
Abstract
Background
Viruses cause significant economic losses to shrimp aquaculture worldwide. In severe cases, they can lead to 100% mortality within a matter of days, hence the aquaculture industry requires antiviral strategies to minimize economic impacts. Currently, a double-stranded RNA (dsRNA)-based platform has been proven effective at a laboratory scale. The bottleneck for its industrialization is the lack of low-cost, efficient and practical delivery approaches. In an effort to bridge the gap between laboratory and farm applications, virus-like particles (VLP) have been used as nanocarriers of dsRNA. However, the implementation of this approach still suffers from high costs and a lengthy procedure, co-expression of subunits of VLP or capsid proteins (CPs) and dsRNA can be the solution for the problem. CP and dsRNA are traditionally expressed in two different E. coli hosts: protease-deficient and RNase III-deficient strains. To condense the manufacturing of dsRNA-containing VLP, this study constructed a novel E. coli strain that is able to co-express viral capsid proteins and dsRNA in the same E. coli cell.
Results
A novel bacterial strain DualX-B15(DE3) was engineered to be both protease- and RNase III-deficiency via P1 phage transduction. The results revealed that it could simultaneously express recombinant proteins and dsRNA.
Conclusion
Co-expression of viral capsid proteins and dsRNA in the same cell has been shown to be feasible. Not only could this platform serve as a basis for future cost-effective and streamlined production of shrimp antiviral therapeutics, it may be applicable for other applications that requires co-expression of recombinant proteins and dsRNA.
Similar content being viewed by others


Background
Viral pathogens have devastated global shrimp aquaculture for decades. It was assessed that shrimp production losses owing to viral diseases were around USD 15 billion over the last decade [1]. Accordingly, practical antiviral strategies are in urgent demand. However, as invertebrates, shrimp lack an adaptive immune system, rendering vaccination ineffective. As a result, many antiviral approaches that have been studied rely on enhancing host innate immunity [2]; examples include oral administration of probiotics [3], peptidoglycans [4] of β-glucan [5] and plant or seaweed extracts [6]. Other studies revealed that shrimp was capable of deploying an antiviral immune response via the RNA interference (RNAi) pathway [7, 8], as evidenced by the finding that injection of double-stranded RNA (dsRNA) constructs with sequences that matched viral mRNA was effective in preventing viral diseases or reducing their severity [9].
RNA interference (RNAi) is an endogenous gene-silencing mechanism that can be repurposed to combat viral infection through sequence-specific recognition and degradation of viral RNA [10]. As one of the most infamous shrimp viruses, RNAi applications against White Spot Syndrome Virus (WSSV) have been extensively investigated [11, 12] and dsRNA targeting the VP28 mRNA, which encodes the virus envelope protein that has been implicated in host entry [13, 14], has become one of the most effective targets [15]. Despite the promising results at a laboratory level, the implementation of the RNAi technology at a farm level is still at its infantile stage due to the lack of effective delivery approaches.
Many dsRNA-packaging strategies have been explored such as chitosans, liposomes and virus-like particles (VLP), which are self-assembled viral capsid proteins (CP) that are devoid of infectious nucleic acids. Among these, the VLP delivery systems have proved promising as their structures and attributes resemble native viruses. The advantages of VLP include the absence of infectious viral genetic materials, lack of infectivity, and specificity to the host cells [16]. Two types of VLP have, thus far, been applied for shrimp aquaculture: infectious hypodermal and hematopoietic necrosis virus (IHHNV) VLP and Macrobrachium rosenbergii nodavirus (MrNV) VLP. These VLPs are non-enveloped and composed of a single type of CP, which allows them to simply be repurposed as a nanocarrier.
In aquaculture, VLP provide potential delivery options because they can be recombinantly expressed and prolong the half-life of their dsRNA cargo by protecting them from nucleases as demonstrated in two previous reports [7, 17,18,19]. In the first example, it was revealed that dsRNA encapsulated in IHHNV-VLP conferred higher protection against YHV in Penaeus vannamei relative to naked dsRNA [19]. In the second example, the finding showed that delivery of dsRNA-VP28 encapsulated in MrNV-VLP gave protection against WSSV challenge in P. monodon: the survival rate of shrimp around 70% was observed in the encapsulated dsRNA treatment group, compared with 53 and 0% in the groups injected with naked dsRNA and phosphate buffer saline (PBS), respectively [7]. In addition to increasing the survival rate of shrimp, the MrNV-VLP itself elicited an innate response in shrimp by upregulation of a number of key immune genes [8].
The gridlock in the production of dsRNA-encapsulating VLP is the lengthy manufacturing process which includes individual expressions of CP and dsRNA in two separate bacterial hosts, followed by a complicated in vitro assembly to insert dsRNA into VLP. To reduce the production cost of the VLP-based delivery method, one possible scenario could be co-expression of VLP and dsRNA in a single culture. Currently, the Escherichia coli BL21(DE3) and HT115(DE3) strains are commonly used to independently express recombinant proteins and dsRNA, respectively. BL21(DE3) is deficient in two major protease-encoding genes but has an active dsRNA-specific RNase III-encoding gene. In contrast, HT115(DE3) [20] lacks the RNase III-encoding gene but contains protease-encoding genes.
This study, therefore, aimed to combine the strengths of these two strains of E. coli into a single platform that can simultaneously express recombinant proteins and dsRNA. The P1 phage system was employed to disrupt the RNase III-encoding gene in the protease-deficient BL21(DE3). To our best knowledge, this is the first attempt to engineer E. coli to be both protease- and RNase III-deficient for co-expression of recombinant proteins and dsRNA.
Results
Engineering of E. coli strain by the P1 phage system
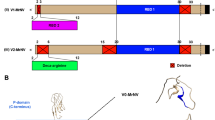
Many gene knockout techniques had been considered. At the end, however, P1 transduction was chosen due to its technical simplicity. The technique exploits the DNA packaging mechanism of P1 bacteriophage. Briefly, during growth and packaging of its own chromosome, P1 bacteriophage occasionally encapsidates DNA from a donor bacterial strain into its capsid, and as a result some of the new phage particles contain donor’s DNA [21] (Fig. 1a and b). Upon infecting a recipient bacterial strain with the P1 donor lysate, the host homologous recombination machinery recombines and integrates the donor-derived DNA pieces into the recipient’s chromosome [22] (Fig. 1d and e). Through this “hitchhiking” mechanism, any DNA sequences from the donor bacterial strain can be transferred to the recipient strain, provided that the cassette-flanking regions and the target site are homologous.

Construction of the RNase III- and protease-deficient DualX-B15(DE3) strain by P1 phage transduction. a Infection of the donor strain HT115(DE3) by P1 bacteriophage. b Random incorporation of the donor genome into viral capsids. c Release of new viral particles upon lysis of the donor cells. d Infection of the recipient strain BL21(DE3) with P1 lysate. e Homologous recombination at the rnc14 gene locus. f Generation of the novel E. coli strain DualX-B15(DE3) with a tetracycline resistant marker (TetR)
There are two possible strategies for generating a new bacterial strain that is both protease- and RNase III-deficient. First, the protease-deficient BL21(DE3) could be rendered RNase III-deficient by mutating the RNase III-encoding gene, rnc14, through a single P1 phage transduction step. Alternatively, at least two P1 phage transduction events would be necessary to eliminate lon and ompT gene activities from the RNase III-deficient HT115(DE3). Therefore, the first approach was utilized in this study due to its convenience.
To knockout the rnc14 gene from the recipient strain BL21(DE3), HT115(DE3) was used as a donor strain. The rnc14 gene of HT115(DE3) is disrupted by Transposon 10 (Tn10) which harbors a tetracycline resistant cassette (TetR). To generate P1 lysate from the HT115(DE3) donor strain, P1 bacteriophage was allowed to infect HT115(DE3), so that they can randomly capture fragments of the HT115 genome, including parts of the Tn10-containing rnc14 gene. Upon infecting the recipient BL21(DE3) with the P1 lysate, homologous recombination will occur between the recipient rnc14 gene and its phage-derived counterpart, concomitantly resulting in the introduction of the tetracycline resistant cassette into the recipient genome and the disruption of the rnc14 gene in the BL21(DE3) recipient genome (Fig. 1).
Selection of the novel E. coli strain
A successful gene knockout was detected by tetracycline resistance acquired by the recipient strain BL21(DE3) during transduction. BL21(DE3) treated with the P1 donor lysate was plated on LB agar plates with or without tetracycline. The growth of colonies on tetracycline-supplemented plates was used as an indicator for desirable recombination that resulted in disruption of the rnc14 gene. By comparing the numbers of colonies on the tetracycline-containing and control plates, the efficiency of selection was determined to be 0.1–1%.
To determine whether the rnc14 gene in the tetracycline-resistant colonies contained any mutation that resulted in loss of function of the rnc14 gene, chromosomal DNA of these colonies were extracted and sent for sequencing using primers that flanked the rnc14 gene. The sequencing result showed that there was an insertion after the 14th codon of the rnc14 gene. This insert coded for 12 amino acids followed by a premature stop codon which shortened the resulting protein from 226 amino acids to 26 amino acids (Fig. 2). The newly engineered strain was dubbed DualX-B15(DE3).

E. coli DualX-B15(DE3) was generated by incorporation of a premature stop codon into the rnc14 gene of the donor strain BL21(DE3). Alignments of the amino acid and nucleotide sequences of the rnc14 gene from the BL21(DE3) (Top) and DualX-B15(DE3) (Bottom) strains from the start codon (ATG). The asterisk at the end of the sequence of DualX-B15(DE3) indicates the premature stop codon (TAA). The nucleotide sequence inserted into the BL21(DE3) genome during P1 phage transduction is shown as blue underlined letters. The chromatogram from Sanger sequencing in the panel below showed the insertion mutation and the premature stop codon in the rnc14 gene
Effect of rnc14 knockout on bacterial growth
To investigate the effect of eliminating the rnc14 gene activity in the novel E. coli strain on bacterial growth, BL21(DE3), HT115(DE3) and DualX-B15(DE3) were transformed with a dsRNA-expression plasmid. The growth of transformants at 37 °C was monitored by measuring optical density (OD) at 600 nm. It was shown that the growth rate of BL21(DE3) was highest, whereas the engineered strain exhibited the slowest growth rate and did not reach the stationary phase by the end of the monitored period (160 min) (Fig. 3).

Knocking out the rnc14 slowed down the growth rate of the newly engineered strain. Growth curve of BL21(DE3), HT115(DE3) and DualX-B15(DE3) are represented in red, blue and green, respectively
Expression of dsRNA in DualX-B15(DE3)
To demonstrate that DualX-B15(DE3) could indeed express dsRNA, expression of dsRNA-GW182 was conducted as a control as this dsRNA had been successfully expressed in our laboratory [23]. After dsRNA expression, the same wet mass of each bacteria culture was used to extract RNA, which was then subjected to an RNase-digestion assay and agarose gel electrophoresis to verify the double-stranded nature of the extracted RNA.
As a positive control, the untreated sample from HT115(DE3) showed a discrete band at 454 bp (Fig. 4). When the same RNA sample was incubated with RNase A, which specifically cleaves single-stranded RNA, the band migrated faster as the loop region of the dsRNA hairpin was hydrolyzed by the enzyme. When the same RNA was treated with dsRNA-specific RNase III, no discrete RNA band was observable. Instead, a dark smear which is indicative of degraded nucleic acids was clearly evident at low molecular weight. Altogether, the three reactions verified that the RNA purified from HT115(DE3) was double-stranded in character. In contrast, no RNA from BL21(DE3) survived the enzymatic treatments, as only dark smears were found in the lanes from those samples. The analysis of RNA samples purified from DualX-B15(DE3) was identical to that of the dsRNA-producing HT115(DE3). Hence, it can be concluded that the newly engineered strain could indeed express dsRNA.

Double-stranded RNA could be expressed in DualX-B15(DE3). RNA analyzed on the gel included untreated samples (U), samples treated with RNase A (A) and samples treated with RNase III (III). Lane M (Marker) is a 2-log DNA ladder. The dsRNA were extracted from 50 mg wet weight of bacteria
To compare the dsRNA-GW182 yields, three bacteria cultures were grown, and RNA was purified from identical wet cell mass. It was revealed that the dsRNA yields for this particular dsRNA construct were 0.92 ± 0.07 μg/mg wet cell mass for HT115(DE3) and 1.63 ± 0.22 μg/mg wet cell mass for DualX-B15(DE3).
Expression of recombinant proteins in DualX-B15(DE3)
To determine whether the newly engineered E. coli strain can express proteins, Glutathione S-Transferase (GST) was used as a control because it can be readily overexpressed in many E. coli strains. To compare GST expression in three different strains of E. coli, the same wet weight of bacterial cells was used for protein analysis. The soluble 26-kDa GST protein was successfully overexpressed in BL21(DE3) and DualX-B15(DE3) as revealed by the presence of the overexpression band at the corresponding molecular weight (Fig. 5a). On the other hand, no equivalent band was detected in the sample from HT115(DE3).

Recombinant proteins could be expressed in DualX-B15(DE3). a SDS-PAGE analysis of GST expressed in HT115(DE3), BL21(DE3) and DualX-B15(DE3) strains. b Western blot of GST expressed in DualX-B15(DE3). Lane M: prestained protein marker; Lane N: cells without the protein-expression plasmid; Lane U: uninduced cells with the protein-expression plasmid; Lane I: induced cells; Lane INS: insoluble fraction; Lane SOL: soluble fraction. The molecular weight of GST is 26 kDa as indicated by a red asterisk
To further confirm that the overexpressed protein band in DualX-B15(DE3) was GST, Western blot was performed using an antibody against GST (Fig. 5b). A band at 26 kDa was observed in both the soluble and insoluble fractions, suggesting that the band in question was indeed GST. Therefore, the protein expression capacity of the parental BL21(DE3) strain was retained in DualX-B15(DE3), uncompromised by the knockout of the rnc14 gene.
To compare the yield of GST expressed from the three bacterial strains, the amount of GST on the SDS-PAGE was estimated by comparing to the intensities of the pre-stained protein markers to that of the GST bands. Both DualX-B15(DE3) and BL21(DE3) expressed approximately 14 μg GST/mg wet cell mass.
In the following co-expression study, bacterial cells without any expression plasmids were used to represent background expression instead of uninduced cells because a small amount of recombinant protein was detected prior to IPTG induction as shown in the Western blot (Fig. 5b).
Co-expression of dsRNA-VP28 and MrNV capsid protein (MrNV-CP)
Having shown that the new bacterial strain could express dsRNA and recombinant proteins individually, the next step was to co-express the two biological molecules in the same host. BL21(DE3), HT115(DE3) and DualX-B15(DE3) were co-transformed with dsRNA-VP28 and MrNV-CP expressing plasmids and co-expression of the recombinant products was induced with IPTG at 25 °C.
Analysis of the resulting RNA revealed that both HT115(DE3) and DualX-B15(DE3) could produce dsRNA-VP28 at the expected size of 615 bp (Fig. 6a). In parallel, SDS-PAGE and Western blot in the presence of an anti-MrNV antibody confirmed the expression of MrNV-CP at 40 kDa in BL21(DE3) and DualX-B15(DE3) (Fig. 6b and c). Overall, the experiment showed that only DualX-B15(DE3) was capable of concurrent expression of both dsRNA-VP28 and MrNV-CP.

dsRNA-VP28 and MrNV-CP could be co-expressed in DualX-B15(DE3). a The RNase digestion assays: untreated (U), RNase A (A) and RNase III (III)-treated samples. Lane M (Marker) is a 2-log DNA ladder. The molecular weight of dsRNA-VP28 is indicated by an arrow. b SDS-PAGE analysis of protein expression in the three different strains of E. coli. A red dot is shown to the right of the expected band of MrNV-CP. c Western blot was performed with anti-MrNV capsid protein monoclonal antibody. Lane M: a pre-stained protein marker; Lane N: cells without the protein-expression plasmid; Lane I: induced cells; Lane P: a positive control (purified MrNV capsid proteins); Lane INS: insoluble fraction; Lane SOL: soluble fraction. The molecular weight of MrNV capsid protein is 40 kDa as indicated by a red asterisk.
Discussion
Previous reports have documented beneficial effects of VLP-encapsulated dsRNA over their unpackaged counterparts in controlling viral infection in shrimp [7, 8, 17, 19]. To increase the practicality of this platform, this study has successfully streamlined the co-expression of CP and dsRNA in the same cell by utilizing the P1 phage system to construct DualX-B15(DE3). The transduction process exhibited 0.1–1% efficiency which is considered typical according to a published report [24].
In this study, the sequence downstream from the premature stop codon that disrupted the rnc14 gene in DualX-B15(DE3) was not further investigated once it was found that the newly engineered strain was able to express dsRNA while retaining its original protein expression capacity. In other words, the lack of the sequencing result did not alter the conclusion that the P1 transduction method knocked out the rnc14 gene without altering the original protein expression genotype.
The fact that DualX-B15(DE3) could express both GST and MrNV-CP indicated that the protein expression capacity of the novel strain is not sequence-specific. While the amount of GST expression in DualX-B15(DE3) and BL21(DE3) appeared to be similar, further experiments are necessary to draw any conclusion about the quantitative nature of protein expression in these two strains.
When expressed in separate hosts, MrNV-VLP have to be disassembled into their capsid proteins (CP) building blocks in vitro in the presence of a chelating agent in order to introduce dsRNA and, then, reassembled upon the removal of the chelator [17]. We hypothesize that by synchronous co-expression of CP and dsRNA, dsRNA-containing VLP can be spontaneously formed based on a previous Cryo-EM report [25] which revealed that when MrNV-CP are expressed, they automatically form VLP that engulf random RNA from the expression host. Further studies may improve dsRNA-encapsulation specificity by introducing a pair of affinity tags into the N-terminus of CP, which protrudes inside the VLP, and the hairpin of the target dsRNA.
Even though DualX-B15(DE3) contains the desirable dsRNA and protein co-expression traits, it seemed that the removal of the rnc14 gene activity via the P1 phage system resulted in the slow growth rate of the newly engineered strain. However, further optimization of growth conditions such as a change of media may curb this shortcoming. Despite this drawback of DualX-B15(DE3), its ability to co-express dsRNA-containing CP in vivo may outweigh its slower growth rate. In addition, based on the two dsRNA constructs tested in this study, it appeared that dsRNA yield in DualX-B15(DE3) was higher than HT115(DE3). However, whether this observation could be universally applied to other dsRNA constructs remained to be validated.
Late during the course of this study, it was revealed that insects do have an adaptive immune response that functions via the RNAi pathways [26]. Related research with shrimp suggests that they may be capable of a similar adaptive immune response. If this turns out to be correct, practical applications for the development of true vaccines in shrimp are likely to be possible and will be based on nucleic acid constructs rather than proteins. A key issue for successful applications would be the mode of preparation and effective delivery of the constructs via feeds. Thus, the results of this study may be even more significant than originally conceived.
Conclusion
By using P1 phage transduction in which HT115(DE3) and BL21(DE3) were used as the donor and recipient strains, respectively, this study engineered a novel protease- and RNase III-deficient E. coli strain, DualX-B15(DE3) which can simultaneously express recombinant proteins and dsRNA.
Methods
Construction of recombinant plasmids
The pET17b-GST was constructed by using Glutathione S-Transferase encoding gene (Genbank accession number M97937.1). This gene was inserted into NdeI and SacI restriction sites of pET17b plasmid. The pET28a-MrNV was synthesized by General Biosystems. To construct the plasmid, a gene encoding an MrNV capsid protein (Genbank accession number EU150129.1) with a C-terminus six-histidine was inserted between NcoI and XhoI restriction sites of an empty pET28a plasmid.
Preparation of donor bacteriophage P1
To prepare P1 lysate from a donor strain, HT115(DE3) was grown overnight with shaking at 37 °C in 5 ml lysogeny broth (LB) with 12.5 μg/ml tetracycline. The overnight culture was diluted 100-fold with 4 ml antibiotic-free LB containing 5 mM CaCl2, 10 mM MgCl2 and 5 mM glucose. The culture was incubated at 37 °C for 1 h with shaking at 250 rpm. Then, 100 μl of E. coli bacteriophage P1 (ATCC® 25404B1™) was added to the donor bacteria culture and incubated at 37 °C with aeration overnight. Afterwards, 50 μl of chloroform was added and mixed by vortex for 30 s. The mixture was centrifuged at 13,200×g for 1 min, before collecting the P1 phage-containing supernatant into a fresh tube.
P1 phage transduction
To infect the recipient strain BL21(DE3) with the P1 lysate, BL21(DE3) was grown overnight at 37 °C in 5 ml antibiotic-free LB broth. Then, 2 ml of the culture was centrifuged at 4500×g. The BL21(DE3)-containing pellet was resuspended in 250 μl of LB broth containing 10 mM MgSO4 and 5 mM CaCl2. Next, 100 μl of the P1 lysate from the previous step was mixed with 100 μl of the resuspended recipient cells and incubated at 37 °C for 30 min on a bench. To chelate calcium and minimize secondary infection of P1 phage [27], 1 ml of LB media containing 0.2 M citrate was added. The mixture was further incubated with shaking at 250 rpm for 1 h at 37 °C, before being centrifuged at 4500×g for 1 min. The resulting sediment was resuspended in 100 μl of a fresh LB media containing 0.2 M citrate. The suspension was equally divided and plated on LB agar plates with and without 12.5 μg/ml tetracycline.
Selection and verification of desirable mutants
Colonies grown on the tetracycline-supplemented plates were selected for further chromosomal DNA extraction by Qiagen genomic DNA extraction kit. The resulting DNA was used as a template DNA to amplify the rnc14 gene by polymerase chain reaction (PCR) using an rnc-KO forward primer (5′-AAA CTG CAG CGA AGC AGT TA-3′) and an rnc-KO reverse primer (5′-TCA TTC CAG CTC CAG TTT TT-3′). The primer sequences were designed based on the E. coli str. K-12 substr. MG1655 genome (GenBank accession number NC_000913.3). The forward primer annealed upstream from the start codon between nucleotide positions 2,704,131 and 2,704,150. The reverse primer annealed adjacent to the stop codon between the nucleotide positions 2,703,383 and 2,703,402. A 50 μl PCR reaction mixture contained a final concentration of 1x reaction buffer, 1.5 mM MgCl2, 0.2 mM dNTPs, 0.5 μM of each forward and reverse primers, 100 ng extracted DNA, and 1 unit of Taq DNA polymerase (Invitrogen). Thermocycling conditions were at 94 °C for 3 min; followed by 30 cycles of 94 °C for 45 s, 55 °C for 30 s, 72 °C for 1 min; then a final extension at 72 °C for 10 min. To identify a mutation, PCR amplicons from wild type and mutant bacteria were purified and sent for sequencing by using the rnc-KO forward and reverse primers (Macrogen).
Determination of the gene knockout effect on bacterial growth
To test the effect of rnc14 knockout on the growth of the novel E. coli strain, the pET28a-GW182 plasmid [23] was transformed into BL21(DE3), HT115(DE3) and DualX-B15(DE3) for simulation of the actual expression condition of double-stranded RNA (dsRNA). The transformants were grown in LB media containing 50 μg/ml kanamycin (for all strains) and 12.5 μg/ml tetracycline (for HT115(DE3) and DualX-B15(DE3)). The overnight cultures were diluted in 15 ml of fresh LB media supplemented with the corresponding antibiotics and OD600 was adjusted to 0.05. Then, they were incubated at 37 °C with shaking at 250 rpm. The growth of bacterial strains was observed by measuring OD600 every 20 min for 160 min by NanoDrop One Spectrophotometer (Thermo Fisher Scientific). Experiments were performed in triplicate to obtain average OD600 at each time point.
Expression of dsRNA
To express dsRNA, the pET28a-GW182 plasmid [23] was transformed into BL21(DE3), HT115(DE3) and DualX-B15(DE3). The transformants were grown in LB media containing 50 μg/ml kanamycin (for all strains) and 12.5 μg/ml tetracycline (for the HT115(DE3) and DualX-B15(DE3)) overnight. The cultures were diluted 100-fold in 25 ml of antibiotic-supplemented fresh LB media and OD600 was adjusted to 0.1. Then, they were incubated at 37 °C with shaking at 250 rpm for 2 h until OD600 reached 0.4. To induce the expression of dsRNA, 1 mM IPTG was added to the cultures for 3 h at 37 °C before the final OD600 was measured by a spectrophotometer. The cultures were harvested by centrifugation at 4500×g for 5 min.
Purification of dsRNA
To extract dsRNA by the ethanol extraction method [28], 50 mg of the resulting wet bacterial pellet was resuspended with 1 ml of 75% ethanol in 1x PBS (Sigma-Aldrich) and incubated at − 20 °C overnight. The mixture was centrifuged at 6000×g for 5 min at 4 °C, resuspended with 0.3 ml of 150 mM NaCl, and incubated at room temperature for 1 h. To isolate the soluble fraction, the suspension was centrifuged at 8000×g for 10 min at 4 °C and dsRNA-containing supernatant was collected.
To compare the yields of dsRNA-GW182 among different E. coli strains, the bacteria culture was done in triplicate. To visualize the extracted RNA, RNA isolates were analyzed by 1.5% agarose gel electrophoresis. Analysis of RNA band intensity was performed using ImageJ. The concentration of dsRNA was calculated by comparing the band intensity with that of the corresponding band in a 2-log DNA marker (New England Biolabs).
RNase treatment of the purified RNA
To verify that the RNA expressed in the engineered strain was in the form of dsRNA, the resulting RNA was divided into three equal parts. One fraction was mixed with 1x RNase A buffer and distilled water for the untreated condition. The other fraction was treated with 0.01 μg/μl RNase A enzyme (New England Biolabs) in 10 μl reaction mixture containing 1x RNase A buffer. The remaining fraction was treated with 1 unit of RNase III enzyme (New England Biolabs) in 10 μl reaction mixture containing 1x RNase III buffer and 1x MnCl2. The reactions were incubated at 37 °C for 5 min before being analyzed by 1.5% agarose gel electrophoresis alongside a 2-log DNA ladder staining with ethidium bromide and then visualized under UV lamp of Gel Documentation (Bio-Rad).
Expression of GST protein
To compare protein expression capacity of the three E. coli strains, the pET17b-GST plasmid was transformed into HT115(DE3), BL21(DE3) and DualX-B15(DE3). The bacteria were grown in LB medium containing 100 μg/ml ampicillin (for all strains) and 12.5 μg/ml tetracycline (for HT115(DE3) and DualX-B15(DE3)) at 37 °C with shaking at 250 rpm until OD600 reached 0.6. Protein expression was induced with IPTG at a final concentration of 1 mM and cells were then incubated at 25, 30 and 37 °C overnight. After centrifugation at 4500×g for 5 min, 50 mg of wet bacterial pellet was resuspended with 1 ml of lysis buffer pH 8 (50 mM NaH2PO4, 150 mM NaCl and 10 mM imidazole). Sonication of the cell suspension was performed at 40% amplitude with 2 cycles of 10 short bursts of 5 s followed by 5 s intervals for cooling (Sonics VCX 750). The lysates were then fractionated by centrifugation at 12,000×g. The total protein concentration was measured by using the Bradford reagent (Bio-Rad). The fractions were analyzed on 12.5% SDS-PAGE and Western blot using a mouse anti-GST antibody with a pre-stained protein ladder (Thermo Scientific).
To compare the GST yields among three E. coli strains, cell lysates were analyzed by SDS-PAGE to detect protein band. Analysis of protein band intensity was performed using ImageJ. The concentration of GST was calculated by comparing the band intensity with that of the corresponding band in 4 μl pre-stained protein ladder (Thermo Scientific).
Co-expression of dsRNA and CP
To co-express dsRNA-VP28 and MrNV-CP in the three bacterial strains, HT115(DE3), BL21(DE3) and DualX-B15(DE3) were co-transformed with the pGEM-VP28 [7] and pET28a-MrNV plasmids for dsRNA and CP expression, respectively. The expression temperatures were varied at 25, 30 and 37 °C. The optimized condition of co-expression was found to be at 25 °C overnight because, while dsRNA-VP28 could be expressed at all of the above temperature (Fig. S1), the best expression temperature of MrNV-CP was 25 °C as previously reported in Jariyapong et. al, 2015 [7]. The bacterial cells were grown in LB medium containing 50 μg/ml ampicillin, 25 μg/ml kanamycin (for all strains) and 6.25 μg/ml tetracycline (for HT115(DE3) and DualX-B15(DE3)) at 37 °C with shaking at 250 rpm until OD600 reached 0.6. Co-expression of dsRNA and CP was induced with IPTG at a final concentration of 1 mM and cells were then incubated at 25 °C overnight before harvest by centrifugation at 4500×g for 5 min.
The cell wet weight used for dsRNA and CP extraction was 100 mg. Half of the bacteria culture was used for protein analysis, while the other half of culture was subjected to dsRNA purification and RNase treatment (see above). To analyze protein expression, the total amount of 7.5 μg total protein determined by Bradford assay was loaded in each lane of gel electrophoresis. The MrNV-CP expression was confirmed by Western blot. For dsRNA-VP28 analysis, the total amount of 2 μg RNA, as determined by a spectrophotometer, was loaded in agarose gel electrophoresis.
Western blot
After SDS-PAGE, proteins were transferred onto a polyvinylidene fluoride (PVDF) membrane by Pierce Power Blotter (Thermo Scientific) for Western blot according to the manufacturer’s protocol. The membranes were immersed in 5% skim milk in PBS to block non-specific antibody binding at room temperature for 1 h with constant shaking. For GST protein detection, the membranes were immersed in a 1:1000 dilution of a mouse anti-GST antibody (Bio-Rad). For MrNV-CP expression, the membranes were immersed in a 1:100 dilution of an anti-MrNV-CP monoclonal antibody [17]. The membranes were washed three times for 5 min each with PBS containing 0.05% Tween-20 at room temperature with shaking. A secondary antibody (horseradish peroxidase conjugated goat anti-mouse IgG) was added at 1:2500 dilution and incubated for 1 h at room temperature with agitation. The membranes were washed three times with PBS containing 0.05% Tween-20. The antigen-antibody reactivity was detected by chemiluminescent method using Clarity Western ECL substrate (Bio-Rad).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- dsRNA:
-
Double-stranded RNA
- CP:
-
Capsid protein
- VLP:
-
Virus-like particles
- RNAi:
-
RNA interference
- GST:
-
Glutathione S-Transferase
- WSSV:
-
White spot syndrome virus
- MrNV:
-
Macrobrachium rosenbergii nodavirus
- IHHNV:
-
Infectious hypodermal and hematopoietic necrosis virus
References
Flegel TW. Historic emergence, impact and current status of shrimp pathogens in Asia. J Invertebr Pathol. 2012;110(2):166–73. https://doi.org/10.1016/j.jip.2012.03.004.
Syed Musthaq SK, Kwang J. Reprint of “Evolution of specific immunity in shrimp - a vaccination perspective against white spot syndrome virus”. Dev Comp Immunol. 2015;48(2):342–53. https://doi.org/10.1016/j.dci.2014.07.016.
Lakshmi B, Viswanath B, Sai Gopal D. Probiotics as antiviral agents in shrimp aquaculture. J Pathogens. 2013;2013:1–13. https://doi.org/10.1155/2013/424123.
Itami T, Asano M, Tokushige K, Kubono K, Nakagawa A, Takeno N, et al. Enhancement of disease resistance of kuruma shrimp, Penaeus japonicus, after oral administration of peptidoglycan derived from Bifidobacterium thermophilum. Aquaculture. 1998;164(1–4):277–88. https://doi.org/10.1016/S0044-8486(98)00193-8.
Thitamadee S, Srisala J, Taengchaiyaphum S, Sritunyalucksana K. Double-dose β-glucan treatment in WSSV-challenged shrimp reduces viral replication but causes mortality possibly due to excessive ROS production. Fish Shellfish Immunol. 2014;40(2):478–84. https://doi.org/10.1016/j.fsi.2014.07.033.
Chakraborty S, Ghosh U, Balasubramanian T, Das P. Screening, isolation and optimization of anti–white spot syndrome virus drug derived from marine plants. Asian Pac J Trop Biomed. 2014;4(Suppl 1):S107–17. https://doi.org/10.12980/APJTB.4.2014C1037.
Jariyapong P, Chotwiwatthanakun C, Direkbusarakom S, Hirono I, Wuthisuthimethavee S, Weerachatyanukul W. Delivery of double stranded RNA by Macrobrachium rosenbergii nodavirus-like particles to protect shrimp from white spot syndrome virus. Aquaculture. 2015;435:86–91. https://doi.org/10.1016/j.aquaculture.2014.09.034.
Jariyapong P, Weerachatyanukul W, Direkbusarakom S, Hirono I, Wuthisuthimethavee S, Chotwiwatthanakun C. Enhancement of shrimp immunity against white spot syndrome virus by Macrobrachium rosenbergii nodavirus-like particle encapsulated VP28 double-stranded RNA. Aquaculture. 2015;446:325–32. https://doi.org/10.1016/j.aquaculture.2015.05.016.
Chaimongkon D, Assavalapsakul W, Panyim S, Attasart P. A multi-target dsRNA for simultaneous inhibition of yellow head virus and white spot syndrome virus in shrimp. J Biotechnol. 2020;321:48–56. https://doi.org/10.1016/j.jbiotec.2020.06.022.
Robalino J, Bartlett T, Shepard E, Prior S, Jaramillo G, Scura E, et al. Double-stranded RNA induces sequence-specific antiviral silencing in addition to nonspecific immunity in a marine shrimp: convergence of RNA interference and innate immunity in the invertebrate antiviral response? J Virol. 2005;79(21):13561–71. https://doi.org/10.1128/JVI.79.21.13561-13571.2005.
Rattanarojpong T, Khankaew S, Khunrae P, Vanichviriyakit R, Poomputsa K. Recombinant baculovirus mediates dsRNA specific to rr2 delivery and its protective efficacy against WSSV infection. J Biotechnol. 2016;229:44–52. https://doi.org/10.1016/j.jbiotec.2016.05.007.
Li H, Yin B, Wang S, Fu Q, Xiao B, Lǚ K, et al. RNAi screening identifies a new toll from shrimp Litopenaeus vannamei that restricts WSSV infection through activating dorsal to induce antimicrobial peptides. PLoS Pathog. 2018;14(9):e1007109. https://doi.org/10.1371/journal.ppat.1007109.
Verbruggen B, et al. Molecular mechanisms of white spot syndrome virus infection and perspectives on treatments. Viruses. 2016;8(1):23.
Sritunyalucksana K, Wannapapho W, Lo CF, Flegel TW. PmRab7 is a VP28-binding protein involved in white spot syndrome virus infection in shrimp. J Virol. 2006;80(21):10734–42. https://doi.org/10.1128/JVI.00349-06.
Nilsen P, Karlsen M, Sritunyalucksana K, Thitamadee S. White spot syndrome virus VP28 specific double-stranded RNA provides protection through a highly focused siRNA population. Sci Rep. 2017;7(1):1028. https://doi.org/10.1038/s41598-017-01181-w.
Ludwig C, Wagner R. Virus-like particles-universal molecular toolboxes. Curr Opin Biotechnol. 2007;18(6):537–45. https://doi.org/10.1016/j.copbio.2007.10.013.
Jariyapong P, Chotwiwatthanakun C, Somrit M, Jitrapakdee S, Xing L, Cheng HR, et al. Encapsulation and delivery of plasmid DNA by virus-like nanoparticles engineered from Macrobrachium rosenbergii nodavirus. Virus Res. 2014;179:140–6. https://doi.org/10.1016/j.virusres.2013.10.021.
Zhu Y-P, Li C, Wan XY, Yang Q, Xie GS, Huang J. Delivery of plasmid DNA to shrimp hemocytes by infectious hypodermal and hematopoietic necrosis virus (IHHNV) nanoparticles expressed from a baculovirus insect cell system. J Invertebr Pathol. 2019;166:107231. https://doi.org/10.1016/j.jip.2019.107231.
Sinnuengnong R, Attasart P, Smith DR, Panyim S, Assavalapsakul W. Administration of co-expressed Penaeus stylirostris densovirus-like particles and dsRNA-YHV-pro provide protection against yellow head virus in shrimp. J Biotechnol. 2018;267:63–70. https://doi.org/10.1016/j.jbiotec.2018.01.002.
Timmons L, Court DL, Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001;263(1–2):103–12. https://doi.org/10.1016/S0378-1119(00)00579-5.
Bergman J, et al. Transduction: The Transfer of Host DNA by Bacteriophages; 2019.
Thomason LC, Costantino N, Court DL. E. coli genome manipulation by P1 transduction. Curr Protoc Mol Biol. 2007Chapter 1: p. Unit;1:17.
Sudsat P, et al., Comparison of Knockdown Efficiency of GW182 by dsRNAs Specific to GW182. Accessed 2-4 May 2017.
Tran F, Boedicker JQ. Genetic cargo and bacterial species set the rate of vesicle-mediated horizontal gene transfer. Sci Rep. 2017;7(1):8813. https://doi.org/10.1038/s41598-017-07447-7.
Ho KL, Gabrielsen M, Beh PL, Kueh CL, Thong QX, Streetley J, et al. Structure of the Macrobrachium rosenbergii nodavirus: a new genus within the Nodaviridae? PLoS Biol. 2018;16(10):e3000038. https://doi.org/10.1371/journal.pbio.3000038.
Tassetto M, Kunitomi M, Andino R. Circulating immune cells mediate a systemic RNAi-based adaptive antiviral response in Drosophila. Cell. 2017;169(2):314–25 e13.
Wenk S, et al. An engineering approach for rewiring microbial metabolism. In: Methods in enzymology: Elsevier; 2018. p. 329–67.
Posiri P, Ongvarrasopone C, Panyim S. A simple one-step method for producing dsRNA from E. coli to inhibit shrimp virus replication. J Virol Methods. 2013;188(1–2):64–9. https://doi.org/10.1016/j.jviromet.2012.11.033.
Acknowledgements
We would like to thank Prof. Dr. Timothy William Flegel for constructive criticism on this manuscript.
Funding
This research was supported by Thailand Graduate Institute of Science and Technology (TGIST), National Science and Technology Development Agency (NSTDA) and Faculty of Science, Mahidol University. The funding body had no role in the design of the study, collection, analysis, data interpretation and writing the manuscript.
Author information
Authors and Affiliations
Contributions
OI conceived the project. OI and TC designed the experiments. KW performed the experiment. All authors analyzed the data. CC and MS interpreted the data. KW, OI and TC drafted and revised the manuscript. OI and KS provided funding. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
dsRNA-VP28 could be expressed at 25 °C, 30 °C and 37 °C. RNA isolates were extracted from E. coli DualX-B15(DE3). RNA analyzed on the gel included untreated samples (U), samples treated with RNase A (A) and samples treated with RNase III (III). Lane M (Marker) is a 2-log DNA ladder. The molecular weight of dsRNA-VP28 was 615 bp as indicated by a triangle.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wuthisathid, K., Chaijarasphong, T., Chotwiwatthanakun, C. et al. Co-expression of double-stranded RNA and viral capsid protein in the novel engineered Escherichia coli DualX-B15(DE3) strain. BMC Microbiol 21, 88 (2021). https://doi.org/10.1186/s12866-021-02148-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-021-02148-8