
Abstract
Background
Impaired respiratory and intestinal microbiome composition is linked to cystic fibrosis lung disease severity. In people with cystic fibrosis (pwCF), regular exercise is recommended to delay disease progression and preserve a stable lung function. An optimal nutritional status is vital for best clinical outcomes. Our study investigated whether regular and monitored exercise and nutritional support promotes CF microbiome health.
Methods
A personalized nutrition and exercise program promoted nutritional intake and physical fitness in 18 pwCF for 12 months. Throughout the study, patients performed strength and endurance training monitored by a sports scientist via an internet platform. After three months, food supplementation with Lactobacillus rhamnosus LGG was introduced. Nutritional status and physical fitness were assessed before the study started, after three and nine months. Sputum and stool were collected, and microbial composition was analyzed by 16S rRNA gene sequencing.
Results
Sputum and stool microbiome composition remained stable and highly specific to each patient during the study period. Disease-associated pathogens dominated sputum composition. Lung disease severity and recent antibiotic treatment had the highest impact on taxonomic composition in stool and sputum microbiome. Strikingly, the long-term antibiotic treatment burden had only a minor influence.
Conclusion
Despite the exercise and nutritional intervention, respiratory and intestinal microbiomes proved to be resilient. Dominant pathogens drove the composition and functionality of the microbiome. Further studies are required to understand which therapy could destabilize the dominant disease-associated microbial composition of pwCF.
Similar content being viewed by others


Background
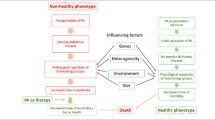
Cystic fibrosis (CF) is an autosomal-recessive genetic disorder. Due to mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, a defective or absent CFTR channel leads to an impaired chloride-sodium balance at cell surfaces, followed by a thickening of mucus in many organs. CF is a multi-organ disease affecting the lungs, the gastrointestinal and the reproductive organs [1].
In the lungs, impaired airway clearance leads to the accumulation of respiratory secretions and subsequent inflammation. This environment promotes the growth of pathogenic microbes such as Pseudomonas aeruginosa, Staphylococcus aureus or Stenotrophomonas maltophilia [2]. Frequent administration of antibiotics to treat progressive, chronic lung infections, generates a significant selective pressure on the lung and the gut microbiome and aggravates microbial dysbiosis [3]. However, even before antibiotic treatment, respiratory [4, 5] and intestinal microbiome dysbiosis is observed in infants and dysfunction in the CFTR channel is the primary associated cause [3, 6,7,8,9].
Abnormal microbiome colonization in the gastrointestinal and respiratory system amplifies undernutrition, growth failure, systemic inflammation and may result in pulmonary infections [10]. Reduced respiratory microbiome diversity and richness and increased dominance of CF-pathogens have been identified to correlate with lung function decline and disease deterioration [11, 12]. Gut dysbiosis is linked to intestinal inflammation and subsequent barrier impairment [9]. Most importantly, gut dysbiosis facilitates airway disease in CF, as distinct intestinal microbial composition patterns are associated with pulmonary infections already in the first year of life [7] Thus, the gut microbiome has direct implications for the course of lung disease in CF. This ability of the gut microbiome to influence lung physiology and pathology and vice versa is referred to as the gut-lung axis [13]. The connection of the gut-lung axis with CF health outcomes underlines the enormous need to decipher strategies to promote intestinal and respiratory microbiome health in CF [13].
Diet and exercise represent two potential means to influence microbiome composition in CF. Diet impacts microbiome composition [14, 15]. Diet high in fiber or in fermented foods might influence microbiome-mediated immune responses [16]. In order to improve health outcomes in CF, modulating the microbiome via diet or probiotic supplementation is a very appealing approach. In pwCF, probiotic intake has been associated with fewer pulmonary exacerbations and may decrease CF-related gastrointestinal inflammation [9, 17,18,19]. However, the evidence of probiotic benefits in CF is still limited. Probiotic studies are needed to provide further insights into the pivotal role of the gut-lung axis in CF [9].
Multiple studies linked physical exercise with gut microbiome modulation (reviewed in [20]). The relation between exercise and gut microbiota composition seems to be bidirectional [21]. Regular physical exercise modulates the gut microbiota composition, and on the other hand, gut microbiota composition impacts the physical exercise capacity of the host [21]. The impact of physical exercise on the respiratory microbiome has not been studied so far.
We offered a personalized nutrition and exercise program to pwCF for the duration of one year. We hypothesized that lifestyle changes, including increased physical activity, nutritional improvements and probiotic supplementation promotes intestinal and respiratory microbiome composition, thus delays disease progression in pwCF.
Results
Study population characteristics and evolution of clinical parameters
Out of 18 recruited pwCF, 15 concluded the study. Cohort characteristics are displayed in Table 1. Primary goal of the exercise intervention was to increase exercise participation in pwCF. This goal was met, as participants increased their training minutes per week about 42% due to the offered program [22].
Clinical assessments and microbiome characterization were performed at baseline (Visit 1), after 3 months (Visit 2) and after 12 months (Visit 3). Cardiopulmonary exercise testing (CPET) was performed to assess cardiorespiratory fitness. VO2peak measured during CPET remained stable, while a decline of lung function was observed in the same period. At Visit 2 & 3 nutritional counseling on the basis of 3-days weighing food protocols was offered to the participants. Fat intake was in 51% and protein intake in 28% of the assessments within the nutritional recommendations for pwCF (35%-45% of total caloric intake from fat and > 20% from protein [23]). A change in this macronutrient intake and eating habits did not occur throughout the study period (Supplementary Table 1). To assess body composition bioelectrical impedance analysis was performed to measure fat free mass (FFM) as a proxy for muscle mass. During the exercise intervention BMI remained stable, FFM increased in contrast.
Microbial communities remain stable during the study period
To determine whether the intervention had an impact on stool and sputum microbiome, we compared overall alpha diversity metrics (diversity, richness, evenness and dominance) between visits. We observed no difference in alpha diversity over time (p > 0.05) (Supplementary Fig. 1).
We confirmed stability of the microbiome composition over the three visits, measured as inter-sample Bray–Curtis dissimilarity and visualized using principal coordinates analysis (PCoA)) (Fig. 1 A-B, Supplementary Fig. 3).

A Composition of sputum microbiome was highly explained by dominant taxa and severity of the disease between patients. Principal Coordinate Analysis (PCoA) based on Bray–Curtis dissimilarity between samples. Points are colored by the dominant taxa detected and the shape indicates the phenotype severity based on ppFEV1. Crosses indicate the centroid of each cluster and the dashed lines the mean distances between clusters. Ellipses represent the 95% confidence interval of intra-cluster variability of sample distance to the centroid. Distances on PCo1 and PCo2 between groups based on dominant taxa were compared by Mann–Whitney U tests with Benjamini–Hochberg correction for multiple testing (Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1). Cluster of Staphylococcus dominated samples were significantly different to clusters with Pseudomonas and Streptococcus dominated microbiomes on PCo1 and PCo2. B Changes in sputum microbiome composition within patients between visits. A shift of dominance between Staphylococcus to Stenotrophomonas was linked to a high Bray–Curtis dissimilarity between visits for patient 9 (P9). For panel B and E each color represents samples from the same patient. C Association of dominant genera to alpha-diversity measures in sputum microbiomes. D Heatmap of the most abundant genera (> 0.5% mean relative abundance) for each cluster of dominant taxa observed on panel A). Dendrogram indicates Ward clustering of samples. E Relationship between dominant taxa in sputum and lung function. Lung function is represented by ppFEV1 at each visit, points are colored by patient and the size represents the relative abundance in percentage at the sample. Dashed read line indicates the threshold of 70% ppFEV1 to separate moderate (< 70%) from mild (> 70%) severity
Dominant taxa and disease severity are the main drivers of community structure
CF severity significantly impacted clustering of sputum or stool microbial compositions, explaining 8% and 7% of variance, respectively (Fig. 1A displays sputum clustering, Supplementary Fig. 3A displays stool clustering, statistical results of PERMANOVA are displayed in Table 2). Highest explanation of variance was explained by dominant taxa, dominant taxa were highly related to changes in microbial compositions and explained 55% and 32% of variance in sputum and stool, respectively (Table 2).
In sputum, Pseudomonas, Staphylococcus, Stenotrophomonas and Streptococcus generated four clusters (Fig. 1A). The intra-cluster variability was measured as the distance to each cluster centroid, the cluster of Streptococcus dominated samples has less variability among them than those clusters dominated by Pseudomonas or Staphylococcus (Supplementary Fig. 2). For stool just Bacteroides seemed to drive the clustering and separated samples from the rest (Supplementary Fig. 3A). BMI, sex, and age did not show a significant effect on microbial composition in sputum, contrary to the idea of the individuality of bacterial composition (Table 2). Visit, as a proxy for the intervention, did not explain changes in microbial composition. Disease severity and dominant taxa explained the composition of predicted functional genes for sputum, but not for stool.
The relative abundance of dominant taxa in sputum as an indicator of poor lung function
Considering the significant impact of dominant taxa in the composition of sputum, we explored whether the relative abundance of Pseudomonas, Staphylococcus, Streptococcus and Stenotrophomonas were linked to the lung function parameters. During the three visits we observed that the dominance of Pseudomonas was linked to ppFEV1 values below 70%, thus related to moderate-severe lung disease. On the contrary, even a high relative abundance of Staphylococcus occurred in patients with ppFEV1 values above 70% (Fig. 1E). Streptococcus dominated lung microbiome in individuals with better lung function, also displaying higher genus richness and diversity (Fig. 1C-E). A strong correlation of lung function parameters and alpha diversity metrics, as in previous studies [11, 24] was not reproducible (Supplementary Figs. 4 and 5 and Supplementary Tables 4 and 5).
Sport intervention lacks impact on microbial composition
We tested whether lung function and/or training parameters including time and frequency could explain BC-dissimilarity as a proxy for changes in individual microbial composition (Supplementary Fig. 6 A and B, Supplementary Tables 2 and 3).
Lung function parameters, but not training frequency or time, significantly explained changes in stool microbiome (ppFEV1:LRT, df = 1, P < 0.001, ppFVC: LRT, df = 1, P < 0.001). For sputum, neither training nor lung function parameters were significant explanatory variables of microbial changes (Supplementary Table 3), suggesting the stability and lower-complexity of sputum compared to stool. Sputum microbial changes only occurred in a small subset of patients, in which composition was strongly and significantly related to the dominant taxa present (Table 2).
Association of microbial features to clinical and nutritional characteristics (Fig. 2)

Association of microbial features to clinical and nutritional covariates from sputum microbiome. The plot shows bacterial taxa, at genus levels, altered significantly (post hoc FDR < 0.05) in abundance and showing a study effect, compared to baseline and follow-up visits. Signed effect sizes are shown through marker color. Direction of cuneiform plot indicates if taxa were enriched or depleted within the covariate groups (e.g. pwCF with oral Macrolide treatment displayed lower abundance of Neisseria, higher ppFVC correlated positively with increased Neisseria etc.). Size of cuneiform plots indicate significance. Covariates not shown had no difference in rank-transformed values (Cliff’s delta = 0). PPI = proton pump inhibitors, inh = inhalative, iv = intravenous
We analyzed the complete dataset to identify associations between clinical data and the abundance of bacterial taxa in the specific microbiomes.
In sputum, a higher abundance of Neisseria and Mogibacterium and other low abundant taxa correlated with higher ppFVC but not ppFEV1(Fig. 2). CFTR-modulator Lumacaftor/Ivacaftor therapy (N = 6) had no relevant effect on sputum bacteria. Inhaled antibiotics (N = 10) did not impact genera abundance in sputum. However, oral macrolides (N = 4) were associated with lower Neisseria abundance. Staphylococcus abundance in sputum was negatively associated with total antibiotic burden. IV-antibiotic burden correlated positively with Pseudomonas abundance in sputum.
In the stool microbiome (Supplementary Fig. 7), Lachnospiraceae UCG:004 showed a significant positive association with higher FFM measurements. We observed that regular intake of the stool softener Polyethylene Glycol (N = 4) was associated with an increased abundance of several genera, like Olsenella or Collinsella. Antibiotic burden did not correlate with specific genera abundance (Supplementary Fig. 7). Among the nutritional characteristics from the cohort, higher fiber intake correlated positively with Solobacterium abundance in the stool. Lactobacillus abundance did not increase during probiotic supplementation (Supplementary Fig. 8).
Recent antibiotic treatment, but not long-term cumulative antibiotic burden, affects diversity and richness
We used generalized linear mixed models (GLMM), adjusting for repeated measures by subject (Patient_number) and visit as random factors, to assess how antibiotic intake impacts the stool and sputum microbial composition. Shannon diversity and genus richness were significantly reduced in stool samples from patients receiving antibiotics in the last 15 days prior to sampling (LRT: χ2 = 6.79 (diversity) and 7.24 (richness), FDR < 0.05*) (Fig. 3A, Supplementary Table 6). In sputum samples this effect did not reach the significance threshold. Bacterial load (in 16S sRNA copy number) of sputum and stool was not reduced by oral or IV-antibiotic treatment taken within the last 2 weeks prior to sampling (Fig. 3A).

A Impact of antibiotic intake within the last 15 days prior sampling on alpha diversity measures and 16S copy number. Asterisks indicate significance for LMMs with Benjamini–Hochberg correction for multiple testing (FDR: ‘*’ < 0.05). B Relation between antibiotic burden and alpha diversity measures in sputum. Antibiotic burden had a significant impact on sputum evenness
Despite not reaching significance, we observed that changes in total antibiotic burden might explain around 10% of the microbial composition changes among stool samples (LRT: χ2 = 1.46, p = 0.226), and in contrast, just around 1% among sputum samples (LRT: χ2 = 0.1674, p = 0.682). Antibiotic burden did not affect alpha diversity measures in stool samples. In sputum, evenness increased with higher antibiotic burden (LRT: χ2 = 9.907, fFDR < 0.05*)(Fig. 3B, Supplementary Table 7). IV-antibiotics alone had no significant impact on diversity measurements, neither in stool nor in sputum.
Discussion
We hypothesized that exercise and nutritional intervention would improve health in pwCF, which could be traced in their microbiomes. So far, microbiome modulation via exercise therapy has not been studied in CF. This study demonstrates the high resilience of the CF respiratory and intestinal microbiota to supportive and antibiotic treatment.
The resilience of the microbiome, defined by the ability to remain or return to a stable state [25], was demonstrated by the lack of significant shifts in intra- or inter-individual microbial composition during our intervention at three sample time points. In a healthy host-microbiome ecosystem, resilience is considered to be beneficial to maintain equilibrium during external perturbations and can confer protection from disease [25]. We demonstrate that the resilience of the CF respiratory microbiome is driven by the high stability and dominance of disease-associated pathogens.
Participants successfully increased weekly training duration and frequency by about 40% due to the offered program [22], which underlines the efficacy of the study. Considering that VO2peak remained stable while lung function declined (as expected in pwCF), highlights further benefit of the sports intervention for the participants. Despite this significant lifestyle change, the respiratory and intestinal microbiomes remained highly resistant to the intensified exercise. This is in contrast to previous studies in prediabetic, diabetic or obese individuals [26, 27], which linked sport to a healthier gut microbiome composition. In those studies, effective sports intervention substantially improved cardiorespiratory fitness or body composition. Our cohort increased fat-free mass by intensified training, but not cardiorespiratory fitness (VO2peak) or lung function. An increase in FFM serves as a proxy for muscle gain [28] and demonstrates the additional efficacy of the offered program. Hence as muscle and fat tissues are less affected by the disease genotype, modulation of those is more feasible for pwCF, whereas VO2peak and lung function are strongly linked to CF lung disease and are less susceptible to exercise. In healthy populations, intensified exercise did not influence gut microbiota. Moitinho-Silva et al. [29] and Wang et al. [30] offered similar training, endurance and strength exercise programs to healthy adult and adolescent participants. Their intervention periods were much shorter (6 or 12 weeks, respectively), but they also found no changes in the gut microbiota related to exercise. One might hypothesize that prolonged intensified exercise and relevant changes in cardiorespiratory fitness are needed to influence the gut microbiota. In contrast, summarizing results of acute exercise interventions, Clauss et al. [20] concluded that intestinal bacterial diversity increased with the intensity of the physical activity performed. This included higher abundance of short-chain fatty acids producing bacteria, which might confer anti-inflammatory benefits to the host. The intensity of the physical activity performed by our study participants might not have been sufficient to modulate microbial composition, due to the underlying disease. The authors also noted a lack of longitudinal studies, especially considering nutritional intake and host characteristics as major confounders. Within our longitudinal study, we could control for confounding effects of nutritional variation, medication and subjects’ characteristics as BMI, FFM and disease severity.
No studies on the impact of exercise on the respiratory microbiome have been published so far. There is well established evidence that disease-associated pathogens are dominating the CF respiratory microbiome [11, 24]. Despite this distinct clustering of dominant taxa, changes and abundance of other genera within the sputum remained highly individual (Fig. 1E). Frey et al. [24] showed similar clustering of CF sputum into Pseudomonas, Staphylococcus, and an oropharyngeal-like flora (OF), including Streptococcus- and Neisseria- dominated communities. As in our cohort, Pseudomonas- and Staphylococcus-dominated microbiomes displayed lower Shannon diversity when compared to the OF microbiome. The OF microbiome can be compared to our cohort’s Streptococcus dominated microbiomes, which displayed higher diversity and species richness (Fig. 1C, D, E), thus implying a healthier respiratory microbiome in those individuals. Similar dominance patterns have also been found in sinus secretion of pwCF with chronic rhinosinusitis [31]. In both studies [24, 31], as in our study, individuals with a Pseudomonas-dominated respiratory microbiome had the poorest lung function (ppFEV1). The overgrowth of pathogens has been linked with disease severity several times [11, 24, 32]. Antibiotic treatment to reduce or eradicate those pathogens from the lungs is the standard of care in CF.
We showed how antibiotic intake affects the microbiome both in sputum and stool only transiently. We found that recent antibiotic treatments decreased the diversity and richness of the intestinal microbiome, and to some extent, of the respiratory microbiome (Fig. 3A).
Interestingly, bacterial biomass (measured in 16S copies) did not change due to antibiotic intake. Hence, the abundance of dominant pathogenic taxa was little affected by antibiotic intake. A reduction of less-abundant “satellite taxa” might explain the decrease of diversity and richness. Similar results were shown by Cuthbertson et al. [33] and Fodor et al. [34], who both analyzed the CF sputum microbial composition during exacerbation and after antibiotic treatment. Nelson et al. showed that inhaled tobramycin affected low abundant genera but not Pseudomonas abundance [35], thus supporting our results. Pseudomonas abundance was stable in our cohort in individuals with continuous inhaled antibiotic treatment.
The observed resilience of the respiratory microbiome to antibiotic treatment is in line with previous studies. Bacci et al. [36] showed in a 15 months-longitudinal study, how the pathogenic microbiome signature of the lung is recovered after antibiotic treatment, demonstrating the ability of the CF respiratory microbiome to return to stable composition after perturbation. In cardiometabolic diseased and healthy adults, a higher number of antibiotic courses within the last 5 years correlated with lower intestinal microbiota diversity [37]. We could not reproduce this observation in pwCF. Long-term antibiotic burden did not affect alpha diversity measures and further emphasizes the resilience of CF microbiomes (Fig. 3B and Supplementary Table 7). This might be explained by an already established dysbiotic composition at study start in the respiratory and intestinal microbiome of our cohort. Thus, we might not detect subtle changes due to antibiotic intake in our cohort. Burke et al. showed that number of iv-antibiotic courses in the year prior sampling can affect intestinal microbiome alpha-diversity and taxonomic abundances in their cohort of pwCF [38]. In addition, they pointed out, how the intense medical treatment of pwCF can promote intestinal dysbiosis. The dysbiotic state of the CF intestinal microbiome has been recently highlighted again [39]. We attempted to modulate intestinal microbiome composition via a probiotic intervention. Intriguingly, despite Lactobacillus intake, the abundance of it remained stable. Previous studies reported that lactic acid bacteria might fail to colonize the human gut depending on the hosts’ baseline gut microbiome characteristics [40]. Further studies are needed to investigate if only certain pwCF might benefit from probiotic supplementation, depending on baseline microbiome structures.
As part of the limitations of the current study, the limited number of pwCF that finalized the program is a major. In view of the expected high degree of compositional variability, we might have failed to detect more subtle signals due to limited sample numbers. Despite the limited cohort size, the repeated sampling allowed us to test our hypothesis with confidence. Even though the taxonomic resolution of 16S rRNA gene amplicon sequencing is limited, we showed that dominant CF-pathogens can be detected at the genus level. In the future, larger cohort studies, deploying whole genome sequencing, should overcome these limitations to study the putative effects of exercise and nutrition on the intestinal and respiratory microbiota in pwCF.
Conclusions
In summary, there is a lack of effective microbiome-modulating therapies to overcome the vigorous pathogen-dominated signature of the CF respiratory microbiome. The recently approved highly effective CFTR-modulator therapeutics might have the potential to surpass the demonstrated resilience of CF microbiomes. Future studies on CF microbiome composition under these therapeutics are needed.
Methods
Study design
An online monitored personalized exercise and nutrition program was offered to pwCF. Participants were enrolled between August 2016 and February 2019 at the University Medical Center in Mainz, Germany. Inclusion criteria were age > 12 years at study start and forced expiratory volume in one sec FEV1% predicted-values (ppFEV1) > 28 and < 100 and/or lung clearance index (LCI) > 9. Patients were excluded if they met orthopedic, cardiovascular or neurological contraindications regarding the sports program. Study start was delayed if a patient suffered severe pulmonary exacerbation or acute infection.
Interventions
The exercise program combined strength and endurance training and was performed in the home-environment of the patients. Participants reported their training progress and received weekly adjusted training recommendations by a sports scientist via an online platform. Details regarding the exercise intervention were recently published by Hillen et al. [22].
For the 3- day weighed food record (3d-WFR), participants weighed their dietary intake for three consecutive days prior V2 and V3. The daily intake of calories and macronutrients were quantified via an electronic dietary assessment tool (PRODI). Based on the results from the 3d-WFR, patients received nutrition counseling from a dietician to improve their diet according to CF-guidelines. Every patient took probiotics (Lactobacillus rhamnosus LGG Infectopharm®) from V2 to V3. When probiotics were part of the patient’s diet, they discontinued their use four weeks before the study started.
Clinical assessments
To monitor participants during the study, 3 clinical visits took place. Visit 1 (V1) at baseline, visit 2 (V2) after 3 months and visit 3 (V3) after 12 months (at the end of the study).
At each visit lung function (ppFEV1 and ppFVC (forced vital capacity in percent-predicted-values)) was assessed via flow- volume spirometry (MasterScope®Body, CareFusion, Germany) in line with the criteria of the American Thorax Society/European Respiratory Society. Cardiopulmonary exercise testing was performed on a treadmill with stepwise increment of speed (km/h) and incline (%) until maximum exhaustion was reached to assess peak oxygen uptake (VO2max). Further details are published by B. Hillen et al. [22].
To better evaluate body mass composition, bioelectrical impedance analysis (BIA) was performed to assess fat free mass. At each visit 3 measurements were taken, means were calculated and fat free mass in kg estimated using the CF-specific equation published by Charatsi et al. [41].
Disease severity was defined as mild (ppFEV1 > 70) and moderate (ppFEV1 40–70), choosing the highest ppFEV1 for each participant within three months prior to the study start. Antibiotic intake was retrospectively assessed for the 12 months prior to the study start and during the study. The number of treatment days and administration routes were collected. Antibiotic burden prior visit was defined by days on antibiotics during the last 365 days before sampling (e.g. 14 days on ceftazidime + colistin = 14 days).
Microbiome sampling, DNA-extraction and 16S sRNA gene sequencing, absolute quantification of bacterial load by qPCR
The intestinal and respiratory microbiome was characterized at each visit (V1-V3). Respiratory microbiome was evaluated from sputum; for intestinal microbiome stool microbiome was inferred as a proxy. Stool sampling was done at home with a provided stool sampling kit, as described in Knoll/Forslund et al. [42]. Collected stool samples were stored at home between 4 °C and 8 °C and transferred to the clinic within 24 h. Spontaneously expectorated sputum was collected into a sterile container during each visit at our CF-department. In the CF-department, both sample types were frozen at -80 °C immediately until further processing. For both sample types no preservation media were used. The number of participant samples analyzed varied depending on sample availability.
DNA was extracted and the V4 region of the 16 s rRNA genes was amplified using the primers 515F and 806R. For all samples, 16S copies/ml of DNA were estimated by qPCR.
For detailed DNA extraction and sequencing methods please refer to the supplement. Sequencing data is available at NCBI BioProject PRJNA810321.
Bioinformatic analysis
Sequences were pre-processed to infer amplicon sequence variants (ASVs) following the DADA2 v1.18 pipeline using R v. 4.03. All the R scripts used for this analysis are available at https://github.com/VictorHJD/exercise-cf-intervention.git. Details on sequence processing and statistical analyses are in the supplementary methods. All analyses were controlled for false discovery rate (FDR), employing the Benjamini–Hochberg procedure.
To assess alpha diversity metrics, we used Chao1, Shannon, Pielou and Berger-Parker indices to estimate richness, diversity, evenness and dominance. Beta diversity was assessed using the Bray–Curtis dissimilarity index between samples. We compared alpha diversity metrics and beta diversity distances between visits and dominant taxa for each sample type via Mann–Whitney U tests. We used Principal Coordinates Analysis (PCoA) for multivariate analysis with the Bray–Curtis distances. Permutational analysis of variance (PERMANOVA) for multivariate effect was performed, stratified for patient ID. We identified the dominant taxa from each patient as the ASV with highest relative abundance of all ASVs at the genus level per sample. Functional profiles were predicted from the 16S rRNA sequences using PICRUSt2 [43].
Generalized linear mixed-effects models (GLMM) were calculated with patient ID and visit (where appropriate) as random factors for different inference analyses. Pairwise comparisons for the interactions were calculated. GLMMs were compared through a likelihood ratio test (LRT).
To assess possible associations between ASVs abundance and clinical characteristics, nutritional information, or medication, we ran a multivariate correlation analysis [44]. In brief, we tested for correlations for each microbial feature with each covariate and used Spearman’s rho as a standardized signed effect estimate. A post-hoc test was done to account for dependency between patient samples: for each of two correlated features, a mixed-effects model was fitted of the rank-transformed variable using the rank of the other as an explanatory variable, with patient ID as a random effect. We reported standardized nonparametric effect sizes (signed) Cliff’s delta metric.
Changes on clinical parameters were analyzed with repeated measures ANOVA.
Availability of data and materials
The datasets supporting the conclusions of this article are available in NCBI BioProject PRJNA810321 https://www.ncbi.nlm.nih.gov/bioproject/PRJNA810321.
Abbreviations
- CF:
-
Cystic fibrosis
- CFTR:
-
Cystic fibrosis transmembrane conductance regulator
- pwCF:
-
People with cystic fibrosis
- ppFEV1:
-
Forced expiratory volume in first second in percent-predicted-values
- ppFVC:
-
Forced vital capacity in percent-predicted-values
- LCI:
-
Lung clearance index
- BMI:
-
Body mass index
- IV:
-
Intravenous
- ASV:
-
Amplicon sequence variant
- SD:
-
Standard deviation
- ANOVA:
-
Analysis of variance
- IQR:
-
Inter quartile range
- qPCR:
-
Quantitative polymerase chain reaction
- PCoA:
-
Principal coordinates analysis
- FDR:
-
False discovery rate
- GLMM:
-
Generalized linear mixed-effects models
- LRT:
-
Likelihood ratio test
References
Elborn JS. Cystic fibrosis. Lancet. 2016;388(10059):2519–31.
Rogers G, Huang YJ. Chronic suppurative lung disease: cystic fibrosis and non-cystic fibrosis bronchiectasis. In: Cox MJ, Ege MJ, von Mutius E, eds. The Lung Microbiome (ERS Monograph). Sheffield, European Respiratory Society, 2019; pp. 158–172 [https://doi.org/10.1183/2312508X.10016218].
Kristensen M, Prevaes SMPJ, Kalkman G, Tramper-Stranders GA, Hasrat R, de Winter-de Groot KM, et al. Development of the gut microbiota in early life: The impact of cystic fibrosis and antibiotic treatment. J Cyst Fibros. 2020;19(4):553–61.
Hahn A, Burrell A, Ansusinha E, Peng D, Chaney H, Sami I, et al. Airway microbial diversity is decreased in young children with cystic fibrosis compared to healthy controls but improved with CFTR modulation. Heliyon. 2020;6(6):e04104. https://doi.org/10.1016/j.heliyon.2020.e04104.
Muhlebach MS, Zorn BT, Esther CR, Hatch JE, Murray CP, Turkovic L, et al. Initial acquisition and succession of the cystic fibrosis lung microbiome is associated with disease progression in infants and preschool children. PLoS Pathog. 2018;14(1):1–20.
Nielsen S, Needham B, Leach ST, Day AS, Jaffe A, Thomas T, et al. Disrupted progression of the intestinal microbiota with age in children with cystic fibrosis. 2016;6(1):24857. Available from: www.nature.com/scientificreports.[cited 2018 Jul 24]
Antosca KM, Chernikova DA, Price CE, Ruoff KL, Li K, Guill MF, et al. Altered stool microbiota of infants with cystic fibrosis shows a reduction in genera associated with immune programming from birth. J Bacteriol. 2019;201(16):e00274-19.
Vernocchi P, del Chierico F, Russo A, Majo F, Rossitto M, Valerio M, et al. Gut microbiota signatures in cystic fibrosis: Loss of host CFTR function drives the microbiota enterophenotype. 2018. https://doi.org/10.1371/journal.pone.0208171
Thavamani A, Salem I, Sferra TJ, Sankararaman S. Impact of altered gut microbiota and its metabolites in cystic fibrosis. Metabolites. 2021;11(2):1–22.
Madan JC. Neonatal gastrointestinal and respiratory Microbiome in cystic fibrosis: potential interactions and implications for systemic health. Clin Ther. 2016;38(4):740–6. https://doi.org/10.1016/j.clinthera.2016.02.008.
Cuthbertson L, Walker AW, Oliver AE, Rogers GB, Rivett DW, Hampton TH, et al. Lung function and microbiota diversity in cystic fibrosis. Microbiome. 2020;8(1):1–13.
Héry-Arnaud G, Boutin S, Cuthbertson L, Elborn SJ, Tunney MM. The lung and gut microbiome: what has to be taken into consideration for cystic fibrosis? J Cyst Fibros. 2019;18(1):13–21. https://doi.org/10.1016/j.jcf.2018.11.003.
Price CE, O’Toole GA. The gut-lung axis in cystic Fibrosis. J Bacteriol Am Soc Microbiol. 2021;203:e0031121.
Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science (1979). 2011/09/03. 2011;334(6052):105–8. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3368382/pdf/nihms378475.pdf
David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–63. https://doi.org/10.1038/nature12820.
Wastyk HC, Fragiadakis GK, Perelman D, Dahan D, Merrill BD, Yu FB, et al. Gut-microbiota-targeted diets modulate human immune status. Cell. 2021;184(16):4137-4153.e14. https://doi.org/10.1016/j.cell.2021.06.019.
di Nardo G, Oliva S, Menichella A, Pistelli R, de Biase RV, Patriarchi F, et al. Lactobacillus reuteri ATCC55730 in cystic fibrosis. J Pediatr Gastroenterol Nutr. 2014;58(1):81–6.
Bruzzese E, Callegari ML, Raia V, Viscovo S, Scotto R, Ferrari S, et al. Disrupted Intestinal Microbiota and Intestinal Inflammation in Children with Cystic Fibrosis and Its Restoration with Lactobacillus GG: A Randomised Clinical Trial. Available from: www.plosone.org
Anderson JL, Miles C, Tierney AC. Effect of probiotics on respiratory, gastrointestinal and nutritional outcomes in patients with cystic fibrosis: A systematic review. J Cyst Fibros. 2017;16(2):186–97. https://doi.org/10.1016/j.jcf.2016.09.004.
Clauss M, Gérard P, Mosca A, Leclerc M. Interplay Between Exercise and Gut Microbiome in the Context of Human Health and Performance. Front Nutr. 2021;8:637010. Available from: www.frontiersin.org
Marttinen M, Ala-Jaakkola R, Laitila A, Lehtinen MJ. Gut microbiota, probiotics and physical performance in athletes and physically active individuals. Nutrients. 2020;12(10):1–39.
Hillen B, Simon P, Schlotter S, Nitsche O, Bähner V, Poplawska K, et al. Feasibility and implementation of a personalized, web-based exercise intervention for people with cystic fibrosis for 1 year. BMC Sports Sci Med Rehabil. 2021;13(1):1–11. https://doi.org/10.1186/s13102-021-00323-y.
Turck D, Braegger CP, Colombo C, Declercq D, Morton A, Pancheva R, et al. ESPEN-ESPGHAN-ECFS guidelines on nutrition care for infants, children, and adults with cystic fibrosis. Clin Nutr. 2016;35(3):557–77. https://doi.org/10.1016/j.clnu.2016.03.004.
Frey DL, Boutin S, Dittrich SA, Graeber SY, Stahl M, Wege S, et al. Relationship between airway dysbiosis, inflammation and lung function in adults with cystic fibrosis. J Cyst Fibros. 2021;20(5):754–60.
Sommer F, Anderson JM, Bharti R, Raes J, Rosenstiel P. The resilience of the intestinal microbiota influences health and disease. Nat Rev Microbiol. 2017;15(10):630–8. https://doi.org/10.1038/nrmicro.2017.58.
Motiani KK, Collado MC, Eskelinen JJ, Virtanen KA, Löyttyniemi E, Salminen S, et al. Exercise training modulates gut microbiota profile and improves endotoxemia. Med Sci Sports Exerc. 2020;52(1):94–104.
Kern T, Blond MB, Hansen TH, Rosenkilde M, Quist JS, Gram AS, et al. Structured exercise alters the gut microbiota in humans with overweight and obesity—A randomized controlled trial. Int J Obes. 2020;44(1):125–35.
Müller MJ, Braun W, Pourhassan M, Geisler C, Bosy-Westphal A. Application of standards and models in body composition analysis. Proc Nutr Soc. 2016;75(2):181–7.
Moitinho-Silva L, Wegener M, May S, Schrinner F, Akhtar A, Boysen TJ, et al. Short-term physical exercise impacts on the human holobiont obtained by a randomised intervention study. BMC Microbiol. 2021;21(1):1–14.
Wang R, Cai Y, Li J, Yau SY, Lu W, Stubbs B, et al. Effects of aerobic exercise on gut microbiota in adolescents with subthreshold mood syndromes and healthy adolescents: A 12-week, randomized controlled trial. J Affect Disord. 2021;293:363–72.
Lucas SK, Feddema E, Boyer HC, Hunter RC. Diversity of cystic fibrosis chronic rhinosinusitis microbiota correlates with different pathogen dominance. J Cyst Fibros. 2021;20(4):678–81. https://doi.org/10.1016/j.jcf.2021.03.022.
Losada PM, Chouvarine P, Dorda M, Hedtfeld S, Mielke S, Schulz A, et al. The cystic fibrosis lower airways microbial metagenome. Available from: http://ow.ly/ZiqUE
Cuthbertson L, Rogers GB, Walker AW, Oliver A, Green LE, Daniels TWV, et al. Respiratory microbiota resistance and resilience to pulmonary exacerbation and subsequent antimicrobial intervention. ISME J. 2016;10(5):1081–91. https://doi.org/10.1038/ismej.2015.198.
Fodor AA, Klem ER, Gilpin DF, Elborn JS, Boucher RC, Tunney MM, et al. The Adult Cystic Fibrosis Airway Microbiota Is Stable over Time and Infection Type, and Highly Resilient to Antibiotic Treatment of Exacerbations. PLoS One. 2012;7(9):e45001.
Nelson MT, Wolter DJ, Weiss EJ, Vo AT. Maintenance tobramycin primarily affects untargeted bacteria in the CF sputum microbiome. Available from: http://thorax.bmj.com/
Bevivino A, Bacci G, Drevinek P, Nelson MT, Hoffman L, Mengoni A. Deciphering the Ecology of Cystic Fibrosis Bacterial Communities: Towards Systems-Level Integration. Trends Mol Med. 2019;25(12):1110–22.
Forslund SK, Chakaroun R, Zimmermann-Kogadeeva M, Markó L, Aron-Wisnewsky J, Nielsen T, et al. Combinatorial, additive and dose-dependent drug–microbiome associations. Nature. 2021;600(7889):500–5.
Burke DG, Fouhy F, Harrison MJ, Rea MC, Cotter PD, O’Sullivan O, et al. The altered gut microbiota in adults with cystic fibrosis. BMC Microbiol. 2017;17(1):1–11.
Marsh R, Gavillet H, Hanson L, Ng C, Mitchell-Whyte M, Major G, et al. Intestinal function and transit associate with gut microbiota dysbiosis in cystic fibrosis. J Cyst Fibros. 2021;(xxxx). https://doi.org/10.1016/j.jcf.2021.11.014
Maldonado-Gómez MX, Martínez I, Bottacini F, O’Callaghan A, Ventura M, van Sinderen D, et al. Stable Engraftment of Bifidobacterium longum AH1206 in the Human Gut Depends on Individualized Features of the Resident Microbiome. Cell Host Microbe. 2016;20:515–26.
Charatsi AM, Dusser P, Freund R, Maruani G, Rossin H, Boulier A, et al. Bioelectrical impedance in young patients with cystic fibrosis: Validation of a specific equation and clinical relevance. J Cyst Fibros. 2016;15(6):825–33.
Knoll RL, Forslund K, Kultima JRJR, Meyer CU, Kullmer U, Sunagawa S, et al. Gut microbiota differs between children with Inflammatory Bowel Disease and healthy siblings in taxonomic and functional composition: a metagenomic analysis. Am J Physiol Gastrointest Liver Physiol. 2017;312(4):G327–39. Available from: http://ajpgi.physiology.org/lookup/doi/https://doi.org/10.1152/ajpgi.00293.2016
Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31(9):814–21. https://doi.org/10.1038/nbt.2676.
Birkner T. metadeconfoundR. 2021. Available from: https://github.com/TillBirkner/metadeconfoundR.
Acknowledgements
We would like to thank all participants and their families, as well as the health care staff of our cystic fibrosis outpatient clinic and the members of the pediatric immunology and infectiology lab for handling/biobanking the probes. Alissa Kemper analyzed clinical data presented in this manuscript for her medical doctoral thesis. We thank Jennifer Nazat Martinez for her insightful comments on the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL. The study was partly funded by Vertex pharmaceuticals—grant fund: CF Circle of Care. VHJD and SKF are supported through the JPI AMR—EMBARK project funded by the Bundesministerium für Bildung, und Forschung under grant number F01KI1909A.
Author information
Authors and Affiliations
Contributions
CRediT author statement: Rebecca L. Knoll: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Data Curation, Writing - Original Draft, Visualization. Víctor Hugo Jarquín-Díaz: Methodology, Validation, Formal analysis, Investigation, Data Curation, Writing - Original Draft, Visualization. Jonas Klopp: Validation, Formal analysis, Investigation, Writing - Review & Editing. Alissa Kemper: Formal analysis, Investigation, Data Curation. Katja Hilbert: Validation, Investigation, Data Curation. Barlo Hillen: Conceptualization, Validation, Investigation, Data Curation, Writing - Review & Editing. Daniel Pfirrmann: Supervision, Project administration. Perikles Simon: Supervision, Project administration, Resources. Viola Bähner: Conceptualization, Investigation, Data Curation. Oliver Nitsche: Conceptualization, Investigation, Data Curation. Stephan Gehring: Validation, Resources, Writing - Review & Editing, Supervision. Lajos Markó: Validation, Writing - Review & Editing, Supervision. Sofia K. Forslund: Conceptualization, Methodology, Validation, Resources, Writing - Review & Editing, Supervision, Project administration, Funding acquisition. Krystyna Poplawska: Conceptualization, Methodology, Validation, Resources, Writing - Review & Editing, Supervision, Project administration, Funding acquisition. All authors reviewed the manuscript. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was performed in accordance with the Declaration of Helsinki and was approved by the ethics committee from the State Chamber of Physicians of Rhineland-Palatinate (RN 837.288.16 (10607)). Written informed consent was provided by all adult subjects, and parents or guardians of underaged participants provided written informed consent on their behalf along with written assent from the underaged participants.
Consent for publication
Not applicable.
Competing interests
Krystyna Poplawska received personal payment for participation on the advisory board in December 2021 (Vertex Pharmaceuticals). All other authors have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Knoll, R.L., Jarquín-Díaz, V.H., Klopp, J. et al. Resilience and stability of the CF- intestinal and respiratory microbiome during nutritional and exercise intervention. BMC Microbiol 23, 44 (2023). https://doi.org/10.1186/s12866-023-02788-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-023-02788-y