacterial products acting as strong stimuli of innate immunity [40]. Therefore, it is tempting to assume that an antibiotic treatment of infection may have an at least temporary effect on the course of EAE. The fact that the control group reached more severe disease scores in both the experiments argues against an aggravation of disease symptoms by E. coli infection followed by antibiotic treatment in the EAE model. The overall mortality of the E. coli infection with the antibiotic treatment protocol employed was approximately 20% in our study. Leaving the infection untreated would have resulted in an unacceptably high mortality. The temporary systemic infection followed by antibiotic treatment mimics the situation in well-treated acutely ill patients. However, the effect of a chronic E. coli on the course of EAE remains to be determined.
In conclusion, in our model of MOG-EAE neither the clinical course nor the neurodegeneration was aggravated by systemic infection with E. coli. Our results suggest that the effect of moderately severe adequately treated systemic infections by E. coli on MS is less pronounced than suspected. Given the lack of efficacy of clinical and experimental trials based on targets identified in LPS studies [12, 41], our data confirm the complexity of systemic infections with viable bacteria compared to LPS-induced inflammation and underline the necessity of adequate infection models for testing the role of systemic infection in the pathogenesis of MS.
Abbreviations
- CNS:
-
central nervous system
- CFU:
-
colony forming units
- EAE:
-
experimental autoimmune encephalomyelitis
- LPS:
-
lipopolysaccharide
- UTI:
-
urinary tract infections
- MOG:
-
myelin oligodendrocyte glycoprotein
- MS:
-
multiple sclerosis
- ON:
-
optic nerve
- Pi:
-
post immunization
- RGCs:
-
retinal ganglion cells
References
Correale J, Ysrraelit MC, Gaitan MI (2009) Immunomodulatory effects of Vitamin D in multiple sclerosis. Brain 132:1146–1160
Jafari N, Hintzen RQ (2011) The association between cigarette smoking and multiple sclerosis. J Neurol Sci 311:78–85
Munoz-Culla M, Irizar H, Otaegui D (2013) The genetics of multiple sclerosis: review of current and emerging candidates. Appl Clin Genet 6:63–73
Gourraud PA, Harbo HF, Hauser SL, Baranzini SE (2012) The genetics of multiple sclerosis: an up-to-date review. Immunol Rev 248:87–103
Tselis A (2012) Epstein-Barr virus cause of multiple sclerosis. Curr Opin Rheumatol 24:424–428
Giovannoni G, Ebers G (2007) Multiple sclerosis: the environment and causation. Curr Opin Neurol 20:261–268
Panitch HS (1994) Influence of infection on exacerbations of multiple sclerosis. Ann Neurol 36(Suppl):S25–S28
Buljevac D, Flach HZ, Hop WC, Hijdra D, Laman JD, Savelkoul HF et al (2002) Prospective study on the relationship between infections and multiple sclerosis exacerbations. Brain 125:952–960
Nogai A, Siffrin V, Bonhagen K, Pfueller CF, Hohnstein T, Volkmer-Engert R et al (2005) Lipopolysaccharide injection induces relapses of experimental autoimmune encephalomyelitis in nontransgenic mice via bystander activation of autoreactive CD4+ cells. J Immunol 175:959–966
Moreno B, Jukes JP, Vergara-Irigaray N, Errea O, Villoslada P, Perry VH et al (2011) Systemic inflammation induces axon injury during brain inflammation. Ann Neurol 70:932–942
Remick DG, Ward PA (2005) Evaluation of endotoxin models for the study of sepsis. Shock 24(Suppl 1):7–11
Dieelberg C, Ribes S, Michel U, Redlich S, Brück W, Nau R et al (2012) Follistatin does not influence the course of Escherichia coli K1 sepsis in a mouse model. Shock 38:615–619
Buljevac D, Verkooyen RP, Jacobs BC, Hop W, van der Zwaan LA, van Doorn PA et al (2003) Chlamydia pneumoniae and the risk for exacerbation in multiple sclerosis patients. Ann Neurol 54:828–831
Du C, Yao SY, Ljunggren-Rose A, Sriram S (2002) Chlamydia pneumoniae infection of the central nervous system worsens experimental allergic encephalitis. J Exp Med 196:1639–1644
Munger KL, Peeling RW, Hernán MA, Chasan-Taber L, Olek MJ, Hankinson SE et al (2003) Infection with Chlamydia pneumoniae and risk of multiple sclerosis. Epidemiology 14:141–147
Nakipoglu GF, Kaya AZ, Orhan G, Tezen O, Tunc H, Ozgirgin N et al (2009) Urinary dysfunction in multiple sclerosis. J Clin Neurosci 16:1321–1324
Hobom M, Storch MK, Weissert R, Maier K, Radhakrishnan A, Kramer B et al (2004) Mechanisms and time course of neuronal degeneration in experimental autoimmune encephalomyelitis. Brain Pathol 14:148–157
Maier K, Kuhnert AV, Taheri N, Sättler MB, Storch MK, Williams SK et al (2006) Effects of glatiramer acetate and interferon-beta on neurodegeneration in a model of multiple sclerosis: a comparative study. Am J Pathol 169:1353–1364
Meyer R, Weissert R, Diem R, Storch MK, de Graaf KL, Kramer B et al (2001) Acute neuronal apoptosis in a rat model of multiple sclerosis. J Neurosci 21:6214–6220
Kumar P, Kretzschmar B, Herold S, Nau R, Kreutzfeldt M, Schütze S et al (2015) Beneficial effect of chronic Staphylococcus aureus infection in a model of multiple sclerosis is mediated through the secretion of extracellular adherence protein. J Neuroinflamm 12:22
Rau CR, Hein K, Sättler MB, Kretzschmar B, Hillgruber C, McRae BL et al (2011) Anti-inflammatory effects of FTY720 do not prevent neuronal cell loss in a rat model of optic neuritis. Am J Pathol 178:1770–1781
Storch MK, Stefferl A, Brehm U, Weissert R, Wallström E, Kerschensteiner M et al (1998) Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathol 8:681–694
Sättler MB, Togni M, Gadjanski I, Sühs KW, Meyer N, Bähr M et al (2008) Strain-specific susceptibility for neurodegeneration in a rat model of autoimmune optic neuritis. J Neuroimmunol 193:77–86
Kira JI (2015) Helicobacter pylori infection might prove the hygiene hypothesis in multiple sclerosis. J Neurol Neurosurg Psychiatry 86(6):591–592. doi:10.1136/jnnp-2014-309759
Sibley WA, Bamford CR, Clark K (1985) Clinical viral infections and multiple sclerosis. Lancet 1:1313–1315
Tauber SC, Nau R, Gerber J (2007) Systemic infections in multiple sclerosis and experimental autoimmune encephalomyelitis. Arch Physiol Biochem 113:124–130
Herrmann I, Kellert M, Schmidt H, Mildner A, Hanisch UK, Brück W et al (2006) Streptococcus pneumoniae infection aggravates experimental autoimmune encephalomyelitis via Toll-like receptor 2. Infect Immun 74:4841–4848
Kalkers NF, Vrenken H, Uitdehaag BM, Polman CH, Barkhof F (2002) Brain atrophy in multiple sclerosis: impact of lesions and of damage of whole brain tissue. Mult Scler 8:410–414
Edwards S, Zvartau M, Clarke H, Irving W, Blumhardt LD (1998) Clinical relapses and disease activity on magnetic resonance imaging associated with viral upper respiratory tract infections in multiple sclerosis. J Neurol Neurosurg Psychiatry 64:736–741
Serres S, Anthony DC, Jiang Y, Broom KA, Campbell SJ, Tyler DJ et al (2009) Systemic inflammatory response reactivates immune-mediated lesions in rat brain. J Neurosci 29:4820–4828
Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH (2005) Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci 25:9275–9284
Nguyen MD, D’Aigle T, Gowing G, Julien JP, Rivest S (2004) Exacerbation of motor neuron disease by chronic stimulation of innate immunity in a mouse model of amyotrophic lateral sclerosis. J Neurosci 24:1340–1349
Ebert S, Goos M, Rollwagen L, Baake D, Zech WD, Esselmann H et al (2010) Recurrent systemic infections with Streptococcus pneumoniae do not aggravate the course of experimental neurodegenerative diseases. J Neurosci Res 88:1124–1136
Kojika M, Sato N, Yaegashi Y, Suzuki Y, Suzuki K, Nakae H et al (2006) Endotoxin adsorption therapy for septic shock using polymyxin B-immobilized fibers (PMX): evaluation by high-sensitivity endotoxin assay and measurement of the cytokine production capacity. Ther Apher Dial 10:12–18
Michie HR, Manogue KR, Spriggs DR, Revhaug A, O’Dwyer S, Dinarello CA et al (1988) Detection of circulating tumor necrosis factor after endotoxin administration. N Engl J Med 318:1481–1486
Cannon JG, Tompkins RG, Gelfand JA, Michie HR, Stanford GG, van der Meer JW et al (1990) Circulating interleukin-1 and tumor necrosis factor in septic shock and experimental endotoxin fever. J Infect Dis 161:79–84
Hesse DG, Tracey KJ, Fong Y, Manogue KR, Palladino MA Jr, Cerami A et al (1988) Cytokine appearance in human endotoxemia and primate bacteremia. Surg Gynecol Obstet 166:147–153
Schutze S, Loleit T, Zeretzke M, Bunkowski S, Brück W, Ribes S et al (2012) Additive microglia-mediated neuronal injury caused by amyloid-beta and bacterial TLR agonists in murine neuron-microglia co-cultures quantified by an automated image analysis using cognition network technology. J Alzheimers Dis 31:651–657
Ebert S, Gerber J, Bader S, Mühlhauser F, Brechtel K, Mitchell TJ et al (2005) Dose-dependent activation of microglial cells by Toll-like receptor agonists alone and in combination. J Neuroimmunol 159:87–96
Nau R, Eiffert H (2002) Modulation of release of proinflammatory bacterial compounds by antibacterials: potential impact on course of inflammation and outcome in sepsis and meningitis. Clin Microbiol Rev 15:95–110
Eskandari MK, Bolgos G, Miller C, Nguyen DT, DeForge LE, Remick DG (1992) Anti-tumor necrosis factor antibody therapy fails to prevent lethality after cecal ligation and puncture or endotoxemia. J Immunol 148:2724–2730
Author’s contributions
Contributed to conception and design: KH, MB, RN, PK. Contributed to acquisition of data: PK, KF, RS, ZM. Contributed to analysis and editing of data: PK, KH, KF, SS, RN. Drafted and/or revised the article: PK, SS, RN, MB, KH. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by the Else-Kröner-Fresenius-Stiftung. KH was funded by the Medical Faculty of the University of Goettingen; Germany. PK was (partly) supported by the Goettingen Graduate School for Neurosciences und Molecular Biosciences (DFG Grant GSC226/2). This publication was supported financially by the open access grant program of the German Research Foundation (DFG) and the open access publication fund of the University of Göttingen. We thank Irina Graf and Birte Könnecke for expert technical assistance.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kumar, P., Friebe, K., Schallhorn, R. et al. Systemic Escherichia coli infection does not influence clinical symptoms and neurodegeneration in experimental autoimmune encephalomyelitis. BMC Neurosci 16, 36 (2015). https://doi.org/10.1186/s12868-015-0172-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12868-015-0172-4