
Abstract
Background
The natural hosts of Shigella are typically humans and other primates, but it has been shown that the host range of Shigella has expanded to many animals. Although Shigella is becoming a major threat to animals, there is limited information on the genetic background of local strains. The purpose of this study was to assess the presence of virulence factors and the molecular characteristics of S. flexneri isolated from calves with diarrhea.
Results
Fifty-four S. flexneri isolates from Gansun, Shanxi, Qinghai, Xinjiang and Tibet obtained during 2014 to 2016 possessed four typical biochemical characteristics of Shigella. The prevalences of ipaH, virA, ipaBCD, ial, sen, set1A, set1B and stx were 100 %, 100 %, 77.78 %, 79.63 %, 48.15 %, 48.15 and 0 %, respectively. Multilocus variable number tandem repeat analysis (MLVA) based on 8 variable number of tandem repeat (VNTR) loci discriminated the isolates into 39 different MLVA types (MTs), pulsed field gel electrophoresis (PFGE) based on NotI digestion divided the 54 isolates into 31 PFGE types (PTs), and multilocus sequence typing (MLST) based on 15 housekeeping genes differentiated the isolates into 7 MLST sequence types (STs).
Conclusions
The findings from this study enrich our knowledge of the molecular characteristics of S. flexneri collected from calves with diarrhea, which will be important for addressing clinical and epidemiological issues regarding shigellosis.
Similar content being viewed by others


Background
Shigellosis or blood dysentery is widespread in underdeveloped or developing regions with poor hygiene and limited access to clean drinking water and has become a serious threat to public health [1, 2]. Shigellosis is caused by nonmotile, facultative anaerobic gram-negative bacilli of the Enterobacteriaceae family, including S. dysenteriae, S. flexneri, S. boydii, and S. sonnei [3,4,5]. Shigella species have high effectiveness in invasive systems that enable bacteria to invade and multiply within the human intestinal epithelia, ultimately leading to severe inflammatory colitis, which is referred to as bacillary dysentery or shigellosis [4].
Various virulence factors located on chromosomes or large virulence inv plasmids are recognized as crucial factors related to the pathogenesis of shigellosis [6]. Moreover, these different virulence factors are associated with the colonization of intestinal cells and intracellular invasion, which may partly explain why various manifestations are detected in the clinic, such as intestinal inflammatory responses and watery diarrhea [1]. Bacterial cell-to-cell movement and dissemination within epithelial cells of the intestine are allowed by the iphH gene, which is encoded by chromosomal DNA and/or recombinant plasmids, while ial, which is encoded by plasmids (invasion-associated loci), enables Shigella bacteria to penetrate intestinal epithelial tissues [7, 8]. The chromosomal genes set1A and set1B encode Shigella enterotoxin 1 (ShET-1) [9, 10], which is easily detected in all S. flexneri 2a isolates. Shigella enterotoxin 2, which is encoded by the gene sen, is located on a large plasmid associated with the virulence of Shigella and is found in most Shigella of different serotypes and in enteroinvasive Escherichia coli (EIEC) [11, 12]. In addition to their enterotoxic activity, ShET-1 and ShET-2 play significant roles in the transport of electrolytes and water in the intestine [12]. VirA located on large virulence plasmids has a great impact on intercellular spreading and invasion [13]. On the other hand, the type III secretion system (T3SS) is regarded as an important component for bacterial entry and is also composed of several proteins, including a needle-shaped oligomer anchored in the protein complex that connects the inner and outer bacterial membranes. The tip of the needle is an oligomer composed of the invasion plasmid antigens ipaB, ipaC, and ipaD [14,15,16]. Furthermore, the upstream ipaB region is often used as a marker to detect the ipaBCD gene.
The natural hosts of Shigella are typically humans and other primates [4], but monkeys, rabbits, calves, fish, chickens and piglets were recently reported to be infected with Shigella and are thus considered new hosts [4, 17,18,19,20,21]. In recent years, S. dysenteriae, S. flexneri, and S. sonnei have been isolated from cows. Although Shigella is becoming a major threat to animals, there is limited information on the genetic background of the isolated strains. Therefore, to identify molecular genotypes and determine the genetic relatedness diversity of local S. flexneri strains, we performed analyses using the multilocus sequence typing (MLST), multilocus variable number tandem repeat analysis (MLVA) and pulsed field gel electrophoresis (PFGE) methods.
Results
Biochemical characterization
A total of 54 S. flexneri from six serotypes, 1a (n = 5), 2a (n = 26), 2b (n = 4), 4a (n = 6), 6 (n = 8), and Xv (n = 5), were analyzed in this study. Based on the results of the biochemical reaction assays, we observed that all 54 S. flexneri isolates possessed 4 typical Shigella biochemical characteristics (Table 1). Among these BTs (biochemical types), BT4 (the ability to ferment glucose, mannitol, arabinose, and melibiose) was the predominant biotype, accounting for 70.37 % (38/54) of all BTs. Furthermore, BT4 was widely found in each serotype, except serotype 6. S. flexneri 2a was distributed among all four biochemical phenotypes and was mainly found in BT4 (22/26, 84.62 %). However, the other five serotype strains had only one or two biochemical phenotypes.
Virulence factors
The frequencies of the virulence factor profiles of the S. flexneri isolates are listed in Fig. 1. A total of seven virulence factors, including ipaH (100 %), virA (100 %), ipaBCD (92.59 %), ial (77.78 %), sen (79.63), set1A (48.15 %) and set1B (48.15 %), were detected in those isolates. None of the studied strains possessed the stx gene. The Shigella enterotoxin genes set1A and set1B were present only in S. flexneri 2a, and all of these serotype isolates were positive for these two genes.

Presence of virulence factors in S. flexneri isolates. Red = present; blue = absent. VT: virulence gene profile type
Regarding the differences in the distributions of the virulence factors, the 54 S. flexneri isolates fell into seven virulence gene profile types (VTs) (Table 2). Among these VTs, VT4 (positive for ipaH, virA, ipaBCD, ial, and sen) and VT6 (positive for ipaH, virA, ipaBCD, ial, sen, set1A, and set1B) were the most common, accounting for 29.63 and 44.44 % of all VTs, respectively. Furthermore, 92.59 % of the isolates carried two or more virulence factors. In addition, the virulence factor types were associated with the S. flexneri serotype. VT1 was found only in 4a, and VT4 was present in isolates from each serotype, except 2a. S. flexneri 2a major belonged to VT6 (24/26, 92.31 %).
MLST-based genotype analysis
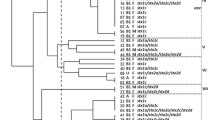
MLST was performed to analyze the genotypic diversity of S. flexneri isolates based on 15 housekeeping genes. The 54 isolates were divided into seven STs: ST68, ST100, ST103, ST120, ST124, ST135 and ST227. Among them, ST227 was novel, while six other STs have previously been reported. These seven STs belonged to several clonal complexes (CCs): CC10 (ST100 and ST103), CC26 (ST68), and others (ST120, ST124, ST135 and ST227). The clustering tree (Fig. 2) based on the MLST data showed that ST68 was a singleton type and that the other six STs contained two or more isolates. The most common ST was ST100 (n = 33, 61.11 %), which included isolates of serotypes 1a, 2a, and Xv. All the isolates of ST124 and ST227 belonged to S. flexneri 6 and 4a, respectively. The cluster tree indicated that isolates belonging to the same serotype were closely clustered based on the province of isolation. In addition, according to the minimum spanning tree (MST) based on the allele, ST100, ST120 and ST135 had closer relationships and differed only in aspC (aspartate aminotransferase), whereas ST68, ST124 and ST227 were very different from the other STs (Fig. 3).

MLST clustering tree of S. flexneri isolates isolated from calves with diarrhea during 2014 to 2016. The 54 isolates were analyzed by 15-allele MLST as described in the Materials and Methods. The scale bar in the top corner of the figure represents the similarity of each strain

Minimum spanning tree of the 54 S. flexneri isolates from calves with diarrhea based on multilocus sequence typing (MLST). The minimum spanning tree was constructed using the 7 identified STs obtained from the 54 isolates using BioNumerics software. Each circle corresponds to a single ST. The shaded zones in different colors correspond to different serotypes. The size of the circle is proportional to the number of isolates, and the color within the circles represents the serotype of the isolates. The corresponding color, serotype, number of isolates and background information are shown to the right of the minimum spanning tree
MLVA-based genotype analysis
MLVA based on eight VNTR loci was performed to further characterize the isolated S. flexneri strains. The copy numbers of the eight VNTR loci are listed in Fig. 4. Overall, the 54 isolates based on their unique MLVA profiles were discriminated into 39 different MLVA types (MTs). Among them, twenty-eight MTs belonged to the singleton type, and the other ten MTs contained no more than three isolates. The MLVA cluster tree of the isolates showed that they were divided into five clusters, designated A to E, with a low coefficient of similarity from 20 to 60 % (Fig. 4). Each cluster was further divided into many subclusters. MLVA can cluster different serotype strains separately and distinguish between the same serotype strains. The main cluster, cluster C, was observed to cluster S. flexneri 2a isolates and further divided into 15 MTs. Additionally, clusters A (except GBSF1502176), D and E clustered only the Xv, 2b, and 6 serotype strains, respectively. The results showed differences based on geographical origin and time span in the same serotype.

Relationship of S. flexneri isolates isolated from calves with diarrhea based on MLVA. Isolates were analyzed using an eight-VNTR locus MLVA scheme. A dendrogram was constructed using UPGMA. The corresponding MLVA types with the copy numbers of the eight VNTRs, serotype, and background information are shown to the right of the dendrogram. The letters A-E represent 5 clusters
PFGE-based genotype analysis
The genotypes and genetic relatedness diversity of 54 S. flexneri isolates were assessed by PFGE. NotI-digested S. flexneri chromosomal DNA generated 31 reproducible unique PFGE patterns (PTs), each with 11–16 bands (Fig. 5). Eleven patterns were represented by more than one isolate, with PT20 (n = 8) containing the most isolates, followed by PT18 (n = 5). The dendrogram of S. flexneri isolates showed low similarity (40-60 %) and could be classified into three gross clusters on the basis of their serotypes: clusters A, B and C. Isolates belonging to the same serotype but recovered in different years showed clear relatedness, as indicated by their grouping in the same clusters. The majority of serotype 2a isolates, with the exception of isolate QYSF1511395, grouped together in cluster B. The QYSF1511395 strain isolated from Qinghai Province clustered independently in cluster C. Isolates 1a, 2b and Xv clustered into cluster B and were closely related to the serotype 2a isolates. However, the isolates of serotypes 4a and 6 were assigned to cluster A with a relatively close relationship, but different serotype strains clustered separately.

Dendrogram of 54 NotI-digested S. flexneri isolates based on cluster analysis of PFGE patterns. A dendrogram was constructed using the UPGMA clustering method. The corresponding antibiotic resistance profile, PFGE pattern and background information for each strain are listed on the right side of the dendrogram. PT: PFGE type
Discussion
Shigella is an important invasive enteric infectious pathogen known for its sporadic, epidemic and pandemic spread [3],remains still a landmark cause of inflammatory diarrhea and dysentery thus poses a serious challenge to public health and is particularly tracked in most middle-income countries and regions with substandard hygiene and poor-quality water supplies [22]. All four types of Shigella can cause shigellosis, but S. flexneri is the most common bacterial preparation used for shigellosis [23]. The traditional hosts of this pathogen are limited to primates; however, the range of hosts has been extended to many animals in recent decades [4]. The symptoms of shigellosis in humans include diarrhea (100 %), headache (100 %), fever (100 %), nausea (99 %), abdominal cramping (97 %), vomiting (95 %), and bloody stools (51 %); however, the symptoms in animals are unclear [24]. A better understanding of the hosts of Shigella is needed to assess their potential effects on animal health; otherwise, preventing Shigella from causing disease is a challenge.
The pathogenesis of Shigella contributes to the organism’s ability to invade, replicate and spread intercellularly within the colonic epithelium. Pathogenic factors cause pathogenic Shigella to invade intestinal epithelial cells, leading to dysentery and other intestinal clinical symptoms in the host [25], and its pathogenesis is often multifactorial and coordinated [26]. Virulence factors have become important indicators of pathogenic bacteria.
Based on the detection of virulence factors, the Shigella isolates used in the present study had vast genetic diversity. Our results showed that ipaH and virA were found in each strain. Arabshahi et al. similarly showed that ipaH was present in all Shigella isolates and that virA was harbored by 88.9 % (8/9) of S. flexneri isolates [27], which agrees with a previous study that demonstrated that ipaH is carried by all four Shigella species as well as by enteroinvasive E. coli (EIEC). Multiple copies (ipaH1.4, ipaH2.5, ipaH4.5, ipaH7.8 and ipaH9.8) on large plasmids and chromosomes may explain why the ipaH gene tested positive in all isolates. Therefore, as a diagnostic tool for detecting Shigella, the ipaH gene is often an appealing target, even in the absence of a plasmid [28]. VirA was initially thought to invade Shigella; however, a structural analysis showed that VirA lacks papain-like protease activity to promote tubulin division. VirA belongs to the GTPase-activating protein family, which is involved in the cleavage of a single membrane into vacuoles. Previous studies have shown that VirA is often present in Shigella and is an important terminal point for bacteria to invade host cells and nucleate actin at one end of bacteria [9, 29].
Expert opinions have suggested that the T3SS is essential for host cell invasion and intracellular survival among virulence factors, whereas IpaB, IpaC, and IpaD are key factors of virulent Shigella [9, 30, 31]. Unlike the ipaH gene, the ial gene is not common. The ipaH gene is located only on the inv plasmid, and compared with the chromosome gene, the stability of the IPAH plasmid for storage/subculturing is poor [6,7,8]. Our results show that the ial gene has high invasiveness in the isolates studied. Therefore, it should be noted that the ial gene is involved in the invasion of intestinal cells and that the higher positivity rate of this gene in S. flexneri might indicate stronger aggressiveness.
The Shigella enterotoxins ShET-1 and ShET-2, which alter electrolyte and water transport in the small intestine, can cause diarrhea and dehydration [22]. ShET-1 is located on chromosomes encoding set1 (A and B subunit) genes, is almost exclusively found in several S. flexneri serotype 2 isolates and is rarely found in other serotypes [32]. Consistent with previous studies, our study showed that set1A and set1B were detected only in the S. flexneri 2a strain. The plasmid encoding ShET-2 (encoded by sen) is an enterotoxin hemolysin that causes an inflammatory response during Shigella invasion [12, 22]. It has been reported that there is a close relationship among sen, set enterotoxins and bloody diarrhea [22], which implies that sen and set enterotoxins are pathogenic factors of bloody diarrhea. However, unlike ShET-1, ShET-2 could be harbored by other species of Shigella.
The molecular characterization of strains is significant for epidemiological studies. However, few reports are available to systematically understand the molecular characteristics of S. flexneri isolated from animals. Several useful genotyping tools with higher discriminatory power than traditional tests, including MLST [33], PFGE [34] and MLVA [35], have recently been applied to explore and analyze the characteristics of Shigella isolates. Phylogenetic analysis, as an important method for supporting strain isolation, is based on differences in strain genetics.
MLST is an important source of sequence data for relative genetics and thus provides a tool for exploring molecular evolutionary methods among bacteria [36]. With the key elements of 15 housekeeping genes and analysis of the EcMLST database, the advantage of MLST is the comparison of data from different laboratories. Our results suggested that the predominant ST was ST 100, which has previously been found in human S. flexneri isolates [37, 38]. Specifically, isolates belonging to the same serotype often showed one ST type, indicating the low discriminative ability of closely related strains within a specific serotype due to the high sequence conservation of housekeeping genes.
Compared with MLST profiles, MLVA and PFGE may be forceful tools that can provide a satisfactory level of discrimination. However, the function of MLVA in the phylogenetic analysis of different bacterial species or serotypes is poorly targeted [39]. Nevertheless, MLVA is an ordinarily used typing tool that has been used to establish genetic relatedness and perform phylogenetic analysis among strains of monomorphic species. In our study, with approximately 20 % similarity, the 54 S. flexneri isolates were divided into 39 different MTs and clustered into 5 groups. Previous studies have also shown the high resolving power of MLVA in closely related strains [40,41,42]. Though applied in a limited collection of S. flexneri isolates, this study indicates the high discriminatory power of the MLVA method for subtyping strains with the same serotype.
With its strong function and widespread use, PFGE is also an applicable typing tool available in the laboratory for discriminating several enteric bacteria, such as Shigella. PFGE has a high degree of intra- and interlaboratory reproducibility when standardized protocols are followed [43]. Thirty-one low homophyly and unique PFGE patterns confirmed the existence of diverse S. flexneri clones and the usefulness of PFGE in local epidemiological studies.
Conclusions
This study demonstrated that spontaneously prevalent S. flexneri in cows shelter the same virulence factors as the prevalent isolates in humans. Therefore, these isolates are a potential threat to public safety. To systematically understand S. flexneri, the PFGE, MLVA and MLST methods were applied to characterize the 54 isolates hereditary. MLVA based on 8 VNTR loci discriminated the 54 isolates into 39 different MTs, PFGE based on NotI digestion ambiguously differentiated the 54 isolates into 31 PTs, MLST based on 15 housekeeping genes differentiated the 54 isolates into 7 STs, and 1 ST (ST227) was novel. Although MLST provided suitable discrimination in S. flexneri subtyping, PFGE and MLVA might both exhibit a higher discriminatory ability. Overall, the data from this study will provide a useful typing resource, which will provide a scientific basis for addressing clinical and epidemiological issues regarding S. flexneri.
Methods
Bacterial isolates and bacteriological examination
Animal-based active surveillance was conducted in 3321 calves with diarrhea from five provinces (Gansun, Shanxi, Qinghai, Xinjiang and Tibet) in northwestern China from 2014 to 2016. All of the isolates were collected directly from fresh stool samples following plating on Salmonella-Shigella (SS) selective agar and confirmation on MacConkey (MAC) agar at 37°C for 24 h. Colorless, semitransparent, smooth, and moist circular plaques were considered presumptive Shigella for biochemical confirmation. Biochemical tests were performed on S. flexneri using API20E test strips (bioMerieux Vitek, Marcy-l’ Etoile, France), and the serotype was tested by a commercially available agglutinating antibody kit (Denka Seiken, Tokyo, Japan) according to the manufacturers’ recommendations. Information on the S. flexneri isolates in this study is listed in Figs. 2 , 4 and 5.
Preparation of DNA templates
The DNA templates for PCR (virulence factors, MLST, MLVA) were directly extracted from bacterial colonies using the boiled lysate method as previously reported [44].
Detection of virulence factors
All 54 strains were tested by PCR for the presence of 8 virulence-associated genes, namely, ipaH, ipaBCD, virA, ial, stx, set1A, set1B, and sen, according to published procedures [15, 45, 46]. PCRs were performed according to published protocols, and the primer sequences are listed in Table S1.
Multilocus sequence typing
All isolates were subjected to MLST according to the protocols described in the EcMLST database (http://www.shigatox.net/ecmlst). The PCR products were bidirectionally sequenced, and the sequences of the 15 housekeeping genes were edited by using SeqMan 7.0. Each unique allele was assigned a different number, and the allelic profile (string of fifteen allelic loci) was used to define the ST of each isolate [47]. Clustering and minimum spanning tree (MST) analyses were used to infer relationships among the isolates using the fingerprint analysis software BioNumerics (version 7.1).
Multilocus variable number tandem repeat analysis
MLVA of 8 VNTR loci (SF3, SF4, SF6, SF7, SF8, SF9, SF10 and SF25) was performed using a previously described method [35]. The forward primer for each primer set was labeled at its 5’ end with the ABI-compatible dyes HEX, 6’-FAM, TAMRA, and ROX (Table S2). In these cases, the loci were individually amplified, with each 20 µL PCR mixture containing 1 µL of each primer, 1 µL of DNA template, 10 µL of Taq MasterMix (Takara, Japan) and deionized water to a final volume of 20 µL. PCR was performed with a denaturing step at 94 °C for 5 min, followed by 30 cycles of amplification at 94 °C for 30 s, 55 °C for 45 s, and 72 °C for 45 s and a final extension at 72 °C for 5 min at the final step.
The PCR products were analyzed by capillary electrophoresis on an ABI Prism 3730 XL Genetic Analyzer with the GeneScan 500 LIZ Size Standard as previously described [48]. The number of repeat units for each allele was calculated from the length of the amplicon. The copy number of each VNTR locus was subjected to cluster analysis using the MST algorithm and the categorical coefficient provided in BioNumerics software. Each unique allelic string was designated a unique MLVA type. A dendrogram was constructed by UPGMA clustering based on categorical coefficient analysis [35, 49].
Pulsed-field gel electrophoresis
DNA fingerprinting was performed by PFGE with the restriction enzyme NotI (TaKaRa; Japan) according to the international standards set by the CDC. PFGE images were photographed with a Universal Hood II (Bio-Rad; USA) and analyzed with BioNumerics using the Dice similarity coefficient, unweighted pair-group method with the arithmetic mean (UPGMA) and 1.0 % band position tolerance. A PFGE type (PT) was defined as a pattern with one or more DNA bands different from other patterns.
Availability of data and materials
The data supporting the findings of this study are contained within the manuscript.
Abbreviations
- VTs:
-
Virulence gene profile types
- BTs:
-
Biochemical types
- MLVA:
-
Multiple-locus variable number tandem repeat analysis
- VNTR:
-
Variable number of tandem repeats
- PFGE:
-
Pulsed-field gel electrophoresis
- MLST:
-
Multilocus sequence typing
References
Dutta S, Jain P, Nandy S, Matsushita S, Yoshida S. Molecular characterization of serologically atypical provisional serovars of Shigella isolates from Kolkata, India. J Med Microbiol. 2014;63(Pt 12):1696–703.
Soltan Dallal MM, Ranjbar R, Pourshafie MR. The study of antimicrobial resistance among Shigella flexneri strains isolated in Tehran, Iran. J Pediatr Infect Dis. 2011;6:125–9.
Ranjbar R, Bolandian M, Behzadi P. Virulotyping of Shigella spp. isolated from pediatric patients in Tehran, Iran. Acta Microbiol Immunol Hung. 2017;64(1):71–80.
Shi R, Yang X, Chen L, Chang HT, Liu HY, Zhao J, et al. Pathogenicity of Shigella in chickens. PLoS One. 2014;9(6):e100264.
Ranjbar R, Behnood V, Memariani H, Najafi A, Moghbeli M, Mammina C. Molecular characterisation of quinolone-resistant Shigella strains isolated in Tehran, Iran. J Glob Antimicrob Resist. 2016;5:26–30.
Shen Y, Qian H, Gong J, Deng F, Dong C, Zhou L, et al. High prevalence of antibiotic resistance and molecular characterization of integrons among Shigella isolates in Eastern China. Antimicrob Agents Chemother. 2013;57(3):1549–51.
Phantouamath B, Sithivong N, Insisiengmay S, Ichinose Y, Higa N, Song T, et al. Pathogenicity of Shigella in healthy carriers: a study in Vientiane, Lao People’s Democratic Republic. Jpn J Infect Dis. 2005;58(4):232–4.
Ashida H, Sasakawa C. Shigella IpaH Family Effectors as a Versatile Model for Studying Pathogenic Bacteria. Front Cell Infect Microbiol. 2016;5:100.
Schroeder GN, Hilbi H. Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion. Clin Microbiol Rev. 2008;21(1):134–56.
Sousa MÂ, Mendes EN, Collares GB, Péret-Filho LA, Penna FJ, Magalhães PP. Shigella in Brazilian children with acute diarrhoea: prevalence, antimicrobial resistance and virulence factors. Mem Inst Oswaldo Cruz. 2013;108(1):30–5.
Farfán MJ, Toro CS, Barry EM, Nataro JP. Shigella enterotoxin-2 is a type III effector that participates in Shigella-induced interleukin 8 secretion by epithelial cells. FEMS Immunol Med Microbiol. 2011;61(3):332–9.
da Cruz CB, de Souza MC, Serra PT, Santos I, Balieiro A, Pieri FA, et al. Virulence factors associated with pediatric shigellosis in Brazilian Amazon. Biomed Res Int. 2014;2014:539697.
Zaidi MB, Estrada-García T. Shigella: a highly virulent and elusive pathogen. Curr Trop Med Rep. 2014;1:81–7.
Büttner D, Bonas U. Who comes first? How plant pathogenic bacteria orchestrate type III secretion. Curr Opin Microbiol. 2006;9(2):193–200.
Faruque SM, Khan R, Kamruzzaman M, Yamasaki S, Ahmad QS, Azim T, et al. Isolation of Shigella dysenteriae type 1 and S. flexneri strains from surface waters in Bangladesh: comparative molecular analysis of environmental Shigella isolates versus clinical strains. Appl Environ Microbiol. 2002;68(8):3908–13.
Marteyn B, Gazi A, Sansonetti P. Shigella: a model of virulence regulation in vivo. Gut Microbes. 2012;3(2):104–20.
Onyango DM, Wandili S, Kakai R, Waindi EN. Isolation of Salmonella and Shigella from fish harvested from the Winam Gulf of Lake Victoria, Kenya. J Infect Dev Ctries. 2009;3(2):99–104.
Priamukhina NS, Kilesso VA, Tikhomirov ED. Animal carriers of Shigella and their possible epidemiological importance. Zh Mikrobiol Epidemiol Immunobiol. 1984;11:20–4.
Maurelli AT, Routh PR, Dillman RC, Ficken MD, Weinstock DM, Almond GW, et al. Shigella infection as observed in the experimentally inoculated domestic pig, Sus scrofa domestica. Microb Pathog. 1998;25(4):189–96.
Zhu Z, Cao M, Zhou X, Li B, Zhang J. Epidemic characterization and molecular genotyping of Shigella flexneri isolated from calves with diarrhea in Northwest China. Antimicrob Resist Infect Control. 2017;6:92.
Zhu Z, Shi Y, Zhou X, Li B, Zhang J. Molecular characterization of fluoroquinolone and/or cephalosporin resistance in Shigella sonnei isolates from yaks. BMC Vet Res. 2018;14(1):177.
Gu B, Fan W, Qin T, Kong X, Dong C, Tan Z, et al. Existence of virulence factors in clinical Shigella sonnei isolates from Jiangsu Province of China: a multicenter study. Ann Transl Med. 2019;7(14):305.
Sangeetha AV, Parija SC, Mandal I, Krishnamurthy S. Clinical and microbiological profiles of shigellosis in children. J Health Popul Nutr. 2014;32(4):580–6.
Ranjbar R, Hosseini MJ, Kaffashian AR, et al. An outbreak of shigellosis due to Shigella flexneri serotype 3a in a prison in Iran. Arch Iran Med. 2010;13(5):413–6.
Ashida H, Ogawa M, Mimuro H, Kobayashi T, Sanada T, Sasakawa C. Shigella are versatile mucosal pathogens that circumvent the host innate immune system. Curr Opin Immunol. 2011;23(4):448–55.
Qu M, Zhang X, Liu G, Huang Y, Jia L, Liang W, et al. An eight-year study of Shigella species in Beijing, China: serodiversity, virulence factors, and antimicrobial resistance. J Infect Dev Ctries. 2014;8(7):904–8.
Arabshahi S, Novinrooz A, Ranjbar R, et al. Molecular characterization of Shigella species isolated from diarrheal patients in Tehran, Iran: phylogenetic typing and its association with virulence gene profiles and a novel description of Shigella invasion associated locus. Eur J Clin Microbiol Infect Dis. 2020;39(9):1727–37.
Vu DT, Sethabutr O, Von Seidlein L, Tran VT, Do GC, Bui TC, et al. Detection of Shigella by a PCR assay targeting the ipaH gene suggests increased prevalence of shigellosis in Nha Trang, Vietnam. J Clin Microbiol. 2004;42(5):2031–5.
Yaghoubi S, Ranjbar R, Dallal MMS, Fard SY, Shirazi MH, Mahmoudi M. Profiling of virulence-associated factors in Shigella species isolated from acute pediatric diarrheal samples in Tehran, Iran. Osong Public Health Res Perspect. 2017;8(3):220–6.
Parsot C. Shigella type III secretion effectors: how, where, when, for what purposes? Curr Opin Microbiol. 2009;12(1):110–6.
Mattock E, Blocker AJ. How do the virulence factors of Shigella work together to cause disease? Front Cell Infect Microbiol. 2017;7:64.
Roy S, Thanasekaran K, Dutta Roy AR, Sehgal SC. Distribution of Shigella enterotoxin genes and secreted autotransporter toxin gene among diverse species and serotypes of shigella isolated from Andaman Islands, India. Trop Med Int Health. 2006;11(11):1694–8.
Cao Y, Wei D, Kamara IL, Chen W. Multi-Locus Sequence Typing (MLST) and Repetitive Extragenic Palindromic Polymerase Chain Reaction (REP-PCR), characterization of shigella spp. over two decades in Tianjin China. Int J Mol Epidemiol Genet. 2012;3(4):321–32.
Xia S, Xu B, Huang L, Zhao JY, Ran L, Zhang J, et al. Prevalence and characterization of human Shigella infections in Henan Province, China, in 2006. J Clin Microbiol. 2011;49(1):232–42.
Wang YW, Watanabe H, Phung DC, Tung SK, Lee YS, Terajima J, et al. Multilocus variable-number tandem repeat analysis for molecular typing and phylogenetic analysis of Shigella flexneri. BMC Microbiol. 2009;9:278.
Hu Y, Xi Z, Liu X, Wang J, Guo Y, Ren D, et al. Identification and molecular characterization of Wolbachia strains in natural populations of Aedes albopictus in China. Parasit Vectors. 2020;13(1):28.
Li S, Sun Q, Wei X, Klena JD, Wang J, Liu Y, et al. Genetic characterization of Shigella flexneri isolates in Guizhou Province, China. PLoS One. 2015;10(1):e0116708.
Cui X, Wang J, Yang C, Liang B, Ma Q, Yi S, et al. Prevalence and antimicrobial resistance of Shigella flexneri serotype 2 variant in China. Front Microbiol. 2015;6:435.
Cheng T, Shi X, Yong W, Wang J, Xie G, Ding J. Molecular typing of Shigella sonnei isolates circulating in Nanjing, China, 2007–2011. J Infect Dev Ctries. 2014;8(12):1525–32.
Filliol-Toutain I, Chiou CS, Mammina C, et al. Global distribution of Shigella sonnei clones. Emerg Infect Dis. 2011;17(10):1910–2.
Ranjbar R, Memariani M, Memariani H. Diversity of variable number tandem repeat Loci in Shigella species isolated from pediatric patients. Int J Mol Cell Med. 2015;4(3):174–81.
Ranjbar R, Memariani M. Multilocus variable-number tandem-repeat analysis for genotyping of Shigella sonnei strains isolated from pediatric patients. Gastroenterol Hepatol Bed Bench. 2015;8(3):225–32.
Ribot EM, Fair MA, Gautom R, Cameron DN, Hunter SB, Swaminathan B, et al. Standardization of pulsed-field gel electrophoresis protocols for the subtyping of Escherichia coli O157:H7, Salmonella, and Shigella for PulseNet. Foodborne Pathog Dis. 2006;3(1):59–67.
Ahmed AM, Shimamoto T, Shimamoto T, et al. Molecular characterization of multidrug-resistant avian pathogenic Escherichia coli isolated from septicemic broilers. Int J Med Microbiol. 2013;303:475–83.
Das A, Natarajan M, Mandal J. The emergence of quinolone resistant Shigella sonnei, Pondicherry, India. PLoS One. 2016;11(8):e0160290.
Vargas M, Gascon J, Jimenez De Anta MT, Vila J. Prevalence of Shigella enterotoxins 1 and 2 among Shigella strains isolated from patients with traveler’s diarrhea. J Clin Microbiol. 1999;37(11):3608–11.
Schaumburg F, Alabi AS, Kaba H, et al. Molecular characterization of Shigella spp. from patients in Gabon 2011–2013. Trans R Soc Trop Med Hyg. 2015;109(4):275–9.
Liang SY, Watanabe H, Terajima J, et al. Multilocus variable-number tandem repeat analysis for molecular typing of Shigella sonnei. J Clin Microbiol. 2007;45(11):3574–80.
Hyytia-Trees E, Smole SC, Fields PA, et al. Second generation subtyping: a proposed PulseNet protocol for multiple-locus variable-number tandem repeat analysis of Shiga toxin-producing Escherichia coli O157 (STEC O157). Foodborne Pathog Dis. 2006;3:118–131.
Acknowledgements
The authors thank all the State Key Laboratory for Infectious Disease Prevention and Control (China CDC) for providing funding and technical support.
Funding
The design of the study, including the collection, analysis, and interpretation of data, was supported by grants from the National Natural Science Foundation of China (No: 31872520) and Drug Development and Clinical Drug Use Posts of the National Beef Yak Industry Technical System (No: CARS-37). The writing and submission of the manuscript was supported by the Natural Science Foundation of Hebei Province (No: C2019402114) and a grant (2018SKLID308) from the State Key Laboratory for Infectious Disease Prevention and Control (China CDC).
Author information
Authors and Affiliations
Contributions
Z.Z. and J.Y.Z. designed the study; Z.Z., M.Z.C., T.H.M., Y.X.S., X.Z.Z., G.H. and B.L. generated and provided the dataset; Z.Z., Q.Q.Z., Y.Y.Z. and W.W.W. performed the experiments, analyzed the data, and wrote the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was carried out in compliance with the ARRIVE guidelines and obtained ethical approval from the Ethics Committee of Lanzhou Institute of Husbandry and Pharmaceutical Sciences of CAAS. Our study was conducted according to the Animal Administration and Ethics Committee of the Lanzhou Institute of Husbandry and Pharmaceutical Sciences of the CAAS (Permit No. SYXK-2014-021). We obtained informed consent from the owners of the animals for use in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Primers used for the detection of virulence genes.
Additional file 2: Table S2.
MLVA primers for S. flexneri.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhu, Z., Wang, W., Cao, M. et al. Virulence factors and molecular characteristics of Shigella flexneri isolated from calves with diarrhea. BMC Microbiol 21, 214 (2021). https://doi.org/10.1186/s12866-021-02277-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-021-02277-0