
Background
Mycoplasma pneumoniae (M. pneumoniae) is an important pathogen of community-acquired pneumonia in children. The factors contributing to the severity of illness caused by M. pneumoniae infection are still under investigation. We aimed to evaluate the sensitivity of common M. pneumoniae detection methods, as well as to analyze the clinical manifestations, genotypes, macrolide resistance, respiratory microenvironment, and their relationship with the severity of illness in children with M. pneumoniae pneumonia in Wuhan.
Results
Among 1,259 clinical samples, 461 samples were positive for M. pneumoniae via quantitative polymerase chain reaction (qPCR). Furthermore, we found that while serological testing is not highly sensitive in detecting M. pneumoniae infection, but it may serve as an indicator for predicting severe cases. We successfully identified the adhesin P1 (P1) genotypes of 127 samples based on metagenomic and Sanger sequencing, with P1-type 1 (113/127, 88.98%) being the dominant genotype. No significant difference in pathogenicity was observed among different genotypes. The macrolide resistance rate of M. pneumoniae isolates was 96% (48/50) and all mutations were A2063G in domain V of 23S rRNA gene. There was no significant difference between the upper respiratory microbiome of patients with mild and severe symptoms.
Conclusions
During the period of this study, the main circulating M. pneumoniae was P1-type 1, with a resistance rate of 96%. Key findings include the efficacy of qPCR in detecting M. pneumoniae, the potential of IgM titers exceeding 1:160 as indicators for illness severity, and the lack of a direct correlation between disease severity and genotypic characteristics or respiratory microenvironment. This study is the first to characterize the epidemic and genomic features of M. pneumoniae in Wuhan after the COVID-19 outbreak in 2020, which provides a scientific data basis for monitoring and infection prevention and control of M. pneumoniae in the post-pandemic era.
Similar content being viewed by others


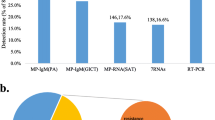
Background
M. pneumoniae belongs to the class of the Mollicutes and is one of the smallest known free-living cell organisms. M. pneumoniae infections affect both the upper and lower respiratory tracts and are endemic and epidemic worldwide in individuals of all ages [1]. In children, M. pneumoniae is a significant cause of community-acquired pneumonia (CAP), accounting for 10%-40% of cases [2]. The main symptoms of M. pneumoniae infection include headache, sore throat, fever, cough, and other common respiratory symptoms [3]. While M. pneumoniae infection can occur at any time of the year, its incidence is higher during late fall or winter [4]. The illness spreads via droplets and tends to spread within families or communities with an incubation period of 2 to 4 weeks. Incidences of M. pneumoniae infection showed three- to five- year epidemic cycles in multiple countries [5, 6], but the mechanism for such epidemic cycle remains unclear. A study argues that minor variation in the duration of immunity at the population level must be considered essential for the M. pneumoniae epidemic cycle [7].
The research on methods for detecting M. pneumoniae infection is important and necessary. Nucleic acid assays, such as qPCR, are prevalently employed in the detection of M. pneumoniae infections, owing to their high sensitivity and specificity [8, 9]. Serological assays are often preferred in clinical settings due to their rapid processing and ability to yield timely diagnostic results. However, it is important to note that, despite their widespread use in China, serological assays exhibit a relatively lower sensitivity compared to other diagnostic approaches [10, 11]. Therefore, in light of these limitations, it becomes imperative to reassess the value of serological testing in the accurate diagnosis of diseases.
M. pneumoniae isolates can be classified into two major genetic groups, designated subtype 1 (P1-type1) and subtype 2 (P1-type2), based on variations in repetitive elements RepMP2/3 and RepMP4 in the P1 protein gene [12]. The prevalence of genotypes varies in different countries and regions, and there are characteristic periodic shifts in genotypes [13, 14]. Multi-locus sequence typing (MLST) is another genotyping method for M. pneumoniae, which divides M. pneumoniae strains into 46 sequence types (STs) based on nucleotide polymorphisms of eight housekeeping genes (pubMLST, https://pubmlst.org/) [15]. The relationship between M. pneumoniae genotypes and the severity of clinical manifestations in infected patients is a widely debated issue.
For the treatment of M. pneumoniae infections in children, macrolides such as azithromycin and clarithromycin are generally considered as the first choice [16]. However, the widespread use of antibiotics has led to the emergence of macrolide-resistant M. pneumoniae (MRMP) [17]. This had led to an increasing number of severe M. pneumoniae pneumonia cases in children, which are more likely to develop serious pulmonary and extrapulmonary complications [18, 19]. Naturally occurring macrolide resistance in M. pneumoniae is due to mutations in various positions in the 23S rRNA, including C2617G, A2063G/C/T, and A2064G/C [19], with the transition mutation A2063G being the most commonly reported. Monitoring the prevalence and mutation types of MRMP will aid in clinical diagnosis and treatment.
In many areas of China, M. pneumoniae epidemic surveillance is not conducted, and there has been little data on M. pneumoniae genotypes, macrolide resistance, and epidemiology in Wuhan. Although the outbreak of M. pneumoniae pneumonia in Wuhan during the winter of 2019 was initially estimated, the monitoring of M. pneumoniae was delayed due to the impact of the coronavirus disease 2019 (COVID-19) outbreak. In recent years, there has been an increase in cases requiring hospitalization for treatment among children with M. pneumoniae infection. Therefore, the objective of this study was to elucidate the characteristics of this M. pneumoniae epidemic in Wuhan, including the incidence, genotype, drug resistance characterization of M. pneumoniae.
Results
Epidemiological characteristics of M. pneumoniae in Wuhan between 2020 and 2022
From October 2020 to March 2022, 1,259 clinical samples from children diagnosed with pneumonia in Wuhan were collected. Of the 1,259 children diagnosed with CAP, 461 (36.6%) cases were determined to be positive for M. pneumoniae via qPCR. After excluding patients with underlying diseases, 371 patients remained for further analysis. In terms of patient age, preschoolers (302/371, 81.4%) accounted for the majority of the M. pneumoniae pneumonia cases. However, the infection of M. pneumoniae in preschoolers, especially children aged 1–24 months, is milder in symptoms, while the infection in school-age children aged 6–11 years is more likely to develop into severe. There was no correlation between the gender of infected children and disease severity (Table 1). In terms of the clinical symptoms of patients, the most common symptoms observed in children included fever (260, 70.3%), paroxysmal cough (324, 87.3%), and in some cases wheezing (77, 20.8%). The median duration of fever was 4 days (Table 1). From the serological test results, it can be observed that patients with severe symptoms showed significantly lower qPCR cycle threshold (Ct) values and higher IgM titers. IgM titers ≥ 320 were considered to be a cause for concern, as patients with such titers are at a higher risk of developing severe disease (Table 1). In terms of the epidemic time of M. pneumoniae pneumonia, this study showed that M. pneumoniae infections have a high incidence during winter and early spring. M. pneumoniae infections can account for 40–50% of community-acquired pneumonia cases from January to March (Fig. 1).

Epidemiological data about M. pneumoniae pneumonia. "MP" refers to patients diagnosed with M. pneumoniae pneumonia, while "All" refers to all patients with pneumonia
The most common extra-pulmonary manifestation was gastrointestinal (20.5%), others included vomit (10.8%), diarrhoea (11.9%) and liver function damage (3.2%). 6.2% of all children had some type of rash. Uncommonly, 14 (3.8%) children have experienced cardiovascular system complications, with three cases of Kawasaki disease and one case of pericardial effusion. Additionally, there were two cases of embolism and one case of encephalitis. Further analysis focusing on extra-pulmonary complications reveals that there are significant differences in gastrointestinal, dermatological, cardiovascular system complications between the mild and severe groups (Table 2).
Evaluation of detection methods for M. pneumoniae infection
Serological testing and qPCR are common laboratory methods for detecting M. pneumoniae and a first-time IgM titer greater than 1:160 is considered as evidence of M. pneumoniae infection. The relationship between Ct values and IgM titers was analyzed, but no significant association was found (Fig. 2a). This finding suggests that IgM titers may not be a sensitive method for diagnosing M. pneumoniae infection, as they are inconsistent with nucleic acid detection results.

Relationships between antibody titers, Ct value and metagenome sequencing coverage of M. pneumoniae samples. a. Distribution of Ct values of samples with different antibody titers. Paired t-tests were used to measure the differences between groups. D40/D80/D160/D320 means Mycoplasma pneumoniae IgM antibody titer. b. Scatter plot of Ct values and 10 × genome coverage of metagenomic-sequenced samples
Metagenomic sequencing has been proven to be a powerful tool for various pathogens' identification. However, it is necessary to evaluate the efficiency of obtaining M. pneumoniae genomes through metagenomic sequencing. The reads were aligned to the reference genome of M. pneumoniae to determine the efficiency of metagenomic sequencing in obtaining the genome. While genome coverage appeared to be higher in patients with severe symptoms, this difference was statistically significant at sequencing depth greater than 30 × (Figure S1 a). Despite the amount of sequenced data is enormous, only a small portion of reads come from M. pneumoniae (Figure S1 b). Meanwhile, except for some samples with extremely low Ct values, the Ct value seemed to have little relationship with the coverage of the M. pneumoniae genome (Fig. 2b). This suggests that performing metagenomic sequencing directly on clinical samples may not be the optimal approach for obtaining the M. pneumoniae genome.
The genomes of M. pneumoniae are highly conserved
To obtain sufficient variation for phylogenetic analysis, we performed pan-genome analysis on 87 genomes of M. pneumoniae. The results showed that the total pan-genome was composed of 937 homologous genes (homologs) with a core genome of 624 (66.6%) conserved homologs presenting in 100% of all M. pneumoniae isolates (Fig. 3a). Examination of the gene accumulation curve (Fig. 3b) revealed that as the number of genomes increased, the number of core genes quickly decreased to a stable level, indicating a highly conserved genome in M. pneumoniae. The total number of genes also approached saturation, indicating that the pan-genome is open but still relatively constrained.

Pan-genomes of M. pneumoniae. a. The pan-genome pie chart shows the breakdown of genes and the number of isolates they are present in. b. Heaps law chart representation regarding conserved genes vs total genes in 87 genomes
Prevalence of M. pneumoniae genotypes in Wuhan
Since there were few studies on M. pneumoniae genotype surveillance in Wuhan previously, it is crucial to understand the epidemic trend of M. pneumoniae genotypes and the formulation of prevention and control measures accordingly.
The results of the phylogenetic analysis showed that the M. pneumoniae epidemic was a mixed infection of two P1 genotypes (Fig. 4). Among the 127 P1-type strains identified, 113 (88.98%) were P1-type 1 and 14 were P1-type 2 (11.02%). Additionally, the MLST genotype of M. pneumoniae was successfully distinguished in 67 samples by mNGS and Sanger sequencing, of which 2 (3%) were ST-7, 5 (7.5%) were ST-14, and 60 (89.5%) were ST-3 (Fig. 4).

Prevalence of M. pneumoniae genotypes of the present epidemic. Maximum likelihood phylogenetic tree of the core genome of 87 M. pneumoniae isolates, of which 80 are from GenBank. The blue branches represent P1-type1 of M. pneumoniae, and the green branches represent P1-type 2. Marked in red are samples or sequence types in this study. The pie chart represents the proportion of M. pneumoniae MLST genotypes. ST, sequence typing
To explore the differences in the pathogenic potential of different genotypes, we counted the number of mild and severe patients with different P1 genotypes. In sample P1-type 1, there are 28 samples from mild patients and 85 samples from severe patients. In sample P1-type 2, there are 3 samples from mild patients and 11 samples from severe patients. The result of Fisher's exact test showed that M. pneumoniae genotype had no effect on disease severity (p-value = 1) (Table 1).
Comparison of microbial composition and diversity in different clinical samples
As we did not observe a correlation between M. pneumoniae genotypes and the severity of illness, we wondered to investigate whether there are differences in the respiratory microbiota of patients with different disease severities. Our results showed that although infected with M. pneumoniae, the top 20 most abundant genera in OP samples were mainly colonized conditional pathogens, such as Streptococcus, Prevotella, Veillonella and Neisseria (Fig. 5a). Alpha diversity analysis did not show any significant difference in the number of microbial genera (Chao1 and observed OTU index) and the within-sample diversity (Shannon and Simpson index) among OP samples (Fig. 5b). Beta diversity analysis also revealed no differences in microbial composition among OP samples (Fig. 5C). However, there were differences in microbial diversity and composition between BALF and OP samples, which were attributed to sampling site (Fig. 5b, c). The relative abundance of genera did not differ significantly between OP samples from mild and severe patients (Figure S2). Linear discriminant analysis (LDA) scores showed Mycoplasma as a marker genus for BALF (Fig. 5d), while biomarkers in OP included genera such as like Streptococcus, Prevotella, Neisseria, Haemophilus, among others. Therefore, it was concluded that the upper respiratory microbiome of M. pneumoniae infected patients was not affected by the severity of symptoms.

Comparison of microbial composition and diversity in different clinical samples. a The microbial composition of 46 individual donors is shown, with only the top 20 microbial genera with the highest abundance displayed. b The alpha diversity of OP microbiota from patients with mild and severe symptoms and BALF in critically ill patients was calculated according to the genus-level Chao1 index, observed OTUs, Shannon index, and Simpson index. Paired t-tests were used to compare the differences between groups. c Beta diversity of OP microbiota from patients with mild and severe symptoms and BALF in critically ill patients. Differences between groups were tested by PERMANOVA. d LEfSe identifies microbial markers in the three clinical subgroups with a linear LDA score greater than 4 considered a biomarker. e The pie chart displays the number of oropharyngeal swab samples with detected resistance genes relative to the total number of samples
Antibiotic resistance mutations in 23S rRNA gene of M. pneumoniae and resistance genes in the microbiome
Macrolide-resistant M. pneumoniae is a potential factor leading to intractable M. pneumoniae pneumonia, and the resistance rate of M. pneumoniae in China is very high. Therefore, monitoring the resistance rate of M. pneumoniae is particularly important. We successfully amplified 23S rRNA sequences from 50 clinical samples, 48 (96%) of which contained the A2063G mutation. Besides, drug resistance mutations at positions 2064 and 2617 were not found in this study.
De novo assembly of other microorganisms from the OP samples also revealed the presence of resistance genes to various antibiotics, including Tetracycline, Macrolide, Amikacin, Gentamicin, Kanamycin, Tobramycin, Lincosamides, Streptogramin, Cephalosporin, and Beta-lactam in many OP samples. However, these antibiotic resistance genes were rarely identified in BALF samples (Fig. 5e, Table S3). The sample shows a remarkably high frequency of resistance genes to three types of antibiotics: Tetracycline, Macrolide, and Beta-lactam. The presence of these resistance genes in other microorganisms may pose challenges for the treatment of M. pneumoniae co-infection. Notably, these resistance genes were frequently detected in genera such as Streptococcus, Enterococcus, Pseudomonas, Escherichia and Prevotella, which were less abundant in BALF and thus rarely detected (Table S4).
Discussion
In mainland China, most studies related to M. pneumoniae have been conducted in Beijing and North China [20, 21], while few have been conducted in other regions. Xue et al. were the first to report the genotype and antimicrobial resistance of M. pneumoniae in 6 regions simultaneously [22]. In this study, we assessed the genotype and macrolide resistance mutation of M. pneumoniae in children in Wuhan for the first time. More importantly, this is the first surveillance data on M. pneumoniae in Wuhan since the COVID-19 outbreak. M. pneumoniae is underestimated because of its self-limiting nature, but it still causes up to 40% of CAP and contributes to the social burden. Our results showed that M. pneumoniae pneumonia accounted for 36.6% of community-acquired pneumonia cases, making it a respiratory pathogen that deserves our close attention. Our study inferred that 88.98% of infected M. pneumoniae strains were classified as P1-type 1 and 11.02% were P1-type 2. This result was similar to a previous study in that 89.1% of M. pneumoniae in Beijing were classified as P1-type 1 in 2018 [22]. This study is the first to investigate M. pneumoniae genotypes in Wuhan, which provides a foundation for future studies on M. pneumoniae genotype shift cycles in Wuhan. What’s more, it remains controversial whether clinical manifestations of lower respiratory tract infections (LRTI) in children differ between the two M. pneumoniae P1 genotypes. Some studies indicated that the two P1 genotypes may have different pathogenic potentials and that LRTI with P1-type 2 strains may have a more severe disease course than those with P1-type 1 strains in children [23, 24]. However, the result in other areas may be quite the opposite [25]. In the present study, we did not find any specific genotype associated with more severe clinical symptoms.
Given that many regions lack systematic monitoring of M. pneumoniae pneumonia, we recommend establishing genotype and macrolide resistance monitoring systems for M. pneumoniae in more cities in China to facilitate the study of genotype switching and macrolide resistance and their relationship with clinical manifestations. Furthermore, the development of a novel method to directly obtain the P1 genotype of M. pneumoniae from clinical samples without sequencing is necessary, which would facilitate the monitoring of M. pneumoniae.
The serological assay is the most commonly used method for detecting M. pneumoniae infection. A fourfold increase in IgG titer in acute and convalescent sera is considered a diagnostic criterion of acute M. pneumoniae respiratory infection, while an initial IgM antibody detection titer greater than or equal to 1:160 serve as a criterion for recent M. pneumoniae infection [26]. If IgM is measured at least 7 days after the onset of symptoms, acute infection can be detected. However, if the test is performed earlier, the result may be negative [27]. It is risky to diagnose acute M. pneumoniae respiratory infection based solely on a single IgM test, and it is also difficult to implement in the practical diagnosis of children. qPCR is suitable for detecting M. pneumoniae infection, while IgM titers greater than 1:160 can be used as a predictor of severity. The traditional way of obtaining M. pneumoniae genome is through culture and whole genome sequencing. However, this approach is time-consuming and has a low yield. Therefore, we attempted to perform metagenomic sequencing directly on clinical specimens. Nevertheless, this method only provided a limited amount of genomic information. Out of the large amount of data produced by sequencing, only a few reads were related to M. pneumoniae. Therefore, this strategy is highly dependent on samples with a high M. pneumoniae abundance to obtain high-quality genomes.
Since the first identification of an MRMP strain in pediatric patients in Japan in 2000 [17], there has been a notable increase in the prevalence of such strains, highlighting a growing concern for public health and antibiotic resistance management. The emergence of MRMP brought challenges in the clinical treatment of M. pneumoniae infection [28]. The highest proportion of MRMP infections was observed in the Western Pacific region worldwide, with an increasing trend observed over the years. China had the highest proportion of MRMP infections within this region, followed by Japan and other countries [29]. Mutation A2063G is the most common mutation in macrolide-resistant M. pneumoniae, with a high prevalence in previous reports [28, 30]. Specific mutations at the V domain of 23S rRNA of M. pneumoniae may determine macrolide resistance phenotypes. For instance, mutations at positions 2063 and 2064 are associated with high-level resistance, while mutations at positions 2067 and 2617 are related to low-level macrolide resistance [31,32,33]. In this study, all the mutations detected in M. pneumoniae were of the A2063G and the proportion of this mutation in these strains was as high as 96%. In addition, multiple antibiotic-resistance genes were detected in colonized microorganisms in the respiratory tract, indicating that the widespread use of antibiotics has resulted in more and more microbial resistance, which may pose increasing challenges for treating respiratory infectious diseases.
It must be acknowledged that this study has certain limitations. Firstly, no specific genotype of M. pneumoniae was found to be associated with more severe clinical symptoms in our analysis, but the small sample size may have constrained our findings. Further research, incorporating larger datasets from various regions, is essential to validate these observations. A notable limitation of this study is the insufficient identification of macrolide-sensitive M. pneumoniae strains, which precludes the analysis of any correlation between macrolide resistance and disease severity. Furthermore, the identification of only one type of resistance mutation limits the ability to compare the pathogenic differences among various resistance mutations.
Conclusion
In conclusion, this study laid a foundation for the cyclic study of M. pneumoniae genotype conversion in Wuhan, and the main circulating M. pneumoniae from 2020 to 2022 is P1-type 1. This study also underscored the efficacy of qPCR as a sensitive method for detecting M. pneumoniae infections. The finding that an IgM titer exceeding 1:160 may serve as a predictive angle for assessing the severity of the illness, adding a valuable perspective to clinical assessment. Significantly, our results revealed no direct correlation between the severity of M. pneumoniae pneumonia and its genotypic characteristics or the respiratory microenvironment, suggesting that other factors may influence disease progression. The high prevalence of macrolide resistance mutations identified presented a growing challenge for treatment, emphasizing the need for continued surveillance and novel therapeutic approaches. These findings contribute to a deeper understanding of M. pneumoniae infections and set the stage for future research into effective prevention and control strategies.
Materials and methods
Patients and specimens
A total of 1259 children admitted to Wuhan Children's Hospital with pneumonia between October 2020 and March 2022 were enrolled in this study. Among them, 511 were clinically diagnosed with M. pneumoniae infection based on the colloidal gold assay for M. pneumoniae IgM titer of ≥ 1:160 [34]. After obtaining written informed consent from patients or their parents, 417 oropharyngeal swabs (OP) and 94 bronchoalveolar lavage fluid samples (BALF) were collected. OP samples were collected from each patient within 24 h after admission. Electronic bronchoscopy was performed when necessary, and BALF samples were collected during these procedures simultaneously within the hospitalization period. The samples were sent to Wuhan Institute of Virology, Chinese Academy of Sciences for analysis. Each OP sample was stored in 3 mL of viral transport media at -80℃ and BALF was directly stored at -80℃ before use. Children with immunodeficiency, chronic lung disease, neurological disorders and gastrointestinal and urinary tract infections were excluded.
Real-time PCR, MLST and 23S rRNA PCR
Nucleic acid was extracted from OP and BALF samples (200 μL) using a BeaverBeads™ Viral DNA/RNA Kit (Suzhou, China) according to the manufacturer’s instructions. qPCR was utilized specifically for the detection of M. pneumoniae, while PCR was performed to amplify the target genes using DNA from OP and BALF samples. The primers used for qPCR [35] and PCR, along with their corresponding target genes, are detailed in Supplementary Table S1. MLST was conducted based on the scheme described in the PubMLST website (https://pubmlst.org/mpneumoniae/) developed by Brown et al. [15]. The primers used for amplifying the 23S rRNA fragment were designed in-house. Each reaction of qPCR was conducted in a final volume of 10 μL containing 2 × SYBR® Green Realtime PCR Master Mix (5 μL), specific primers (10 μM × 0.4 μL), 1–2 μL of template DNA, and ddH2O up to 10 μL. The amplification conditions were as follows: 95℃ for 30 s; 40 cycles of 95℃ for 5 s, 55℃ for 10 s and 72℃ for 15 s. Each reaction of PCR was conducted in a final volume of 50 μL containing 2 × TsingKe® Master Mix (Beijing, China), specific primers (10 μM × 1 μL), 1–2 μL of template DNA, and ddH2O up to 50 μL. The amplification conditions were as follows: 95℃ for 3 min; 30 cycles of 94℃ for 30 s, 55℃C for 30 s and 72℃ for 30–80 s; with a final extension step of 72℃ for 5 min. All amplification products were purified using TIANgel Midi Purification Kit (DP209) (Beijing, China) and sequenced in Tsingke Biotech (Beijing, China).
Metagenome sequencing, classifications and microbiome analysis
DNA library was constructed using VAHTS Universal Plus DNA Library Prep Kit for Illumina® ND617 (Nanjing, China). Library circularization and next-generation sequencing (NGS) of all samples were performed using the MGI T7 platform. 46 samples with Ct value below 27.5 were selected for metagenome next-generation sequencing (mNGS) and paired-end 150-bp reads were used. Reads classification from metagenomic sequencing was conducted using Kraken2 v2.1.2 [36] software with the kraken2 nt database, applying a confidence threshold of 0.05. The report files of taxonomic classifications were visualized by the Pavian software tool [37]. Alpha diversity analysis was performed using QIIME [38], and the beta diversity analysis was performed in R using the vegan package [39]. Inter-group microbial differential analysis was performed using LEfSe v1.1.2 [40], and an linear discriminant analysis (LDA) score greater than 4 was considered to indicate significant differences.
Pan-genome and phylogenetic analysis
Paired-end reads were mapped to either the M129 or FH reference genome (NCBI accession numbers: NC_000912.1 and CP010546.1, respectively) using Snippy v4.6.0 (https://github.com/tseemann/snippy) with default parameters. When mapping reads to a reference genome, Snippy could generate a consensus sequence of the reference genome. We annotated the consensus sequences with high sequencing depth (> 10) and coverage (> 95%) using Prokka v1.14.5 [41]. A pan-genome was constructed using draft assemblies of the 87 M. pneumoniae isolates, which included 78 genomes from GenBank and 9 consensus sequences generated in this study. Genomic feature files output from Prokka in gff3 format for each strain were used for the pan-genome analysis using Roary v3.13.0 [42]. Roary was run with the "-e" flag to generate a multi-FASTA alignment of core genes using PRANK and a minimum blastp identity value of 95%. Summary of the pan-genome composition provided by Roary was visualized in Fig. 3A using the open source Python script 'roary_plots.py' (available at https://github.com/sanger-pathogens/Roary/tree/master/contrib/roary_plots). Gene diversity estimation and visualization presented in Fig. 3B was performed using the native Rscript from Roary (create_pan_genome_plots.R). Alignment of core genes was used to build a maximum likelihood (ML) phylogenetic tree by IQ-tree2 v2.2.0_beta with ModelFinder (model K3Pu + F + I + G4) [43, 44]. The ML tree was visualized by FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
De novo assembly and detection of resistant genes
mNGS reads were de novo assembled using Megahit v1.2.9 [45]. Mass screening for antimicrobial resistance was performed on the contigs assembled by Megahit using AMRFinderPlus v3.10.30 [46]. Following this, the frequencies of these antimicrobial resistance genes across all samples were calculated.
Statistical analysis
The data were expressed as the mean and standard deviation or as a rate (%). Group comparisons were conducted using t-test, PERMANOVA or linear discriminant analysis (LDA). For categorical variables, two-sided proportions test or Fischer's exact test was used. A p-value of less than 0.05 was considered statistically significant. LDA was performed by LEfSe and other statistical analyses were performed by using R v4.1.3 [47].
Availability of data and materials
The bioproject number for the raw sequencing data reported in this paper is PRJCA012901 (available at http://bigd.big.ac.cn).
References
Atkinson TP, Balish MF, Waites KB. Epidemiology, clinical manifestations, pathogenesis and laboratory detection of Mycoplasma pneumoniae infections. FEMS Microbiol Rev. 2008;32:956–73.
Cillóniz C, Torres A, Niederman M, van der Eerden M, Chalmers J, Welte T, et al. Community-acquired pneumonia related to intracellular pathogens. Intensive Care Med. 2016;42:1374–86.
Kutty PK, Jain S, Taylor TH, Bramley AM, Diaz MH, Ampofo K, et al. Mycoplasma pneumoniae Among Children Hospitalized With Community-acquired Pneumonia. Clin Infect Dis. 2018.
Wang X, Li M, Luo M, Luo Q, Kang L, Xie H, et al. Mycoplasma pneumoniae triggers pneumonia epidemic in autumn and winter in Beijing: a multicentre, population-based epidemiological study between 2015 and 2020. Emerg Microbes Infect. 2022;11:1508–17.
Lind K, Benzon MW, Jensen JS, Clyde WA. A seroepidemiological study of Mycoplasma pneumoniae infections in Denmark over the 50-year period 1946–1995. Eur J Epidemiol. 1997;13:581–6.
Lenglet A, Herrador Z, Magiorakos AP, Leitmeyer K, Coulombier D, European Working Group on Mycoplasma pneumoniae surveillance C. Surveillance status and recent data for Mycoplasma pneumoniae infections in the European Union and European Economic Area, January 2012. Eurosurveillance. 2012;17.
Omori R, Nakata Y, Tessmer HL, Suzuki S, Shibayama K. The determinant of periodicity in Mycoplasma pneumoniae incidence: an insight from mathematical modelling. Sci Rep. 2015;5:14473.
Loens K, Ieven M. Mycoplasma pneumoniae: Current Knowledge on Nucleic Acid Amplification Techniques and Serological Diagnostics. Front Microbiol. 2016;7.
Zhao F, Liu L, Tao X, He L, Meng F, Zhang J. Culture-Independent Detection and Genotyping of Mycoplasma pneumoniae in Clinical Specimens from Beijing. China PLoS ONE. 2015;10:e0141702.
Zhang Y, Huang Y, Ai T, Luo J, Liu H. Effect of COVID-19 on childhood Mycoplasma pneumoniae infection in Chengdu. China BMC Pediatr. 2021;21:202.
Chen J, Zhang J, Lu Z, Chen Y, Huang S, Li H, et al. Mycoplasma pneumoniae among Chinese Outpatient Children with Mild Respiratory Tract Infections during the Coronavirus Disease 2019 Pandemic. Microbiol Spectr. 2022;10:e0155021.
Spuesens EBM, Oduber M, Hoogenboezem T, Sluijter M, Hartwig NG, van Rossum AMC, et al. Sequence variations in RepMP2/3 and RepMP4 elements reveal intragenomic homologous DNA recombination events in Mycoplasma pneumoniae. Microbiology. 2009;155:2182–96.
Kenri T, Suzuki M, Sekizuka T, Ohya H, Oda Y, Yamazaki T, et al. Periodic Genotype Shifts in Clinically Prevalent Mycoplasma pneumoniae Strains in Japan. Front Cell Infect Microbiol. 2020;10:385.
Morozumi M, Tajima T, Sakuma M, Shouji M, Meguro H, Saito K, et al. Sequence Type Changes Associated with Decreasing Macrolide-Resistant Mycoplasma pneumoniae. Japan Emerg Infect Dis. 2020;26:2210–3.
Brown RJ, Holden MTG, Spiller OB, Chalker VJ. Development of a Multilocus Sequence Typing Scheme for Molecular Typing of Mycoplasma pneumoniae. J Clin Microbiol. 2015;53:3195–203.
Tsai T-A, Tsai C-K, Kuo K-C, Yu H-R. Rational stepwise approach for Mycoplasma pneumoniae pneumonia in children. J Microbiol Immunol Infect. 2021;54:557–65.
Okazaki N, Narita M, Yamada S, Izumikawa K, Umetsu M, Kenri T, et al. Characteristics of Macrolide-Resistant Mycoplasma pneumoniae Strains Isolated from Patients and Induced with Erythromycin In Vitro. Microbiol Immunol. 2001;45:617–20.
Zhou Y, Zhang Y, Sheng Y, Zhang L, Shen Z, Chen Z. More Complications Occur in Macrolide-Resistant than in Macrolide-Sensitive Mycoplasma pneumoniae Pneumonia. Antimicrob Agents Chemother. 2014;58:1034–8.
Waites KB, Xiao L, Liu Y, Balish MF, Atkinson TP. Mycoplasma pneumoniae from the Respiratory Tract and Beyond. Clin Microbiol Rev. 2017;30:747–809.
Zhao F, Liu G, Cao B, Wu J, Gu Y, He L, et al. Multiple-Locus Variable-Number Tandem-Repeat Analysis of 201 Mycoplasma pneumoniae Isolates from Beijing, China, from 2008 to 2011. J Clin Microbiol. 2013;51:636–9.
Wang Y, Xu B, Wu X, Yin Q, Wang Y, Li J, et al. Increased Macrolide Resistance Rate of M3562 Mycoplasma pneumoniae Correlated With Macrolide Usage and Genotype Shifting. Front Cell Infect Microbiol. 2021;11:675466.
Xue G, Li M, Wang N, Zhao J, Wang B, Ren Z, et al. Comparison of the molecular characteristics of Mycoplasma pneumoniae from children across different regions of China. PLoS ONE. 2018;13:e0198557.
Rodman Berlot J, Krivec U, Mrvič T, Kogoj R, Keše D. Mycoplasma pneumoniae P1 Genotype Indicates Severity of Lower Respiratory Tract Infections in Children. J Clin Microbiol. 2021;59:e00220-e221.
Rodman Berlot J, Krivec U, Praprotnik M, Mrvič T, Kogoj R, Keše D. Clinical characteristics of infections caused by Mycoplasma pneumoniae P1 genotypes in children. Eur J Clin Microbiol Infect Dis. 2018;37:1265–72.
Fan L, Li D, Zhang L, Hao C, Sun H, Shao X, et al. Pediatric clinical features of Mycoplasma pneumoniae infection are associated with bacterial P1 genotype. Exp Ther Med. 2017;14:1892–8.
Subspecialty Group of Respiratory Diseases, The Society of Pediatrics, Chinese Medical Association, Editorial Board, Chinese Journal of Pediatrics. [Guidelines for management of community acquired pneumonia in children (the revised edition of 2013) (I)]. Zhonghua Er Ke Za Zhi. 2013;51:745–52.
Talkington DF, Shott S, Fallon MT, Schwartz SB, Thacker WL. Analysis of eight commercial enzyme immunoassay tests for detection of antibodies to Mycoplasma pneumoniae in human serum. Clin Diagn Lab Immunol. 2004;11:862–7.
Chen Y-C, Hsu W-Y, Chang T-H. Macrolide-Resistant Mycoplasma pneumoniae Infections in Pediatric Community-Acquired Pneumonia. Emerg Infect Dis. 2020;26:1382–91.
Kim K, Jung S, Kim M, Park S, Yang H-J, Lee E. Global Trends in the Proportion of Macrolide-Resistant Mycoplasma pneumoniae Infections: A Systematic Review and Meta-analysis. JAMA Netw Open. 2022;5:e2220949.
Tanaka T, Oishi T, Miyata I, Wakabayashi S, Kono M, Ono S, et al. Macrolide-Resistant Mycoplasma pneumoniae Infection, Japan, 2008–2015. Emerg Infect Dis. 2017;23:1703–6.
Bébéar CM, Pereyre S. Mechanisms of drug resistance in Mycoplasma pneumoniae. Curr Drug Targets Infect Disord. 2005;5:263–71.
Bébéar C, Pereyre S, Peuchant O. Mycoplasma pneumoniae: susceptibility and resistance to antibiotics. Future Microbiol. 2011;6:423–31.
Principi N, Esposito S. Macrolide-resistant Mycoplasma pneumoniae: its role in respiratory infection. J Antimicrob Chemother. 2013;68:506–11.
Li J, Luu LDW, Wang X, Cui X, Huang X, Fu J, et al. Metabolomic analysis reveals potential biomarkers and the underlying pathogenesis involved in Mycoplasma pneumoniae pneumonia. Emerg Microbes Infect. 2022;11:593–605.
Meng FL, He LH, Gu YX, Zhang JZ, Zhao F. A real-time PCR assay for detection of Mycoplasma pneumoniae. Disease Surveillance. 2013;28:209–12.
Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20:257.
Breitwieser FP, Salzberg SL. Pavian: interactive analysis of metagenomics data for microbiome studies and pathogen identification. Bioinformatics. 2020;36:1303–4.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6.
Oksanen J, Simpson GL, Blanchet FG, Kindt R, Legendre P, Minchin PR, et al. vegan: Community ecology package. manual. 2022.
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60.
Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9.
Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3.
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9.
Li D, Luo R, Liu CM, Leung CM, Ting HF, Sadakane K, et al. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods. 2016;102:3–11.
Feldgarden M, Brover V, Gonzalez-Escalona N, Frye JG, Haendiges J, Haft DH, et al. AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci Rep. 2021;11:12728.
R Core Team. R: A language and environment for statistical computing. manual. Vienna, Austria; 2022.
Acknowledgements
We thank Lei Zhang of the core facility and technical support at Wuhan Institute of Virology for assistance with experimental platform, and Liangyu Li, Wenxue Yang, Mengchan Hao, Yanhai Wang for their help in the experiment.
Funding
The study was supported by grants from Natural Science Fund of Hubei Province [2023AFB221, 2021CFA012]; Knowledge Innovation Program of Wuhan Basi Research (2022020801010569); Health Commission of Hubei Province [WJ2021M262] and the National Natural Science Foundation of China [31970548].
Author information
Authors and Affiliations
Contributions
This project was designed by YY, XL, DL. Samples were collected and prepared by YL, HD, QD. Experiments were conducted by MX, YL with assistance of XT, JG. The data analysis was performed by MX, LM with assistance of HL. The manuscript was prepared by MX, YY and revised by YS, YY, XL, DL. All authors read and commented on the paper.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Medical Ethics Committee of Wuhan Children’s Hospital (Wuhan Maternal and Children Healthcare Hospital) (No. 2021R005-E01). Informed consent was obtained from all the participants and parent and/or legal guardian of participants for this study. All procedures performed in this study involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Participants' confidentiality was strictly preserved. All data were anonymized and only accessible to the research team. No identifiable information is published.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xu, M., Li, Y., Shi, Y. et al. Molecular epidemiology of Mycoplasma pneumoniae pneumonia in children, Wuhan, 2020–2022. BMC Microbiol 24, 23 (2024). https://doi.org/10.1186/s12866-024-03180-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-024-03180-0