
Abstract
A molecular surveillance of tick-borne diseases was performed in Hulunbuir City, Inner Mongolia. A total of 149 ticks including three species (Ixodes persulcatus, Haemaphysalis concinna, and Dermacentor silvarum) were collected. As many as 11 tick-borne bacterial pathogens were identified in them. Some of them have high positive rates. For example, Candidatus Rickettsia tarasevichiae was detected with a high prevalence of 72.48%, while Candidatus Lariskella sp. was detected in 31.54% of ticks. For both Rickettsia raoultii and Anaplasma phagocytophilum, two distinct genotypes were identified based on their phylogenetic trees based on 16S rRNA, gltA, and groEL sequences. Remarkable genetic diversity was also observed for 16S and flaB genes of Borreliella garinii, an agent of Lyme disease. Rickettsia heilongjiangensis causing Far-Eastern spotted fever (2.68%, 4/149), Ehrlichia muris causing human ehrlichiosis (4.70%, 7/149), Borrelia miyamotoi causing relapsing fever (2.01%, 3/149), and Borreliella afzelii causing Lyme disease (2.01%, 3/149) were also detected. Additionally, a previously uncharacterized Anaplasma species closely related to Anaplasma ovis was identified. Herein we name it “Candidatus Anaplasma mongolica”. Based on these results, we propose that Yakeshi City might be a potential hotspot of tick-borne diseases.
Similar content being viewed by others

Introduction
Emerging tick-borne diseases pose a threat to public health worldwide. To date, at least 124 tick species have been recorded in mainland China, including 113 hard tick species in seven genera and 11 soft tick species in two genera [1]. Accordingly, these tick species have been reported to carry at least 103 tick-borne agents, most of which were detected in the past two decades [1]. The order Rickettsiales and the genus Borrelia are the largest groups of tick-borne bacterial pathogens. Rickettsiales mainly composed of genera Rickettsia, Anaplasma, and Ehrlichia. It includes a large number of important tick-borne pathogens, such as Rickettsia rickettsii, Rickettsia raoultii, Ehrlichia chaffeensis, Anaplasma phagocytophilum, Anaplasma capra, and Candidatus Neoehrlichia mikurensis. Until 2021, at least 21 Rickettsia, nine Anaplasma, eight Ehrlichia, and one Candidatus Neoehrlichia species (or variants) were detected in ticks from China. Of those, A. phagocytophilum, E. chaffeensis, and R. raoultii parasitize the largest number of tick species (reported in 22, 16, and 15 tick species, respectively) [1]. Human cases infected by these pathogens were also frequently reported. From 2009 to 2010, 46 human cases infected with A. phagocytophilum were laboratory-confirmed in Beijing, Hebei, and Shandong provinces, North China [2]. In addition to the well-known pathogens, some newly identified members of Rickettsiales are increasingly reported to infect humans. As recently as in 2022, Candidatus Midichloria mitochondrii, previously known as an endosymbiont of hard ticks, was detected in blood samples of 34.1% of humans with a tick bite history, suggesting that the genetic diversity and human pathogenicity of various Rickettsiales bacteria still warrant further explorations [3]. The genus Borrelia is closely related to lots of human diseases. Since 2014, the genus Borrelia was divided into two genera: the genus Borrelia containing the members of the relapsing fever Borrelia (Borrelia miyamotoi, Borrelia theileri, Borrelia persica, etc.), and the genus Borreliella containing the Lyme disease Borrelia (Borrelia burgdorferi sensu lato, Borrelia afzelii, etc.) [4]. Of those, Lyme borreliosis caused by the Lyme disease group is considered the most common tick-borne disease in the Northern Hemisphere. Approximately 476,000 human cases were reported in the USA each year [5]. In China, at least nine genospecies of B. burgdorferi have been documented, most of which were detected in ticks [6].
The Inner Mongolia Autonomous Region located in the north of China is rich in various wildlife and has a diverse ecosystem. Due to the vast territory (1, 183, 000 km2) and unique geographical/ecological features, this is one of the several regions that harbor the most abundant tick species in China [1]. Accordingly, it is one of the major epidemic areas of tick-borne infectious diseases in China including tick-borne encephalitis, anaplasmosis, rickettsiosis, Lyme disease, and babesiosis [7]. Although lots of studies have been performed in ticks from Inner Mongolia, Rickettsiales bacteria and other tick-borne pathogens in many tick species and many cities in this area are still not extensively characterized. In this study, we collected three tick species from Hulunbuir City in northeast Inner Mongolia and studied the potential human pathogens in them.
Methods
Sample collection and DNA extraction
In 2018, ticks were collected in the Ilekd Village, Wunuer Town, Yakeshi County-Level City of Hulunbuir City, Inner Mongolia (Fig. 1). This location is located in forest areas of the Greater Khingan Range, with an average altitude of approximately 850 m. The ticks were carefully removed from the body surface of free-ranging goats and cattle using tweezers and then transported alive to Wuhan Center for Disease Control and Prevention. Based on the taxonomic characters described in previous literature, the tick species were initially determined by morphological observation of their palp, scutum, anal groove, and shape of basis capitulum. All the ticks were observed by a stereoscopic microscope (Olympus SZX 16, Olympus Corporation, Tokyo, Japan). For further confirmation, ticks were randomly selected from each species, and the COI (Cytochrome oxidase I) sequences were PCR amplified and sequenced (primers shown in reference [8]) after DNA extraction.

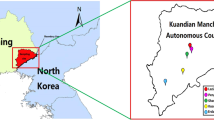
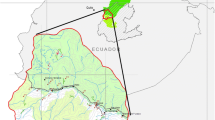
A map showing the location where the ticks were collected
Before DNA extraction, each tick was washed twice using phosphate buffer saline (PBS) to exclude possible environmental contamination, and then individually ground into homogenate manually in a mortar with 100 μL PBS. Total DNA was extracted from the homogenates using a Mollusc DNA Extraction Kit (Omega Bio-Tek, USA) following the instructions. To test the quality of DNA extraction, the DNA concentration was measured by spectrophotometric analysis (Thermo Scientific Nanodrop, Delaware, USA).
Molecular detection and identification of the pathogens
The DNA samples were screened for the presence of Rickettsia, Anaplasmataceae (Anaplasma spp., Ehrlichia spp., etc.), and Borrelia bacteria. Primers used were shown in previous reports [9,10,11], generating PCR products with a size of approximately 900 bp, 450 bp, and 400 bp, respectively. The Rickettsia and Anaplasmataceae were detected targeting the 16S rRNA gene, while Borrelia was screened targeting the flaB (flagellin B) gene. For precise identification, the gltA and groEL (60 kDa chaperonin) sequences were amplified from the Rickettsia, Ehrlichia, and Anaplasma strains detected in this study using primers as shown [9, 10]. Furthermore, 16S rRNA sequences (approximately 1200 bp) were amplified from the Borrelia strains [11], and gltA sequences (approximately 400 bp) were amplified from the Candidatus Lariskella strains [9].
Genetic and phylogenetic analysis
To calculate the nucleotide similarities and determine their species, all the nucleotide sequences recovered in this study were manually aligned with reference sequences in the GenBank Database by BLASTn. The representative reference sequences were downloaded from the database. After alignment by Mega 6.0, the nucleotide sequences were phylogenetically analyzed based on the maximum likelihood (ML) method by PhyML3.0 [12]. All the phylogenetic trees were visualized and edited using FigTree v1.4.3.
Results
Sample collection and species identification
In May 2018, a total of 149 ticks were collected from the body surface of ten cattle and eight goats in the Yakeshi County-Level City of Hulunbuir City, Inner Mongolia (49.17°N, 120.40°E). Based on morphological observation, three species were identified: 99 Ixodes persulcatus, 24 Haemaphysalis concinna, and 26 Dermacentor silvarum (Fig. 2). For further confirmation, the COI sequences of all three tick species have nucleotide similarities higher than 99% with reference sequences.

The photographs of ticks under a stereoscopic microscope. A Ixodes persulcatus. B Dermacentor silvarum. C Haemaphysalis concinna
Detection and analysis of the Rickettsia spp.
Based on the PCR results and sequencing of 16S rRNA sequences, a total of three Rickettsia species including four genotypes were identified. Candidatus Rickettsia tarasevichiae was detected in all I. persulcatus (99/99, 100%), 7 of 24 H. concinna (29.17%), and 2 of 26 D. silvarum (7.69%) (Table 1). All the 16S sequences are 100% identical to Ca. Rickettsia tarasevichiae isolate Dog-145, Ca. Rickettsia tarasevichiae isolate Bayan-68, and Ca. Rickettsia tarasevichiae isolate Mulan-11, which were all detected in the Heilongjiang Province of Northeast China. Similarly, the gltA (1007 bp) sequences show highest 99.90% nucleotide similarity to these strains, and the groEL (1060 bp) sequences have 99.65–99.88% (coverage 76–80%) to previously uploaded sequences (Accession numbers: ON863711, OP722688, MN450404, and MN450402).
Two genotypes of R. raoultii were detected only in D. silvarum, with positive rates of 50.00% (13/26) and 26.92% (7/26), respectively. All three genes of type I (strains N78, N83, and N95) were 100% identical to Rickettsia conorii subsp. raoultii strain IM16. In contrast, the 16S sequences of type II were 100% identical to Rickettsia conorii subsp. raoultii isolate Tomsk, R. conorii strain Malish_7, and R. massiliae MTU5, while their gltA sequences were 100% to that of R. raoultii isolate Binxian-91. As shown in Fig. 3, these strains clearly divided into two distinct clades.

Phylogenetic trees based on the nucleotide sequences of 16S rRNA, gltA, and groEL genes of Rickettsia spp.
Four H. concinna ticks were tested positive for R. heilongjiangensis. All their 16S rRNA and gltA sequences show 100% and 99.90% identities to both R. heilongjiangensis CH8-1 and R. japonica strain YH_M. For the groEL sequences, they are 100% identical to those of R. heilongjiangensis CH8-1 and R. heilongjiangensis HCN-13, but only 99.53% to R. japonica strains. These results confirmed that these strains should be classified as R. heilongjiangensis.
Detection and analysis of the Anaplasma spp.
Based on analysis of the 16S sequences, a total of three Anaplasma species were detected: A. phagocytophilum, A. bovis, and an unclassified Anaplasma sp., showing 100%, 100%, and 99.87% to A. phagocytophilum str. JM, A. bovis clone Am-Hc60, and A. centrale isolate LP10, respectively. Notably, based on gltA and groEL sequences, the A. phagocytophilum strains divided into two types in the phylogenetic trees: A. phagocytophilum N3 represent type I while strains N54, N55, and N136 belong to type II (Fig. 4). The gltA sequences of type II are 100% identical to Anaplasma sp. KhabIx detected in the Russian Far East, and only 82.73–88.11% to A. phagocytophilum strains. Similarly, the groEL sequence of type I (strain N3) was 100% identical to A. phagocytophilum isolate Ip11, but sequences of type II have 98.59–100% similarities to A. phagocytophilum strains identified in Tomsk and Omsk in Russia. These results showed the genetic diversity of A. phagocytophilum in this area. Out of our expectation, the gltA sequence of Anaplasma sp. N127 was 100% identical to Anaplasma sp. BL126-13, but its groEL sequence has a long genetic distance to that of Anaplasma sp. BL126-13 (Fig. 4). In contrast, it was 100% identical to the groEL sequence of Anaplasma sp. clone B251. The 23S sequence of Anaplasma sp. N127 was also obtained, showing highest 96.79% identity to A. ovis str. Haibei and 94.81% to A. marginale str. Florida (Fig. S1). We propose it as a novel species, namely “Candidatus Anaplasma mongolica”.

Phylogenetic trees based on the nucleotide sequences of 16S rRNA, gltA, and groEL genes of Anaplasma spp.
Detection and analysis of the Ehrlichia sp. and Lariskella sp.
Seven I. persulcatus ticks were tested positive for Ehrlichia, and all of them were identified as Ehrlichia muris. In addition to the 16S rRNA (456 bp) sequences which show 100% to E. muris strains, the gltA (986 bp), and groEL (1121 bp) sequences were also successfully obtained. The gltA sequences have highest 99.47–99.80% identities to E. muris strains in rodents (isolate Khab-85_Mruf, AS145) and I. persulcatus ticks (isolate Omsk-563_Ip) from Japan and Russia. The groEL sequences are also highly homologous to E. muris strains from Japan and Russia, with nucleotide identities of 99.29–100% (Fig. 5). To date, there is only one E. muris sequence in the GenBank Database from mainland China.

Phylogenetic trees based on the nucleotide sequences of 16S rRNA, gltA, and groEL genes of Ehrlichia muris
Unexpectedly, Candidatus Lariskella sp. belonging to the family Candidatus Midichloriaceae, order Rickettsiales, was detected in as many as 47 of the 99 Ixodes persulcatus ticks (47.47%) using the primers screening Anaplasmataceae. All the 16S rRNA sequences are 100% identical to each other and have 98.83–100% identity to Candidatus Lariskella arthropodarum, 98.82–99.06% to Candidatus Lariskella guizhouensis we previously reported. Interestingly, their gltA sequences (400 bp) are only highly homologous to Ca. Lariskella guizhouensis, with similarities of 99.50–99.73% (Fig. 6).

Phylogenetic trees based on the nucleotide sequences of 16S rRNA and gltA genes of Candidatus Lariskella sp.
Detection and analysis of the Borrelia sp. and Borreliella spp.
One Borrelia sp. (Borrelia miyamotoi) and two Borrelialla spp. (Borreliella afzelii and Borreliella garinii) were detected. As shown in Table 1, all of them were detected in I. persulcatus, with positive rates of 3.03% (3/99), 3.03% (3/99), and 8.08% (8/99), respectively. The flaB and 16S sequences of B. miyamotoi strains were all closely related to B. miyamotoi strain Yekat-31 from Russia, with nucleotide similarities of 98.72% and 100%, respectively. Notably, phylogenetic analysis indicated that both the flaB and 16S gene sequences of B. garinii showed remarkable genetic diversity (shown in Fig. 7).

Phylogenetic trees based on the nucleotide sequences of flaB and 16S rRNA genes of Borrelia miyamotoi and Borreliella spp.
Co-infections of tick-borne pathogens
Because all I. persulcatus ticks were positive for Ca. Rickettsia tarasevichiae, ticks infected with A. phagocytophilum (four strains), Ca. Anaplasma mongolica (one strain), E. muris (seven strains), Candidatus Lariskella sp. (47 strains), Borrelia (three strains), and Borrelialla (11 strains) strains detected in I. persulcatus were all co-infected with Ca. Rickettsia tarasevichiae. Of those, one tick infected with B. miyamotoi is co-infected with Ca. Rickettsia tarasevichiae and Candidatus Lariskella sp., while one infected with B. aafzelii is co-infected with Ca. Rickettsia tarasevichiae and A. phagocytophilum (type II). Furthermore, in the nine ticks infected with B. garinii, two are co-infected with Ca. Rickettsia tarasevichiae and Candidatus Lariskella sp., and one is co-infected with Ca. Rickettsia tarasevichiae and Ca. Anaplasma mongolica. No co-infection was observed in H. concinna and D. silvarum.
Discussion
Inner Mongolia has been recognized as an endemic region of tick-borne diseases. To date, numerous studies have been carried out on tick-borne pathogens circulating in this area [7, 13,14,15,16,17,18]. However, most of these studies only focused on one or two pathogens, and exhaustive investigations on their genetic characteristics are still very few. In this study, we detected and identified as many as 11 tick-borne bacterial pathogens in three tick species from Hulunbuir City, Inner Mongolia. The abundance of tick-borne pathogens, as well as the high positive rates and remarkable genetic diversity of some pathogens, clearly suggest the risk to public health in this area. Furthermore, apparent host specificity was observed for most of these pathogens. For example, seven bacterial species were only detected in I. persulcatus. It was consistent with the previous report that I. persulcatus is only second to H. longicornis as the carrier of tick-borne agents [1].
All three Rickettsia species in this study are well-recognized human pathogens. Candidatus Rickettsia tarasevichiae is first detected in I. persulcatus ticks from Russia in 2003 [19]. After then, tens of human cases infected by this Rickettsia were reported in Heilongjiang (northeast China) and Henan (eastern central China) provinces, with the major symptoms of fever, malaise, and anorexia [20, 21]. As previously reported, the tick hosts of Ca. Rickettsia tarasevichiae are I. persulcatus, I. sinensis, H. longicornis, H. concinna., and D. silvarum [1]. It is of note that in this study, the positive rate in different tick hosts varies dramatically, suggesting that the distribution and density of I. persulcatus may be the major risk factors for Ca. Rickettsia tarasevichiae infections. Rickettsia heilongjiangensis and R. raoultii are both spotted fever group Rickettsia. As the etiologic agent of Far-Eastern spotted fever, R. heilongjiangensis has been detected in multiple tick species. However, it is most frequently reported in H. concinna ticks, which is consistent with our results [22]. Interestingly, we found two genotypes of R. raoultii in D. silvarum. One is closely related to Rickettsia conorii subsp. raoultii strain IM16. In contrast, the 16S, gltA, and groEL genes of the other type all show different positions in the phylogenetic trees. Rickettsia raoultii infections in humans have been occasionally reported in China. In 2018, 26 human cases collected in three Medical Centers from Henan, Shandong, and Inner Mongolia were determined infected with R. raoultii [23]. Most of the patients only showed common nonspecific manifestations, such as fever and malaise. Our result revealed the genetic diversity of R. raoultii in this area. Their virulence and infectivity still need further exploration.
Three Anaplasma species were identified in this study. Except for A. bovis, all of them were detected in I. persulcatus ticks, indicating their host specificity. Of those, A. phagocytophilum is considered the most important human-pathogenic Anaplasma species. Human cases infected by A. phagocytophilum have been reported in multiple provinces of China [2, 24, 25]. In this study, two genotypes of A. phagocytophilum were identified. Although their 16S sequences were 100% identical, they separated into two distinct clades in the phylogenetic trees based on gltA and groEL sequences. This result may suggest the long-term evolution and recombination of A. phagocytophilum in this area. Notably, the type I strain was detected in an I. persulcatus tick from a goat, while the type II strains were all detected in I. persulcatus ticks from cattle. It is of interest whether there are any relationships between the animal hosts and the genetic types of A. phagocytophilum. In addition, a previously uncharacterized Anaplasma species closely related to A. ovis was detected. Herein we name it “Candidatus Anaplasma mongolica”.
Ehrlichia muris is an agent of human ehrlichiosis. In 2009, an E. muris–like agent was identified as a causative agent of human ehrlichiosis in the United States [26]. Although there have been several reports of E. muris in ticks from northeast China [27], only one single sequence was available in the GenBank Database to date. Our result may provide some information on the distribution and genetic characteristics of E. muris in China. Although no human cases infected by E. muris have been reported in China, our data suggest the potential risk of ehrlichiosis in this area. Out of our expectation, Candidatus Lariskella sp. belonging to the family Candidatus Midichloriaceae, the order Rickettsiales, was detected. In 2004, Candidatus Lariskella arthropodarum has been detected in acutely febrile patients who have been bitten by Ixodes ticks in the Far East of Russia, suggesting that it may be a potential tick-borne human pathogen [28]. In 2022, we detected Candidatus Lariskella sp. in Ixodes ticks from Guizhou Province, Southwest China, and obtained the gltA and groEL sequences of the genus Ca. Lariskella for the first time. We name it “Candidatus Lariskella guizhouensis” [9]. However, in this study, the 16S sequences of Ca. Lariskella strains we detected were 100% identical to Ca. Lariskella arthropodarum, while the gltA sequences were 100% identical to Ca. Lariskella guizhouensis in the absence of those of Ca. Lariskella arthropodarum strains. Based on the current data, it is hard to determine whether the detected strains should be classified as Ca. Lariskella arthropodarum or Ca. Lariskella guizhouensis. Actually, it is also quite possible that these two species be the same species.
Borrelia miyamotoi is an etiologic agent of relapsing fever. In 2021, an investigation performed in the same area reported B. miyamotoi infections in both ticks and humans [16]. Our result confirmed the circulation of B. miyamotoi in this area. In addition, B. afzelii and B. garinii were also detected. Borreliella garinii is the agent of Lyme disease widely distributed in China. To date, it has been reported in Heilongjiang, Jilin, Liaoning, Hebei, Inner Mongolia, Gansu, Zhejiang, and Xinjiang provinces in China, mainly located in north China [29, 30]. In this study, a high prevalence (8.08%) of B. garinii was observed in I. persulcatus ticks. Additionally, the detected strains showed considerable genetic polymorphism. Although Lyme disease is rarely reported in this area, our results suggest that local people may be at risk of Lyme disease infection.
To be noticed, environmental factors may affect the distribution of tick species, thus affecting the diversity and abundance of tick-borne pathogens. In this study, all the ticks were collected in forest areas of the Greater Khingan Range, Northeast China. Accordingly, most of the ticks (I. persulcatus and H. concinna) collected in this study ecologically fit biogeographic zones covered by coniferous forests with strong seasonality in temperature. As previously reported, I. persulcatus and H. concinna harbor an extremely high variety of tick-borne agents [1]. In this study, the remarkable diversity and abundance of tick-borne pathogens were identified, which is highly consistent with previous studies. This result also suggests that similar biogeographic zones may also warrant surveillance of tick-borne pathogens.
There are some limitations in this study. First, all the ticks were collected from a few domestic animals in one site, and the sample size was also small. Therefore, the tick species and the pathogens they carried may not be representative of this area. Second, because all the ticks were removed from domestic animals, it is possible that the detected pathogens are from the blood meal of ticks instead of the ticks themselves. In further study, testing tick-borne pathogens in host-seeking ticks and domestic animals may provide more useful information.
Conclusion
In conclusion, 11 tick-borne bacterial pathogens were identified in Yakeshi City, Inner Mongolia. Some of them showed a high prevalence (Ca. Rickettsia tarasevichiae) and diverse genotypes (R. raoultii and A. phagocytophilum). These data suggest that Yakeshi City might be a potential hotspot of tick-borne diseases.
Availability of data and materials
All sequence files are available from the GenBank database (OR226549-OR226557, OR226561-OR226566, OR228866-OR228882, OR237106-OR237117, OR284777-OR284814, OR287185-OR287190, OR339536-OR339545) (Details shown in Table S1).
References
Zhao GP, Wang YX, Fan ZW, Ji Y, Liu MJ, Zhang WH, et al. Mapping ticks and tick-borne pathogens in China. Nat Commun. 2021;12(1):1075. https://doi.org/10.1038/s41467-021-21375-1.
Zhang L, Wang G, Liu Q, Chen C, Li J, Long B, et al. Molecular analysis of Anaplasma phagocytophilum isolated from patients with febrile diseases of unknown etiology in China. PLoS One. 2013;8(2):e57155. https://doi.org/10.1371/journal.pone.0057155.
Sgroi G, Iatta R, Lovreglio P, Stufano A, Laidoudi Y, Mendoza-Roldan JA, et al. Detection of endosymbiont Candidatus Midichloria mitochondrii and tick-borne pathogens in humans exposed to tick bites, Italy. Emerg Infect Dis. 2022;28(9):1824–32. https://doi.org/10.3201/eid2809.220329.
Adeolu M, Gupta RS. A phylogenomic and molecular marker based proposal for the division of the genus Borrelia into two genera: the emended genus Borrelia containing only the members of the relapsing fever Borrelia, and the genus Borreliella gen. nov. containing the members of the Lyme disease Borrelia (Borrelia burgdorferi sensu lato complex). Antonie Van Leeuwenhoek. 2014;105(6):1049–72. https://doi.org/10.1007/s10482-014-0164-x.
Bobe JR, Jutras BL, Horn EJ, Embers ME, Bailey A, Moritz RL, et al. Recent progress in Lyme disease and remaining challenges. Front Med. 2021;8:666554.
Che TL, Jiang BG, Xu Q, Zhang YQ, Lv CL, Chen JJ, et al. Mapping the risk distribution of Borrelia burgdorferi sensu lato in China from 1986 to 2020: a geospatial modelling analysis. Emerg Microbes Infect. 2022;11(1):1215–26. https://doi.org/10.1080/22221751.2022.2065930.
Liu D, Wulantuya, Fan H, Li X, Li F, Gao T, et al. Co-infection of tick-borne bacterial pathogens in ticks in Inner Mongolia, China. PLoS Negl Trop Dis. 2023;17(3):e0011121. https://doi.org/10.1371/journal.pntd.0011121.
Martínez-de la Puente J, Martínez J, Ferraguti M, Morales-de la Nuez A, Castro N, Figuerola J. Genetic characterization and molecular identification of the bloodmeal sources of the potential bluetongue vector Culicoides obsoletus in the Canary Islands. Spain Parasit Vectors. 2012;5:147. https://doi.org/10.1186/1756-3305-5-147.
Lu M, Meng C, Zhang B, Wang X, Tian J, et al. Prevalence of spotted fever group Rickettsia and Candidatus Lariskella in Multiple Tick Species from Guizhou Province, China. Biomolecules. 2022;12(11):1701. https://doi.org/10.3390/biom12111701.
Lu M, Tian J, Wang W, Zhao H, Jiang H, Han J, et al. High diversity of Rickettsia spp., Anaplasma spp., and Ehrlichia spp. in ticks from Yunnan Province, Southwest China. Front Microbiol. 2022;13:1008110. https://doi.org/10.3389/fmicb.2022.1008110.
Loh SM, Gofton AW, Lo N, Gillett A, Ryan UM, Irwin PJ, et al. Novel Borrelia species detected in echidna ticks, Bothriocroton concolor, in Australia. Parasit Vectors. 2016;9(1):339. https://doi.org/10.1186/s13071-016-1627-x.
Guindon S, Delsuc F, Dufayard JF, Gascuel O. Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol. 2009;537:113–37. https://doi.org/10.1007/978-1-59745-251-9_6.
Gui Z, Cai H, Qi DD, Zhang S, Fu SY, Yu JF, et al. Identification and genetic diversity analysis of Rickettsia in Dermacentor nuttalli within Inner Mongolia, China. Parasit Vectors. 2022;15(1):286. https://doi.org/10.1186/s13071-022-05387-4.
Gaowa, Wulantuya, Sato K, Liu D, Cui Y, Yin X, et al. Surveillance of Borrelia miyamotoi-carrying ticks and genomic analysis of isolates in Inner Mongolia, China. Parasit Vectors. 2021;14(1):368. https://doi.org/10.1186/s13071-021-04809-z.
Jiao J, Lu Z, Yu Y, Ou Y, Fu M, Zhao Y, et al. Identification of tick-borne pathogens by metagenomic next-generation sequencing in Dermacentor nuttalli and Ixodes persulcatus in Inner Mongolia, China. Parasit Vectors. 2021;14(1):287. https://doi.org/10.1186/s13071-021-04740-3.
Gao Y, Lv XL, Han SZ, Wang W, Liu Q, Song M. First detection of Borrelia miyamotoi infections in ticks and humans from the northeast of Inner Mongolia, China. Acta Trop. 2021;217:105857. https://doi.org/10.1016/j.actatropica.2021.105857.
Batu N, Wang Y, Liu Z, Huang T, Bao W, He H, et al. Molecular epidemiology of Rickettsia sp. and Coxiella burnetii collected from Hyalomma asiaticum in Bactrian camels (Camelus bactrianus) in inner Mongolia of China. Ticks Tick Borne Dis. 2020;11(6):101548. https://doi.org/10.1016/j.ttbdis.2020.101548.
Yin X, Guo S, Ding C, Cao M, Kawabata H, Sato K, et al. Spotted fever group rickettsiae in Inner Mongolia, China, 2015–2016. Emerg Infect Dis. 2018;24(11):2105–7. https://doi.org/10.3201/eid2411.162094.
Shpynov S, Fournier PE, Rudakov N, Raoult D. “Candidatus Rickettsia tarasevichiae” in Ixodes persulcatus ticks collected in Russia. Ann N Y Acad Sci. 2003;990:162–72. https://doi.org/10.1111/j.1749-6632.2003.tb07358.x.
Liu W, Li H, Lu QB, Cui N, Yang ZD, Hu JG, et al. Candidatus Rickettsia tarasevichiae infection in eastern central China: a case series. Ann Intern Med. 2016;164(10):641–8. https://doi.org/10.7326/M15-2572.
Jia N, Zheng YC, Jiang JF, Ma L, Cao WC. Human infection with Candidatus Rickettsia tarasevichiae. N Engl J Med. 2013;369(12):1178–80. https://doi.org/10.1056/NEJMc1303004.
He M, Zhang L, Hu H, Liu X, Zhang C, Xin Y, et al. Complete genome sequencing and comparative genomic analyses of a new spotted-fever Rickettsia heilongjiangensis strain B8. Emerg Microbes Infect. 2023;12(1):2153085. https://doi.org/10.1080/22221751.2022.2153085.
Li H, Zhang PH, Huang Y, Du J, Cui N, Yang ZD, et al. Isolation and identification of Rickettsia raoultii in human cases: a surveillance study in 3 medical centers in China. Clin Infect Dis. 2018;66(7):1109–15. https://doi.org/10.1093/cid/cix917.
Tsai KH, Chung LH, Chien CH, Tung YJ, Wei HY, Yen TY, et al. Human granulocytic anaplasmosis in Kinmen, an offshore island of Taiwan. PLoS Negl Trop Dis. 2019;13(9):e0007728. https://doi.org/10.1371/journal.pntd.0007728.
Gaowa, Wulantuya, Yin X, Cao M, Guo S, Ding C, et al. Case of human infection with Anaplasma phagocytophilum in Inner Mongolia, China. Jpn J Infect Dis. 2018;71(2):155–7. https://doi.org/10.7883/yoken.JJID.2017.450.
Pritt BS, Sloan LM, Johnson DK, Munderloh UG, Paskewitz SM, McElroy KM, et al. Emergence of a new pathogenic Ehrlichia species, Wisconsin and Minnesota, 2009. N Engl J Med. 2011;365(5):422–9. https://doi.org/10.1056/NEJMoa1010493.
Wei F, Song M, Liu H, Wang B, Wang S, Wang Z, et al. Molecular detection and characterization of zoonotic and veterinary pathogens in ticks from northeastern China. Front Microbiol. 2016;7:1913. https://doi.org/10.3389/fmicb.2016.01913.
Mediannikov OIu, Ivanov LI, Nishikawa M, Saito R, Sidel'nikov IuN, Zdanovskaia NI, Mokretsova EV, Tarasevich IV, Suzuki H. Microorganism “Montezuma” of the order Rickettsiales: the potential causative agent of tick-borne disease in the Far East of Russia. Zh Mikrobiol Epidemiol Immunobiol. 2004;(1):7–13.
Hao Q, Hou X, Geng Z, Wan K. Distribution of Borrelia burgdorferi sensu lato in China. J Clin Microbiol. 2011;49(2):647–50. https://doi.org/10.1128/JCM.00725-10.
Fang LQ, Liu K, Li XL, Liang S, Yang Y, Yao HW, et al. Emerging tick-borne infections in mainland China: an increasing public health threat. Lancet Infect Dis. 2015;15(12):1467–79. https://doi.org/10.1016/S1473-3099(15)00177-2.
Acknowledgements
NA.
Funding
This work was funded by the National Key Research and Development Program of China (No. 2021YFC2301202) and the Medical youth top talent project of Hubei (EWT2019048).
Author information
Authors and Affiliations
Contributions
Conceptualization, H.Z., M.L., and K.L.; Formal analysis, K.L.; Funding acquisition, J.T. and K.L.; Investigation, J.T., J.L, X.C., M.L., and X.G.; Methodology, M.L., and K.L.; Project administration, J.T., and K.L.; Resources, J.T.; Supervision, K.L.; Writing – original draft, K.L.; Writing – review & editing, H.Z., and K.L.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study protocol was approved by the Animal Ethics Committee of the National Institute for Communicable Disease Control and Prevention, the Chinese CDC (Approval No. 2021–011). Informed consent was obtained from the owners of the animals. The studies in this manuscript adhere to the ARRIVE guidelines for the reporting of animal experiments. All methods were performed in accordance with the relevant guidelines and regulations.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Phylogenetic trees based on the nucleotide sequences of 23S rRNA gene of Candidatus Anaplasma mongolica.
Additional file 2: Table S1.
Genbank numbers of Rickettsia, Anaplasma, Ehrlichia, Borrelia, Borreliella, and Candidatus Lariskella sequences recovered in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tian, J., Liu, J., Zhao, H. et al. Molecular surveillance reveals a potential hotspot of tick-borne disease in Yakeshi City, Inner Mongolia. BMC Microbiol 23, 359 (2023). https://doi.org/10.1186/s12866-023-03110-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-023-03110-6