
Abstract
The growing interest in the therapeutic application of bacteriophages leads to a drastic increase in the number of sequenced genomes. Luckily, recent insights in phage taxonomy facilitate the classification of phages in a comprehensive and data-driven manner as recently proposed by the International Committee on Taxonomy of Viruses. In this research, we present the taxonomical classification of a novel, narrow host range Xanthomonas phage FoX4, isolated from a Brussels sprouts field in Belgium infested with Xanthomonas campestris pv. campestris. The phage has a limited ability to lyse a bacterial culture, yet adsorbs efficiently to its host. Based on its genome sequence and low similarity to previously described phages, the phage comprises the novel phage genus Foxquatrovirus.
Similar content being viewed by others
Introduction
Viruses are considered the most abundant entities in the world with an estimated 1031 particles, including bacteriophages, i.e. viruses infecting bacterial hosts [1]. An important group of bacterial viruses belong to the class of the Caudoviricetes, the tailed bacteriophages. Within this group, three virion morphologies can be distinguished: myoviruses with contractile tails, podoviruses with a short tail structure and siphoviruses with long non-contractile tails [2]. Traditionally, these morphotypes were translated into three phage families. Recent sequencing technologies, however, have revolutionized our understanding of the complexity and diversity of these viruses. New insights in the taxonomy of the tailed phages were gathered and showed that the complexity reached far beyond the diversity based on particle morphology. As such, major shifts in phage taxonomy have recently been proposed by the International Committee on Taxonomy of Viruses (ICTV) suggesting a.o. the abolishment of the traditional Myoviridae , Podoviridae and Siphoviridae families and raising subfamilies to the level of families [3]. This creates a big opportunity in the genomics of bacteriophages and the reorganization of unclassified viruses.
According to a review conducted by Zrelovs and colleagues, the total number of complete phage genomes counted over 7,718 in 2020. This number is an underrepresentation of the current figures, especially since the interest in phages as therapy or biocontrol strategies in different fields such as human medicine and agriculture is increasing [4]. More than half of the total complete genomes were previously classified as Siphoviridae [4]. However, phages with the distinct siphovirus morphology are also found in other families such as the Demerecviridae and Drexlerviridae, further supporting the abolishment of the morphotype-based family distinction in phage taxonomy. As a result, the classification of many siphoviruses is now incomplete.
The overall growing interest in phage biocontrol is especially true for crop protection. Lately, the number of phages reported that infect plant pathogenic bacteria has increased drastically [5,6,7]. Multiple bacterial genera and species are targeted and crop specific application strategies are developed. One particular bacterial species of interest is Xanthomonas campestris pv. campestris (Xcc). This bacterium is the causal agent of black rot disease in brassica crops and arugula [8, 9]. Xcc is a Gram-negative bacterium that is known to grow epiphytically on leaves of crucifers and infects the plant through hydathodes or wounds. Infected weeds and crop residues have been described to function as a reservoir from where the bacteria can spread [8, 10,11,12]. Back in 1924, the first field trials with phages were conducted to control black rot disease [13]. In more recent years, several authors have revived the research on the development of a phage-based biocontrol strategy. Nagai and colleagues described for example the application of an Xcc phage pXcpSFC211 (pXS) together with a non-pathogenic Xanthomonas strain reducing disease severity between 2.7% and 18.9% compared to 43.3% and 93.7% in the controls. They further showed symptoms reductions by 24% in field conditions [14]. Similarly, our group published the potential of two Xcc phages in seed decontamination, bioassays in greenhouse conditions and field trials [5]. Papaianni and colleagues did not only show the potential of their Xcc phage as a biocontrol agent, but also showed its synergy with hydroxyapatite and long fatty acids to reduce Xcc biofilms [15, 16]. They further showed that cabbages do react on the presence of phage by themselves as they demonstrated decreased levels of amino acids- and nitrogen-containing compounds. Additionally, they demonstrated that in the presence of both the phage and the bacterium, significant differences are observed in the metabolic profiles, suggesting a response of the plant in the presence of phage [17]. Combined, these results illustrate the potential of bacteriophages as a valid biocontrol strategy.
In this research, we characterized novel Xanthomonas phage FoX4. We investigated the host range of the phage using an extensive bacterial collection isolated in Flanders (Belgium) along with the infection characteristics of the phage. Furthermore, based on whole genome sequencing, we analyzed the phylogeny of the phage and confirmed the annotation of the structural cassette using a gel-free mass spectrometry analysis.
Materials and methods
Phage isolation and transmission electron micrographs
FoX4 was isolated as described elsewhere [5]. In short, Xcc strain GBBC 1484 was grown to an optical density (600 nm; OD600) of 0.3 in 4 mL tubes. Xcc infested soil collected from infested fields was added to the growing bacterial culture (0.5 g). The mixture was incubated overnight at 25 °C, centrifuged (4,000 rpm, 10 min, 4 °C), filtered using 0.45 µm filters and spotted on a bacterial lawn of GBBC 1484. Lysis zones were picked with sterile toothpicks and plated using the double agar technique. Single plaques were picked up three consecutive times to obtain a monoclonal phage stock. Phages were propagated as previously described [18]. Transmission electron micrographs were made as described by Martino and colleagues [19].
Characterizing the phage infection
The host range of FoX4 was determined by assessing its ability to form plaques on a particular strain. We used our previously reported strain collection [5]. Phages were spotted (3 µL) in a dilution series (106 – 105 – 104 PFU/mL) on a bacterial lawn (200 µL of overnight culture in 4 mL of soft agar) of a specific strain and incubated overnight at 25 °C. Strains were considered susceptible to the phage if individual plaques were observed. The experiment was conducted in duplicate and strains that showed plaques in both assays were considered susceptible to the phage.
The lysis activity of the phage in liquid broth was asses by measuring the optical density of an exponentially growing bacterial culture (GBBC1412 at OD600 of 0.3) infected with phage at different multiplicities of infection (MOIs) (0.1 – 1) for nine hours. The adsorption rate of FoX4 was determined as previously described [20].
Genome sequencing, annotation and proteome analysis
Phage DNA was isolated from the virions as described by Sambrook and Russel (2001). In short, polyethylene glycol (PEG8000)-precipitated phage stocks were treated with DNase I and RNase A for 30 min at 37 °C. Next, 10% SDS, 50 mM EDTA and proteinase K were added and incubated for one hour at 56 °C to break down the virions. A phenol/chloroform extraction followed by an ethanol precipitation was performed to clean up the phage DNA. The phage DNA was sequenced on an Illumina MiSeq platform (VIB Nucleomics Core, Belgium) and analyzed as previously described [5]. In short, reads were trimmed with Trimmomatic v0.39 and assembled using Unicycler v0.5.0 [21, 22]. The quality of the assembly was visualized by Bandage v0.9.0 [23]. Phage genomes were automatically annotated using the RASTtk pipeline [24] and manually curated. By means of primer walking, the physical ends of the genome were determined. Phage structural proteins were extracted by methanol-chloroform extraction as described previously, followed by in-gel trypsinization [25, 26]. Eluted peptide mixtures were analyzed with liquid chromatography tandem mass spectrometry (LC–MS/MS) on an Ultimate 3000 RSLC nano-LC (Thermo Fisher Scientific, Bremen, Germany) in-line coupled to a Q Exactive mass spectrometer (Thermo Fisher Scientific) at the VIB Proteomics Core (Belgium). Peptides were identified using the MaxQuant algorithm (version 2.0.2.0). Spectra were searched against the annotated phage proteome.
Results
Isolation and microbiological characterization of Xanthomonas phage FoX4
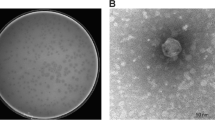
Soil collected from the roots of symptomatic Brussels sprouts was incubated with Xcc GBBC 1484, the strain isolated from the same sample. Small turbid plaques were obtained with a diameter of approximately 1 mm. Based on transmission electron micrographs, FoX4 has a typical siphovirus morphology (Fig. 1).

Transmission electron micrograph of FoX4 showing a typical siphovirus morphology. Scale bar represents 50 nm
The phage had a relatively narrow host range and infects 27% of our collection of 69 diverse strains (Supplementary Table 1). There appeared to be no relation between the susceptibility of a strain and the crop from which it was isolated. Despite its narrow host range, FoX4 could infect one of the Xanthomonas campestris pv. raphani (Xcr) strains in collection, GBBC 1468, suggesting shared phage susceptibility determinants between Xcc and Xcr for this phage. Furthermore, FoX4 infected the historical reference strains LMG 8052 and LMG 566, isolated in 1986 and 1941, respectively.
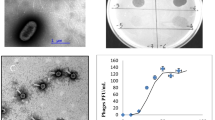
We further investigated the efficacy of FoX4 to lyse a bacterial culture and its adsorption kinetics (Fig. 2). At an MOI of 0.1, FoX4 only displayed a minor influence on the bacterial growth compared to the negative control. When a tenfold higher phage concentration was applied, a reduction of the bacterial growth was observed after 120 min with a clear decrease of the OD after 350 min. Despite its relatively limited lysis activity in an exponentially growing culture, FoX4 adsorbed rather fast to its host. After one minute, 92% of phage particles are adsorbed to the bacterial cells resulting in an adsorption constant of 5.69 × 10–9 mL/min.

Infection characteristics of phage FoX4. A. Killing curve of FoX4 with two different MOIs (0.1 – 1), for which the optical density at 600 nm was monitored over a time span of 550 min on Xcc strain GBBC 1484. Error bars show the standard deviation. B. Adsorption curve of FoX4 on Xcc strain GBBC 1484 showing the percentage of non-adsorbed phages over a time span of 8 min. Error bars represent the standard deviation
FoX4 is an orphan phage of a new phage genus
Whole genome sequencing and de novo assembly of the genome demonstrated that the phage had a relatively low similarity to other phages in NCBI. Based on a VipTree analysis, its closest neighbors were Burkholderia phage BceS AH2 along with Xylella phages Sano and Salvo and Burkholderia phage BcepNazgul. We ran a GRAViTy analysis on the genome as described by Turner et al. 2021 showing that FoX4 clusters together with other siphoviruses [3] (Supplementary Figure 1). We selected the genomes of phages sharing between 0.05 and 0.1 CGJ (composite generalized Jaccard) distance to investigate the most related genomes (Supplementary Figure 2). Based on this relatedness, we distinguished six groups sharing over 0.15 CGJ within this specific cluster. We further zoomed in on the most related phage genomes to FoX4 (> 0.4 CGJ), which were incorporated in a VIRIDIC analysis (Fig. 3). Here, we could clearly see that within this particular subcluster as determined by the GRAViTy analysis, the percentage identity was relatively low. FoX4 can therefore be considered an orphan phage, distantly related to, but only sharing low sequence identities with, BcepNazgul (Nazgulvirus genus), BceS AH2 (Ahduovirus genus), Sano and Salvo (Sanovirus genus). Within this subcluster though, we could distinguish the Chivirus genus as well with main representative Salmonella phage chi. Altogether, these data suggest that FoX4 represents a new phage genus Foxquatrovirus, which was ratified by the ICTV.

Heatmap of the phage genomes predicted to be in the same subfamily as FoX4 based on a VIRIDIC analysis. Genome similarities above 70% represent one phage genus. The Foxquatrovirus genus is most related to the Sanovirus, Nazgulvirus and Ahduovirus genera. Most dominantly in our analysis is the Chivirus genus
Genome architecture of FoX4 and confirmation of the structural cassette by mass spectrometry
When comparing the genomes of FoX4 with its closest relatives, we observed a highly similar genome architecture between the different phages (Fig. 4). Four gene clusters could be distinguished which were shared among all relatives. Notably, the early genes were transcribed on the reverse strand as well as the structural and lysis cassette. Just like its relatives, FoX4 showed potential relicts of a temperate lifestyle encoded in its genome with recombination-related proteins and Cro-like repressors.

Genome maps of FoX4 and its closest relatives Xylella phages Sano and Salvo and Burkholderia phages AH2 and BcepNazgul. ORFs associated with replication are given in blue, lysis in red, the structural cassette in green and hypothetical proteins in white. Sequence similarity between the genomes was assessed with BLASTn
Based on mass spectrometry, we determined the full structural cassette of FoX4 (Table 1). As such, we confirmed that gp18-21, gp25-26, gp28-34, gp36, gp43 and gp44, encoded within the putative structural cassette, indeed code for proteins of the FoX4 virion. Six previously hypothetical proteins can now be annotated as part of the structural cassette encoding virion-associated proteins. Interestingly, peptides from gp60 were also found in the sample. As this gene is not embedded in the structural cassette, it is difficult to conclude whether it is truly part of the virion. Maybe more likely, this recombination protein is produced in large quantities during the infection cycle and therefore detected in the mass spectrometry analysis.
Discussion
As the interest in the application of phages for biological control of bacteria increases and the cost of sequencing further decreases, the number of accessible phage genomes will also keep on increasing. This creates challenges yet unique opportunities to fleece out the taxonomical relatedness of bacteriophages based on genome sequence. In this research, we investigated the microbiological and genomic characterization of Xanthomonas phage FoX4. In our isolation procedure, we chose to use a soil-based isolation as crop residues and infested soil are believed to function as a reservoir for Xcc [8]. The field sampled for this study was used for crucifer production in consecutive years, creating a unique opportunity to sample infested soil. Similarly, Papaianni and colleagues isolated Xcc phages from the rhizosphere of Kohlrabi [16]. Yet, Xcc phage Carpasina was isolated from the leaves of infected brassica leaves [27], also demonstrating the potential of phage isolations from leaves rather than soil. [27].
Siphovirus FoX4 has a narrow host range infecting only a minor subset of the strains in our collection. Interestingly, the host range of FoX4 is highly similar to Xcc phage FoX6 as previously reported by our group [5]. A more detailed analysis of the receptor of FoX6 showed that this phage most likely recognizes lipopolysaccharides (LPS) on the Xcc cell wall. As FoX4 appears to infect the same strains as FoX6, chances are high that FoX4 and FoX6 require similar phage susceptibility determinants. Yet, there is a difference in the host range between FoX4 and FoX6 as FoX4 can infect historical reference strains while these strains were resistant to FoX6. This example not only demonstrates the intimate interaction between the phage and its bacterial host, but also the need for a detailed host range analysis in order to design phage biocontrol applications. Additionally, in recent years, diverse types of phage resistance mechanisms were reported that interrupt phage infection. We are only beginning to understand the distribution of these mechanisms among bacterial strains and how they impact phage infection [28].
Based on its genome, FoX4 can be considered an orphan phage and representative of a new phage genus [3]. The closest relatives, Xylella phages Sano and Salvo, and Burkholderia phages AH2 and BcepNazgul are quite distant based on the genome sequence only sharing 23.46, 24.15, 22.24 and 19.64% of Blastn identity, respectively. These phages represent three different phage genera. Despite this low identity, our analysis showed that there is indeed relatedness between the aforenoted phage genera.
Data availability
Genome data for FoX4 is accessible on NCBI under the accession number MT161385.1.
References
Suttle CA. Viruses in the sea. Nature. 2005;437(7057):356–61.
Ackermann HW. 5500 Phages examined in the electron microscope. Arch Virol. 2007;152(2):227–43.
Turner D, Kropinski AM, Adriaenssens EM. A roadmap for genome-based phage taxonomy. Viruses. 2021;13:506.
Zrelovs N, Dislers A, Kazaks A. Motley crew: overview of the currently available phage diversity. Front Microbiol. 2020;11:579452.
Holtappels D, Fortuna KJ, Moons L, Broeckaert N, Bäcker LE, Venneman S, et al. The potential of bacteriophages to control Xanthomonas campestris pv. campestris at different stages of disease development. Microb Biotechnol. 2022;15(6):1762–82.
Svircev A, Roach D, Castle A. Framing the future with bacteriophages in agriculture. Viruses. 2018;10(5):218.
Jones JB, Svircev AM, Obradović AŽ. Crop use of bacteriophages. In: Harper DR, et al, editors. Bacteriophages: Crown; 2021. p. 839–56.
Vicente JG, Holub EB. Xanthomonas campestris pv. Campestris (cause of black rot of crucifers) in the genomic era is still a worldwide threat to brassica crops. Mol Plant Pathol. 2013;14(1):2–18.
Rosenthal ER, Ramos Sepulveda L, Bull CT, Koike ST. First report of black rot caused by Xanthomonas campestris on Arugula in California. Plant Dis. 2018;102(5):1025.
Ryan RP, Vorhölter FJ, Potnis N, Jones JB, van Sluys MA, Bogdanove AJ, et al. Pathogenomics of Xanthomonas: Understanding bacterium-plant interactions. Nat Rev Microbiol. 2011;9(5):344–55.
An SQ, Potnis N, Dow M, Vorhölter FJ, He YQ, Becker A, et al. Mechanistic insights into host adaptation, virulence and epidemiology of the phytopathogen Xanthomonas. FEMS Microbiol Rev. 2019;44(1):1–32.
Ignatov A, Sechler A, Schuenzel EL, Agarkova I, Oliver B, Vidaver AK, et al. Genetic diversity in populations of Xanthomonas campestris pv. campestris in cruciferous weeds in central coastal California. Phytopathology. 2007;97(7):803–12.
Mallmann W, Hemstreet C. Isolation of an inhibitory substance from plants. Agric Res. 1924;28:599–602.
Nagai H, Miyake N, Kato S, Maekawa D, Inoue Y, Takikawa Y. Improved control of black rot of broccoli caused by Xanthomonas campestris pv. campestris using a bacteriophage and a nonpathogenic Xanthomonas sp. strain. J Gen Plant Pathol. 2017;83(6):373–81.
Papaianni M, Ricciardelli A, Fulgione A, d’Errico G, Zoina A, Lorito M, et al. Antibiofilm activity of a trichoderma metabolite against Xanthomonas campestris pv. campestris, alone and in association with a Phage. Microorganisms. 2020;8(5):620.
Papaianni M, Cuomo P, Fulgione A, Albanese D, Gallo M, Paris D, et al. Bacteriophages promote metabolic changes in bacteria biofilm. Microorganisms. 2020;8(4):480.
Papaianni M, Paris D, Woo SL, Fulgione A, Rigano MM, Parrilli E, et al. Plant dynamic metabolic response to bacteriophage treatment after Xanthomonas campestris pv. campestris infection. Front Microbiol. 2020;11:732.
Rombouts S, van Vaerenbergh J, Volckaert A, Baeyen S, de Langhe T, Declercq B, et al. Isolation and characterization of Pseudomonas syringae pv. porri from leek in Flanders. Eur J Plant Pathol. 2016;144(1):185–98.
Martino G, Holtappels D, Vallino M, Chiapello M, Turina M, Lavigne R, et al. Molecular characterization and taxonomic assignment of three phage isolates from a collection infecting Pseudomonas syringae pv. actinidiae and P. syringae pv. phaseolicola from Northern Italy. Viruses. 2021;13(10):2083.
Holtappels D, Kerremans A, Busschots Y, van Vaerenbergh J, Maes M, Lavigne R, et al. Preparing for the KIL: receptor analysis of Pseudomonas syringae pv. porri phages and their impact on bacterial virulence. Int J Mol Sci. 2020;21(8):2930.
Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 2017;13(6):e1005595.
Wick RR, Schultz MB, Zobel J, Holt KE. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics. 2015;31(20):3350–2.
Brettin T, Davis JJ, Disz T, Edwards RA, Gerdes S, Olsen GJ, et al. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep. 2015;5:8365.
Shevchenko A, Tomas H, Havliš J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 2007 1:6. 2007;1(6):2856–60.
Wagemans J, Tsonos J, Holtappels D, Fortuna K, Hernalsteens JP, de Greve H, et al. Structural analysis of jumbo coliphage phAPEC6. Int J Mol Sci. 2020;21(9):3119.
da Silva FP, Xavier A da S, Bruckner FP, de Rezende RR, Vidigal PMP, Alfenas-Zerbini P. Biological and molecular characterization of a bacteriophage infecting Xanthomonas campestris pv. campestris, isolated from brassica fields. Arch Virol. 2019;164(7):1857–62.
Doron S, Melamed S, Ofir G, Leavitt A, Lopatina A, Keren M, et al. Systematic discovery of antiphage defense systems in the microbial pangenome. Science. 2018;359(6379):eaar4120.
Acknowledgements
The authors thank Dr. J. Van Vaerenbergh and Dr. S. Rombouts for their support in this research.
Funding
This research was supported by the European Union’s Horizon H2020 Research and Innovation Program (grant agreement N.773567) and by the ‘Vlaams Agentschap Innoveren en Ondernemen’(VLAIO) agriculture programme (LA) grant IWT.150914. DH held a predoctoral scholarship from the ‘Fonds voor Wetenschappelijk Onderzoek Vlaanderen’ (FWO) (strategic basic research grant 1S02520N).
Author information
Authors and Affiliations
Contributions
D.H., R.L. and J.W. designed the research plan; D.H., K.F and M.V. performed the experiments; D.H. wrote the first draft of this manuscript; all authors reviewed and corrected the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent to publish
Not applicable.
Competing interests
Not applicable.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
Cite this article
Holtappels, D., Fortuna, K.J., Vallino, M. et al. Isolation, characterization and genome analysis of an orphan phage FoX4 of the new Foxquatrovirus genus. BMC Microbiol 22, 304 (2022). https://doi.org/10.1186/s12866-022-02719-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-022-02719-3