
Abstract
Background
The blacklegged tick, Ixodes scapularis, is the primary vector of the Lyme disease spirochete Borrelia burgdorferi in North America. Though the tick is found across the eastern United States, Lyme disease is endemic to the northeast and upper midwest and rare or absent in the southern portion of the vector’s range. In an effort to better understand the tick microbiome from diverse geographic and climatic regions, we analysed the bacterial community of 115 I. scapularis adults collected from vegetation in Texas and Massachusetts, representing extreme ends of the vector’s range, by massively parallel sequencing of the 16S V4 rRNA gene. In addition, 7 female I. scapularis collected from dogs in Texas were included in the study.
Results
Male I. scapularis ticks had a more diverse bacterial microbiome in comparison to the female ticks. Rickettsia spp. dominated the microbiomes of field-collected female I. scapularis from both regions, as well as half of the males from Texas. In addition, the male and female ticks captured from Massachusetts contained high proportions of the pathogens Anaplasma and Borrelia, as well as the arthropod endosymbiont Wolbachia. None of these were found in libraries generated from ticks collected in Texas. Pseudomonas, Acinetobacter and Mycobacterium were significantly differently abundant (p < 0.05) between the male ticks from Massachusetts and Texas. Anaplasma and Borrelia were found in 15 and 63% of the 62 Massachusetts ticks, respectively, with a co-infection rate of 11%. Female ticks collected from Texas dogs were particularly diverse, and contained several genera including Rickettsia, Pseudomonas, Bradyrhizobium, Sediminibacterium, and Ralstonia.
Conclusions
Our results indicate that the bacterial microbiomes of I. scapularis ticks vary by sex and geography, with significantly more diversity in male microbiomes compared to females. We found that sex plays a larger role than geography in shaping the composition/diversity of the I. scapularis microbiome, but that geography affects what additional taxa are represented (beyond Rickettsia) and whether pathogens are found. Furthermore, recent feeding may have a role in shaping the tick microbiome, as evident from a more complex bacterial community in female ticks from dogs compared to the wild-caught questing females. These findings may provide further insight into the differences in the ability of the ticks to acquire, maintain and transmit pathogens. Future studies on possible causes and consequences of these differences will shed additional light on tick microbiome biology and vector competence.
Similar content being viewed by others


Background
Ticks transmit a greater variety of human and animal pathogens than any other disease vector and have significant public health impacts worldwide [1,2,3,4,5,6,7]. The blacklegged tick, Ixodes scapularis, is the major vector of the Lyme disease agent, Borrelia burgdorferi, in North America [8]. This tick also carries a number of other pathogens that cause human disease, including agents of anaplasmosis, babesiosis and Powassan encephalitis [9,10,11,12,13,14,15,16].
With more than 30,000 reported cases per year and an estimated 10-fold greater burden than the reported case counts, Lyme disease is the most common vector-borne illness in the U.S. [1, 17,18,19]. Despite a broad geographic distribution of I. scapularis across the eastern United States, Lyme disease cases are concentrated in the northeastern and upper midwestern states, whereas the disease is very rare or absent in the southern portion of the vector’s range [9, 19]. The prevalence of B. burgdorferi among I. scapularis in the northeastern U.S. has been reported to be as high as 30–50% [20,21,22], while it is rarely (< 1%) detected in the ticks from the southern United States [23,24,25]. Over the past two decades, the incidence of Lyme disease has increased in numbers and geographical area across the eastern U.S., which coincides with a significant northward range expansion of I. scapularis in the northeastern and midwestern regions [9, 26].
Many factors, including the density of host-seeking B. burgdorferi-harboring ticks, availability of the B. burgdorferi competent hosts, tick-behavior, seasonal activity of the ticks, and environmental variables influence the risk of Lyme disease [9, 27,28,29,30,31,32,33], yet the reasons behind the regional distribution of Lyme disease are not fully understood. In the upper midwestern and northeastern U.S., all active stages of I. scapularis can be encountered by humans during warm season of the year. But, in the southeastern U.S., human encounter occurs primarily with the adults I. scapularis ticks as immature ticks rarely seeks hosts in the region [34]. Recent studies have demonstrated that the resident microbial community of ixodid ticks can influence reproductive fitness and physiological processes of the tick and the acquisition, establishment and transmission of certain tick-borne pathogens [4, 35,36,37,38,39]. The microbial community of I. scapularis ticks has increasingly been studied in recent years [40,41,42,43]. In the U.S., the microbiome of Ixodes ticks varies with sex, species and geography [44]. By contrast, in Canada the microbiomes of I. scapularis ticks from eastern and southern Ontario do not differ significantly with regard to geographic origin, sex or life stages [40]. These contradicting reports highlight the need for additional studies considering the potential role that geography and related ecological and environmental factors may have in shaping the microbiome of ixodid ticks and disease transmission. More recently, we have demonstrated that the composition of the endogenous tick microbial community in colony-reared I. scapularis can be influenced by the environmental temperature [45]. With that goal in mind, we investigated the bacterial microbiomes of I. scapularis adults collected from natural vegetation from Texas and Massachusetts, representing opposing ends of the vector’s range and possessing distinct climates, by sequencing the hypervariable region 4 (V4) of the 16S ribosomal RNA (rRNA) gene using an Illumina MiSeq platform. Adult ticks were chosen to provide a fair comparison of the tick microbiomes from two regions with different geography and climate, and to provide ample DNA per sample without the need for pooling of multiple, smaller life stages.
Host blood meal has been shown to affect microbial diversity in I. pacificus [39], a closely related species to I. scapularis, with potential consequences for vector competence. To investigate how host blood meal affects microbiome, we also analysed the bacterial microbiomes of dog-fed female I. scapularis ticks during this study.
Results
16S V4 sequencing results
From the 122 I. scapularis samples (115 questing I. scapularis adult ticks collected from Texas and Massachusetts plus 7 female ticks collected from dogs in Texas, see Table 2 in Methods section for details) 12,204,399 quality-filtered reads (average per sample = 100,036; standard deviation = 24,411; range = 29,611- 167,874) were generated. This library included 6544 reads generated from negative controls (one blank extraction control and another no-template PCR negative control). The number of reads (for a particular OTU) that were present in the negative controls was subtracted from the libraries of the samples. Additionally, for genus level data analysis, 0.085% of the reads from each sample were considered as zero to minimize putative background contamination (i.e. if an OTU was abundant at less than 0.085% in a given sample, it was removed from the downstream analyses). All libraries generated from tick samples had adequate depth for further analysis, as evident from the mean Good’s coverage of 99.9% (range = 99.9–100%). Additionally, rarefaction curves of the number of observed OTUs plotted at a depth from 1000 to 30,000 sequences reaching plateau ~ 25,000 reads (Additional file 1: Figure S1) suggested sufficient sample coverage to proceed further.
Bacterial composition of I. scapularis
Proteobacteria dominated the I. scapularis microbiomes in both locations under study. Proteobacteria were prevalent at 87.2% mean relative abundance across field-collected ticks from Texas, with 73% (3.5–96.5%) prevalence in males and 100% (99.9–100%) prevalence in females. The prevalence of Proteobacteria in female ticks collected from dogs in Texas was 93% (86–100%). Proteobacteria were prevalent at 84.8% across ticks from Massachusetts, with 71.9% (0–99.9%) prevalence in males and 98.5% (11.1–88.9%) in females. Other common phyla in the microbiomes of the Texas ticks (both field-collected males and the females from dogs) included various proportions of Actinobacteria, Bacteroidetes, and Firmicutes. In addition to these non-proteobacterial phyla found in the Texas ticks, Massachusetts ticks were represented by the Spirochaetes, albeit with higher abundance in males (63% of them with ≥1% relative abundance) compared to that of the females (37%) (see Additional file 1: Figure S2).
In field-collected Texas ticks, Rickettsia spp. almost entirely dominated the microbiome of all the female ticks (Fig. 1). Although 66 taxa, including 48 genera, had an abundance of ≥1% in at least one of the 25 male samples from Texas, about half of them were also entirely dominated by Rickettsia. The remaining Texas male ticks (n = 13) had a more complex microbiome comprised of Mycobacterium (mean = 23%), Acinetobacter (22%), Sphingomonas (5%), Methylobacterium (4%), Corynebacterium (2%), Staphylococcus (2%), Escherichia (2%), Rickettsia (2%), Sphingobium (2%), Rhizobium (1%), Pseudomonas (1%), and others. There was the lack of variation in microbiome composition of field-collected Texas ticks between the collection years (Kruskal-Wallis test p > 0.05 for both male and female ticks (see Fig. 1) thus justified lumping ticks of the same sex but different years into the same category.

Genus-level relative abundance of bacteria in I. scapularis ticks collected from Texas and Massachusetts. Field-collected male (a) and female (b) ticks from TX are compared with the males (c) and females (d) from MA, as well as the females collected from dogs (e) in TX. Each bar represents an individual whole tick (identified on the x-axis) where different colors indicate the percentage of 16S V4 rRNA gene sequences assigned to each genus (whenever possible) based on 97% identity to taxa in the Greengenes reference database. Only the top-15 dominant taxa are shown for the male ticks, while all taxa ≥1% relative abundance are shown for the females. (f = family unclassified)
Libraries generated from ticks collected in woodlands in Massachusetts showed a similar dependence on the sex of the host. Rickettsia dominated the female microbiome (mean relative abundance = 97%, range = 69–100%), followed by Borrelia (overall mean < 2%), Wolbachia (1%), Anaplasma (0.1%), and others (Fig. 1). While Rickettsia was present in all female ticks from Massachusetts, Borrelia and Anaplasma were detected in 60 and 10% of the female samples, respectively. The mean relative abundance of Borrelia and Anaplasma among the positive female samples was 2% (0.2–1%) and 1% (2–79%), respectively. Wolbachia, with a mean relative abundance of 1.1% (0–27%), was found in more than 25% of the females from the region. Microbiomes of male ticks collected in Massachusetts were represented by multiple genera, including Pseudomonas (overall mean relative abundance = 23%), Borrelia (23%), Rickettsia (20%), Anaplasma (9%), Bradyrhizobium (5%) Wolbachia (4%), Ralstonia (4%), Acinetobacter (1%), and others. Altogether, 17 genera and 7 additional unclassified taxa (4 unclassified families, one unclassified at class-level and two unclassified at the phylum-level) had 1% or greater abundance in at least one of the 32 libraries generated from male ticks collected in Massachusetts. Borrelia (mean relative abundance among positive samples = 35%, range = 0.1–84%) was found in 66% of the Massachusetts males, while Anaplasma (50%, 2–80%) was detected in about 19% of the males. Moreover, Wolbachia was also found in about 28% of the male ticks from Massachusetts.
Significant differences were found in the mean relative abundance of certain genera in males from Texas and Massachusetts including, Pseudomonas (Kruskal-Wallis test p = 0.0001), Acinetobacter (p = 0.006) and Mycobacterium (p = 0.004). Additionally, Anaplasma, Borrelia and Wolbachia bacteria were found in both male and female ticks from MA, but not from those in TX.
Libraries generated from female ticks collected from dogs in Texas contained many genera that were rare or absent in libraries generated from female ticks collected from foliage in Texas and Massachusetts. These actively feeding ticks did carry Rickettsia (mean abundance = 53%), but the abundances of Pseudomonas (20%), Bradyrhizobium (7%), Sediminibacterium (5%), Ralstonia (4%), and Acinetobacter (2%) were much higher than in the microbiomes of questing ticks collected from foliage (Fig. 1).
Bacterial composition in I. scapularis females after removal of Rickettsia
Because the microbiomes of female ticks were entirely dominated by amplicons likely derived from the rickettsial endosymbiont known to occur in this species, and because this endosymbiont resides primarily in the ovaries [46], we removed Rickettsia sequences from the female data sets to further explore the inherent (predominately) gut microbiome of the female ticks, as described in Thapa et al. (2018) [45]. After in-silico removal of Rickettsia sequences from the female dataset, none of the field-collected female samples from Texas had sufficient sequences remaining to pass the inclusion criteria (as described in the beginning of the results section) needed to proceed for further analysis. Five female samples from Massachusetts also did not meet the inclusion criteria for further analysis after removing Rickettsia. Of the remaining 25 female samples, all collected from Massachusetts, only 13 had more than 1000 reads (mean = 7439, range = 1149–29,487) after deleting Rickettsia sequences. In-silico removal of Rickettsia from the Massachusetts female datasets revealed the previously hidden bacterial composition (Fig. 2), where the presence of Borrelia was prominent compared to the full female profiles (i.e. Rickettsia included). In contrast to a very low distribution of Borrelia in their full profiles (range = 0.5–11%, mean = 3%), the relative abundance of Borrelia in 11 Rickettsia-deleted female samples (two of the 13 samples under analysis were negative for these bacteria) ranged from 45 to 100% (mean = 79%) (Fig. 2).

Composition of bacterial taxa identified in field-collected female I. scapularis ticks from Massachusetts with Rickettsia (upper panel) and after in-silico removal of Rickettsia (lower panel). Taxa are classified to the genus level whenever possible and each bar represents an individual tick. Only ticks with at least 1000 non-rickettsial sequences are shown. See text for details
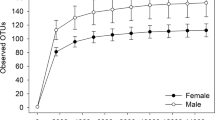
Alpha diversity
Prior to diversity analyses, subsampling at the minimum sequencing depth (25,059 sequences per sample) was performed to normalize the number of sequences in each sample [47]. Regardless of their geographical origin, field-collected male ticks exhibited significantly higher bacterial richness (number of OTUs observed) than that of the females (Wilcoxon rank-sum test p < 0.0001). However, female ticks collected from dogs in Texas had a significantly higher number of observed OTUs in comparison to the foliage-associated female ticks from either Texas or Massachusetts (FDR corrected Wilcoxon rank-sum test p < 0.0001 for all comparisons) (see Fig. 3). Similar results were found with ACE (abundance-based coverage estimator) value and Chao1 estimator (See Additional file 1: Figure S3). The Shannon diversity index of male ticks from both sites was significantly higher than the females (Wilcoxon rank-sum test p < 0.0001 for both sites compared separately) (Fig. 3), and female ticks collected from foliage in both Texas and Massachusetts had a lower bacterial diversity in comparison to the female ticks collected from dogs in Texas. Further multiple comparison analyses revealed no significant differences in the bacterial diversity of female ticks collected from dogs with that of the male ticks collected from foliage in both states.

Alpha diversity measures for male and female ticks
Beta diversity
While cluster analyses did not show a clear separation of the ticks for all samples, the majority of the male I. scapularis ticks collected from Massachusetts clustered separately from others, as did half the males from Texas in an unweighted PCoA plot of axis-1 vs axis-2. PCoA of unweighted UniFrac distances of bacterial communities showed that the first two axes (PCo1 and PCo2) explained 10.5 and 3.7% of the variation in the data, respectively (Fig. 4). PERMANOVA analysis of Unweighted UniFrac distances revealed a significant difference in the microbiome composition of male and female ticks from both collection sites (Adonis p = 0.001). The plot also demonstrated that the male samples from TX clustered separately when compared to the males from Massachusetts (p = 0.001), except one outlier from MA within the cluster of TX males. Female ticks collected from dogs in Texas formed a coherent cluster close to the field-captured males from Massachusetts. No clear clustering was observed in a PCoA plot of the weighted UniFrac distance metrics based on the collection site or sex of the tick (See Additional file 1: Figure S4).

PCoA plot using unweighted UniFrac distance of the bacterial composition in male and female ticks. Texas males are clustered separately from the Massachusetts male ticks; Female ticks collected from dogs in Texas (TX female (dog)) also clustered separately from the field-collected female ticks from both Texas and Massachusetts. Ellipses indicate 95% confidence intervals. Each dot represents the bacterial microbiome of an individual tick
Co-infection of Borrelia and Anaplasma in Massachusetts ticks
Anaplasma and Borrelia were detected exclusively in libraries generated from ticks collected in Massachusetts. Anaplasma-Borrelia appeared together in 7 of 62 (11%) field-collected I. scapularis from Massachusetts (Table 1).
Microbiome of Borrelia-positive and Borrelia-negative I. scapularis ticks
Although the distribution of individual bacterial taxa varied among male and female ticks from Massachusetts, there was no significant difference in bacterial composition between Borrelia-positive and Borrelia-negative groups in both male (PERMANOVA of UniFrac distance, Adonis p = 0.25) and female (p = 0.26). (Additional file 1 Figure S5). However, Borrelia-negative males from Massachusetts had a different community structure of bacteria when compared to the Texas males based on PERMANOVA test of the unweighted UniFrac distance metrics (Adonis p = 0.001). In terms of female ticks, Rickettsia was almost exclusively dominant in both regions, but Massachusetts females contained Borrelia, Anaplasma, and Wolbachia, while these bacteria were absent from the ticks collected in Texas.
Microbiome comparisons of the colony-reared and field-collected I. scapularis ticks
We also compared the baseline microbiome data of the colony-reared I. scapularis ticks from our previous publication [45] to that of the microbiome data obtained from the field-collected ticks in the present study. In the case of the male ticks, relative abundance of Bacteroidetes and Firmicutes was significantly different in colony-reared ticks than that of the wild-caught ticks from Texas or Massachusetts (BH corrected Dunn’s Kruskal Wallis test p < 0.05 for all comparisons). In female ticks, Proteobacteria dominated the microbiome of both colony-reared and field-collected ticks (questing and dog fed) (See Additional file 1: Figure S6). Similar to the Texas ticks, Borrelia, Anaplasma, and Wolbachia were not found in the colony-reared ticks purchased from the Tick Rearing Facility at Oklahoma State University (OSU). Bacterial diversity in colony-reared male ticks found to differ significantly from that of the wild-caught males from TX or MA (unweighted UniFrac PERMANOVA p = 0.001 for both comparisons). The colony-reared female ticks also differed significantly in beta diversity (as measured by unweighted UniFrac distances) when compared to that of the female ticks collected from vegetation in TX and MA (p = 0.001). However, the colony-reared I. scapularis females were no more diverse than the female ticks collected from dogs (p = 0.06).
Discussion
The dominance of Proteobacteria in I. scapularis ticks from both Texas and Massachusetts agrees with a previous study [44] of wild-caught ticks from several U.S. states that also found > 80% of the reads could be assigned to Proteobacteria. Other phyla found in this study, including Spirochaetes, were also previously reported in wild-caught ticks [44]. Our finding of exclusive abundance (100%) of Rickettsia in all field-collected female ticks from Texas, and a very high dominance (97%) in females from Massachusetts, is consistent with previous reports for the microbiome of I. scapularis [41,42,43,44] and high prevalence of Rickettsia in larvae and nymphal I. scapularis [6, 22].
The high numbers of Rickettsia likely reflect a mutualism between this endosymbiont and the host, and most likely belong to the endosymbiont Rickettsia buchneri [46]. R. buchneri has been shown to provide a source of vitamins to the tick [48]. The genus Rickettsia also contains many potentially pathogenic species, including Rickettsia rickettsii, R. japonica, R. akari [49] and R. parkeri [50], but these bacteria are not known to be vectored by I. scapularis. By contrast, high prevalence of R. buchneri endosymbionts in female ticks is generally associated with the ovaries [41, 51]. As Rickettsia was also highly prevalent in male ticks, our findings suggest that Rickettsia resides in other body parts of the male ticks. This is consistent with previous reports of R. parkeri detected in male tissues of Amblyomma maculatum [52].
The complexity of the microbiomes of male ticks collected in both Massachusetts and Texas may reflect acquisition from the environment, as relatives of many genera found in the guts of male ticks are considered free-living (not host-associated) bacteria. Differences, such as the relative abundance of Pseudomonas (MA = 23% vs TX = 1%), Acinetobacter (MA = 1% vs TX =22%), Mycobacterium (MA = 0.4% vs TX = 23%) and the exclusivity of Borrelia and Anaplasma to MA, suggests a geographical and/or ecological variation of the microbiota in these ticks with public health consequences. Our findings of slightly higher abundance of Borrelia or Anaplasma in male ticks from Massachusetts compared to the female ticks suggest the possible roles of the underlying microbial community in the male ticks for pathogen acquisition. However, it should also be noted that some differences between males and females may be artifacts arising from differences in sequencing depth between males and rickettsia-subtracted females. Caution is therefore warranted in interpretation of these differences. In addition, the large variation between microbiomes of wild-caught Texas male ticks suggests the possibility of two distinct microbiomes. However, all the ticks were collected from the same habitat in Texas during 2016 and 2017, and the pattern is similar for both collection years. The variations between the microbiomes of Texas male ticks might have to do with prior host blood meal. The mean abundance of Borrelia, which could include pathogenic B. burgdorferi and B. miyamotoi¸ was higher in males (35%) in comparison to the females (< 2%) and in the range of previous studies [20, 41, 44]. Xu et al. (20) study tested the ticks via qPCR while our study is based on 16S sequencing. So, the methodological differences could also have contributed to the relatively high levels of Borrelia and Anaplasma detected in I. scapularis ticks collected from Massachusetts. In comparison to the traditional PCR-based approaches used previously [20], the 16S rRNA gene sequencing used here cannot discriminate between species. It is highly likely that the samples that yielded Borrelia 16S rRNA gene sequences are due to B. burgdorferi (the causative agent of Lyme disease), but could be partially due to B. miyamotoi (relapsing fever group bacterium). B. miyamotoi has also been identified in this area, albeit at substantially lower numbers, with 2.3% of ticks tested from Cape Cod in 2016 found positive for this bacterium [53]. Furthermore, not all I. scapularis samples yielding Borrelia reads from 16S rRNA Illumina sequencing produce amplicons in PCR testing of the B. burgdorferi specific ospC gene [44]. Similar results of discordance between traditional PCR assays and Illumina MiSeq sequencing was also observed in another study on the A. americanum tick [54]. However, our findings of about 63% Borrelia in I. scapularis ticks collected from North Truro in Cape Cod, Massachusetts is in line with the findings of Xu et al. (2016) [20], who also reported that 62.5% of I. scapularis ticks tested from Nantucket county in Massachusetts were B. burgdorferi positive, and also consistent with unpublished work conducted by our laboratory using nested PCR methods (data not shown).
The Anaplasma-Borrelia co-infection rate of 11% in the ticks from Massachusetts we report was substantially higher than a previous study [20] on human-biting I. scapularis from Massachusetts, where 1.8% of the ticks were coinfected by B. burgdorferi and A. phagocytophilum. The higher rate of co-infection in this study could be attributed to the overall higher prevalence of Borrelia.
The detection of Wolbachia in more than 25% of the ticks from Massachusetts was not expected. Wolbachia are known to exhibit endosymbiotic mutualism with insects [55, 56], and has been previously reported in other ticks [57, 58], but not I. scapularis. Although Wolbachia has been known to induce resistance to dengue virus when introduced into Aedes aegypti mosquitoes [56] and other insects [55], Plantard et al. (2012) showed that Wolbachia in the I. ricinus tick, a major European vector of the Lyme disease agent, is due to the presence of the endoparasitoid wasp Ixodiphagus hookeri, and not representative of a true endosymbiont of the tick [59]. Thus, the prevalence of Wolbachia reported here is likely not a true mutualism with I. scapularis, but rather may indicate the presence of an unidentified parasite.
The higher bacterial richness in the microbiome of male ticks compared to female ticks, regardless of the geographical origin reflects dominance of Rickettsia in female ticks. Furthermore, a significantly higher Shannon diversity in male ticks suggests that the community of male ticks were more diverse as well as even, compared to the females.
The complexity of libraries generated from female I. scapularis ticks collected from dogs in Texas, in comparison to the wild-caught females from both states, suggests that the tick microbiome may shift as the result of a recent blood meal. The microbiomes of the female ticks that originated from dogs in Texas closely matches that of the male ticks from Massachusetts in terms of diversity but not in community membership, further supporting the idea that the bacterial microbiomes of female I. scapularis ticks vary with their sample source. One possible explanation could be that recent blood feeding led to increased abundance of midgut bacteria, lessening the overall relative impact of the rickettsial endosymbiont on subsequent analyses.
Difference in the composition and diversity of the microbiome of colony-reared I. scapularis ticks in comparison to the wild-caught ticks could be attributed to multiple factors, including the type of previous blood meal, and the environmental/ecological parameters. The difference in microbiomes of I. scapularis ticks from Massachusetts and Texas, including the ticks fed on dogs in Texas may, also reflect the seasonal effects on the tick microbiome. Indeed, we have previously shown that the environmental temperature can influence the endogenous tick microbial community composition in colony-reared I. scapularis [45].
Conclusions
Analyses of the microbiomes of field-collected adult I. scapularis ticks from Texas and Massachusetts demonstrated that the bacterial microbiota of the ticks varies by sex and geographic origin. The main findings of this study are that sex plays a larger role than geography in shaping the composition/diversity of the I. scapularis microbiome, but that geography affects what additional taxa are represented (beyond Rickettsia) and whether pathogens are found. In addition, the microbiome of dog-fed female I. scapularis ticks is more complex than those of the wild-caught females.
Taken together, our findings may provide further insight into the sexual and regional differences in the ability of the ticks to acquire, maintain and transmit pathogens. Future studies on functional and mechanistic aspects of the tick microbiome, including possible causes (such as the ecological factors) and consequences of these differences will help us better understand the microbiome biology of the ticks and vector competence. These efforts may ultimately aid development of strategies to control the risk and transmission of tick-borne diseases.
Methods
Tick sampling and processing
During 2016 and 2017, a total of 115 adult I. scapularis were collected in the Davy Crockett National Forest near Kennard, Texas and from the North Truro area in the Barnstable county of Cape Cod, Massachusetts. Due to the difference in activity levels of ticks in different local environments, sample collection in Massachusetts was done during the late spring while ticks from Texas were collected during autumn. I. scapularis is endemic to both Cape Cod, located in the northeast U.S. [20, 60], and Trinity county in Texas, part of the southeastern U.S. [9]. Standard flagging technique was used for tick sampling, which consisted of walking down trails dragging a 1 m2 piece of white cloth attached to a pole gently over and around the vegetation where ticks were likely to be present. All encountered ticks were collected with fine-tipped tweezers and placed into sterile collection vials containing cotton fabric for housing. Ticks were categorized by location (TX or MA) and sex (male or female). All ticks were then preserved at ˗20 °C until DNA extraction. In addition, seven I. scapularis females collected from dogs (pulled off with tweezers) in North Texas were included in the study. These dog-fed ticks were unengorged to partially engorged. Details of the collection sites and dates are provided in Table 2
.
DNA extraction
All tick samples were treated in sequence with 10% sodium hypochlorite and molecular biology grade water to reduce surface contamination. Sterilization techniques using sodium hypochlorite solution have previously been demonstrated to significantly eliminate the bacteria and DNA on the tick surface [61]. Each whole tick was then cut into sections with a sterile scalpel on a glass microscope slide to disintegrate the thick cuticle layer and all sections were used during DNA extraction as previously described [45]. Briefly, all resultant sections of a tick were placed in a 2-ml screw-capped FastPrep tube (MP Biomedicals, LLC., Santa Ana, CA) containing 550 μl CSPL® buffer (Omega Bio-tek, Norcross, GA) and 8–10 sterile 2.8 mm ceramic beads (MoBio Laboratories Inc., Carlsbad, CA). Following pulverization (3 cycles of 7 m/s for 60s) in a FastPrep-24™ 5G Instrument (MP Biomedicals, LLC.), each sample was incubated at 56 °C for 2 h. Total DNA was then extracted from 122 individual ticks using a Mag-Bind® Plant DNA Plus Kit (Omega Bio-tek) as per the manufacturer’s instructions. A blank extraction control with reagents and beads was also prepared for each lot of DNA extractions. The extracted genomic DNA was quantified with a Nanodrop spectrophotometer (Invitrogen, Carlsbad, CA) and stored at − 20 °C until further processing.
Tick mitochondrial 16S rRNA gene amplification
Each DNA extract was first assessed by PCR to amplify the tick mitochondrial 16S rRNA gene as a sample positive control, as previously described [45] using 16S-1 and 16S + 2 primers [62].
Bacterial 16S rRNA gene amplification
DNA was amplified in duplicates by PCR using 515F/806R primers that target the hypervariable region four (V4) of the bacterial 16S rRNA gene. The primer set (forward: 5′-GTGCCAGCMGCCGCGGTAA-3′ and reverse 5′-GGACTACHVGGGTWTCTAAT-3′) had overhanging Illumina sequencing adaptors. Earth Microbiome Project (EMP) 16S Illumina Amplification Protocol was followed [63] with minor modifications as described below. In brief, a master mix solution was prepared per 25-μl PCR reaction volume with 2.5-μl 10X Accuprime™ PCR Buffer II (Invitrogen, Carlsbad, CA), 2.5-μl of 1.6 mg/ml Bovine Serum Albumin (New England Biolabs, Inc., Ipswich, MA), 1-μl 50 mM MgSO4, 0.5-μl 10 μM forward primer, 0.5-μl 10 μM reverse primer, 0.1-μl of 5 U/μl Accuprime™ Taq DNA Polymerase High Fidelity, 10-μl (43–554 ng) of template DNA and 7.9-μl molecular biology grade water. PCR was carried out in a BioRad C1000 Touch™ thermal cycler with the following cycling parameters: an initial denaturation at 94 °C for 2 min followed by 30 cycles (35 cycles for all male samples, with few exceptions, 40 cycles) consisting of denaturation at 94 °C for 30 s, annealing at 55 °C for 40 s, and extension at 68 °C for 40 s, with a final extension at 68 °C for 5 min and a 4 °C indefinite hold. Amplicon quality was evaluated by visualizing under UV light after separation in 1.5% agarose gel after electrophoresis. No template negative controls were used during the PCR runs.
16S rRNA gene library preparation and sequencing
PCR amplicons in duplicate sets were combined for each sample. Purification of the PCR products were performed using AMPure XP magnetic beads, and 16S libraries for a total of 122 samples were prepared following the Illumina 16S metagenomic sequencing library preparation protocol with the following minor modifications: the reaction mixture for index PCR (per 50 μl reaction) consisted of 5-μl 10X Accuprime™ PCR Buffer II, 5-μl Nextera XT Index Primer 1, 5-μl Nextera XT Index Primer 2, 0.2-μl Accuprime™ Taq DNA Polymerase High Fidelity (5 U/μl), 5-μl PCR product and 29.8-μl molecular biology grade water. Amplification was performed in a BioRad C1000 Touch™ thermocycler with the following program: 94 °C for 3 min followed by 8 cycles of 94 °C for 30 s, 55 °C for 30 s, 68 °C for 30 s, and 68 °C for 5 min, followed by holding at 4 °C. The final purified PCR products were quantified using a Qubit® 2.0 fluorometer (Thermo Fisher Scientific Inc., Carlsbad, CA) and libraries were pooled in equimolar amounts. The pooled library (10 pM final concentration) was loaded in the MiSeq Reagent Kit v2 (Illumina Inc., San Diego, CA) and paired-end (2 × 250) sequencing (500 cycles) was performed on an Illumina MiSeq instrument per manufacturer’s recommendations. Two negative controls (a blank extraction control and a no-template PCR negative control) were sequenced alongside of the samples to monitor for background contamination. A 5% PhiX DNA was also included in the run as an internal control.
Data analysis
Data were analyzed in the Windows version of Microsoft Excel 2016 (Microsoft Corporation, Redmond, WA), XLSTAT-Ecology (Addinsoft SARL, NY), and R software (version 3.4.1) as well as in mothur software [64]. GraphPad Prism (v5.04) was also used to generate visuals. Raw sequences generated out of the Illumina MiSeq Instrument were processed using mothur v1.36.1. Quality filtered merged reads were aligned to the SILVA database (Release 132) [65] and chimeras were removed using the UCHIME [66] algorithm. Sequences within a 97% identity threshold were binned into operational taxonomic units (OTUs) [67] and taxonomic groups were assigned by comparison to the Greengenes reference database v13.8.99 [68, 69].
Rickettsia sequences were removed from the dataset using the remove.lineage command in mothur, as described in Thapa et al. (2018) [45]. Relative abundances of bacterial taxa were then compared between groups based on location (Texas vs Massachusetts), sex (male vs female), and source (vegetation vs dogs). Taxa with < 1% relative abundance in all samples were grouped together into ‘< 1% abundant taxa’ category for visual representation. Alpha diversity within samples was calculated using Observed OTUs, ACE value, Chao1 estimator, and Shannon index [70] in the data set rarefied at the lowest sequencing depth of 25, 059 reads/sample. Beta-diversity beween samples was quantified by weighted and unweighted UniFrac distance matrices and the bacterial community structure was visualized using principal coordinates analysis (PCoA) plots. Statistical analyses of the differently abundant taxa among groups were performed using the Kruskal Wallis test. Comparison between groups was performed using the Wilcoxon rank-sum test. Permutational multivariate analysis of variance (PERMANOVA) was used to determine the differences in microbial community composition within and among the groups using the ‘Vegan’ (v2.5.3) and ‘PhyloSeq’ (v1.24.2) R-packages. If appropriate, a post-hoc correction using the Benjamini-Hochberg method [71], which takes into account the false discovery rate (FDR) [72], was applied for multiple comparison testing [73]. The level of significance used in these analyses was 0.05.
Availability of data and materials
All raw sequence data generated and/or analysed during this study are available in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) with the accession number SRP144771 (https://www.ncbi.nlm.nih.gov/sra/SRP144771) under the BioProject PRJNA464062.
Abbreviations
- ACE:
-
abundance-based coverage estimator
- CDC:
-
Centers for Disease Control and Prevention
- MA:
-
Massachusetts
- OTU:
-
Operational taxonomic unit
- PCoA:
-
Principal coordinate analysis
- TX:
-
Texas
References
Jongejan F, Uilenberg G. The global importance of ticks. Parasitology. 2004;129(Suppl):S3–14.
Yuan DT. A metagenomic study of the tick midgut. Thesis (MS). Houston: The University of Texas; 2010.
Adrion ER, Aucott J, Lemke KW, Weiner JP. Health care costs, utilization and patterns of care following Lyme disease. PLoS One. 2015;10(2):e0116767.
Narasimhan S, Fikrig E. Tick microbiome: the force within. Trends Parasitol. 2015;31(7):315–23.
Zhang X, Meltzer MI, Pena CA, Hopkins AB, Wroth L, Fix AD. Economic impact of Lyme disease. Emerg Infect Dis. 2006;12(4):653–60.
Clay K, Fuqua C. The tick microbiome: diversity, distribution and influence of the internal microbial community for a blood-feeding disease vector. In: Workshop report: critical needs and gaps in understanding prevention, amelioration, and resolution of Lyme and other tick-borne diseases, the short-term and long-term outcomes. Washington, DC: National Academies Press; 2010. p. A193–214.
Rochon K, Scoles GA, Lysyk TJ. Dispersion and sampling of adult Dermacentor andersoni in rangeland in Western North America. J Med Entomol. 2012;49(2):253–61.
Burgdorfer W, Barbour AG, Hayes SF, Benach JL, Grunwaldt E, Davis JP. Lyme disease-a tick-borne spirochetosis. Science (New York, NY). 1982;216(4552):1317–9.
Eisen RJ, Eisen L, Beard CB. County-scale distribution of Ixodes scapularis and Ixodes pacificus (Acari: Ixodidae) in the continental United States. J Med Entomol. 2016;53(2):349–86.
Piesman J, Eisen L. Prevention of tick-borne diseases. Annu Rev Entomol. 2008;53:323–43.
Ebel GD. Update on Powassan virus: emergence of a north American tick-borne flavivirus. Annu Rev Entomol. 2010;55:95–110.
Prusinski MA, Kokas JE, Hukey KT, Kogut SJ, Lee J, Backenson PB. Prevalence of Borrelia burgdorferi (Spirochaetales: Spirochaetaceae), Anaplasma phagocytophilum (Rickettsiales: Anaplasmataceae), and Babesia microti (Piroplasmida: Babesiidae) in Ixodes scapularis (Acari: Ixodidae) collected from recreational lands in the Hudson Valley region, New York State. J Med Entomol. 2014;51(1):226–36.
Spielman A, Clifford CM, Piesman J, Corwin MD. Human babesiosis on Nantucket Island, USA: description of the vector, Ixodes (Ixodes) dammini, n. sp. (Acarina: Ixodidae). J Med Entomol. 1979;15(3):218–34.
Scoles GA, Papero M, Beati L, Fish D. A relapsing fever group spirochete transmitted by Ixodes scapularis ticks. Vector Borne Zoonotic Dis. 2001;1(1):21–34.
Platonov AE, Karan LS, Kolyasnikova NM, Makhneva NA, Toporkova MG, Maleev VV, Fish D, Krause PJ. Humans infected with relapsing fever spirochete Borrelia miyamotoi, Russia. Emerg Infect Dis. 2011;17(10):1816–23.
Krause PJ, Narasimhan S, Wormser GP, Rollend L, Fikrig E, Lepore T, Barbour A, Fish D. Human Borrelia miyamotoi infection in the United States. N Engl J Med. 2013;368(3):291–3.
Hinckley AF, Connally NP, Meek JI, Johnson BJ, Kemperman MM, Feldman KA, White JL, Mead PS. Lyme disease testing by large commercial laboratories in the United States. Clin Infect Dis. 2014;59(5):676–81.
Nelson CA, Saha S, Kugeler KJ, Delorey MJ, Shankar MB, Hinckley AF, Mead PS. Incidence of clinician-diagnosed Lyme disease, United States, 2005-2010. Emerg Infect Dis. 2015;21(9):1625–31.
Lyme disease: data and statistics. [https://www.cdc.gov/lyme/stats/index.html]. Accessed 17 Dec 2017.
Xu G, Mather TN, Hollingsworth CS, Rich SM. Passive surveillance of Ixodes scapularis (say), their biting activity, and associated pathogens in Massachusetts. Vector Borne Zoonotic Dis. 2016;16(8):520–7.
Adelson ME, Rao RV, Tilton RC, Cabets K, Eskow E, Fein L, Occi JL, Mordechai E. Prevalence of Borrelia burgdorferi, Bartonella spp., Babesia microti, and Anaplasma phagocytophila in Ixodes scapularis ticks collected in northern New Jersey. J Clin Microbiol. 2004;42(6):2799–801.
Moreno CX, Moy F, Daniels TJ, Godfrey HP, Cabello FC. Molecular analysis of microbial communities identified in different developmental stages of Ixodes scapularis ticks from Westchester and Dutchess counties, New York. Environ Microbiol. 2006;8(5):761–72.
Eisen L, Eisen RJ, Mun J, Salkeld DJ, Lane RS. Transmission cycles of Borrelia burgdorferi and B. bissettii in relation to habitat type in northwestern California. J Vector Ecol. 2009;34(1):81–91.
Clark KL, Oliver JH Jr, James AM, Durden LA, Banks CW. Prevalence of Borrelia burgdorferi sensu lato infection among rodents and host-seeking ticks in South Carolina. J Med Entomol. 2002;39(1):198–206.
Maggi RG, Reichelt S, Toliver M, Engber B. Borrelia species in Ixodes affinis and Ixodes scapularis ticks collected from the coastal plain of North Carolina. Ticks Tick-Borne Dis. 2010;1(4):168–71.
Kiersten JK, Grace MF, Joseph DF, Paul SM. Geographic distribution and expansion of human Lyme disease, United States. Emerg Infect Dis. 2015;21(8):1455.
Estrada-Pena A, Ayllon N, de la Fuente J. Impact of climate trends on tick-borne pathogen transmission. Front Physiol. 2012;3:64.
Monaghan AJ, Moore SM, Sampson KM, Beard CB, Eisen RJ. Climate change influences on the annual onset of Lyme disease in the United States. Ticks Tick-Borne Dis. 2015;6(5):615–22.
Ostfeld RS, Brunner JL. Climate change and Ixodes tick-borne diseases of humans. Philos Trans R Soc Lond Ser B Biol Sci. 2015;370(1665):20140051.
Diuk-Wasser MA, Hoen AG, Cislo P, Brinkerhoff R, Hamer SA, Rowland M, Cortinas R, Vourc'h G, Melton F, Hickling GJ, et al. Human risk of infection with Borrelia burgdorferi, the Lyme disease agent, in eastern United States. Am J Trop Med Hyg. 2012;86(2):320–7.
Kelly RR, Gaines D, Gilliam WF, Brinkerhoff RJ. Population genetic structure of the Lyme disease vector Ixodes scapularis at an apparent spatial expansion front. Infect Genetics Evol. 2014;27:543–50.
Arsnoe IM, Hickling GJ, Ginsberg HS, McElreath R, Tsao JI. Different populations of blacklegged tick nymphs exhibit differences in questing behavior that have implications for human Lyme disease risk. PLoS One. 2015;10(5):e0127450.
Spielman A, Wilson ML, Levine JF, Piesman J. Ecology of Ixodes dammini-borne human babesiosis and Lyme disease. Annu Rev Entomol. 1985;30:439–60.
Eisen RJ, Eisen L, Ogden NH, Beard CB. Linkages of weather and climate with Ixodes scapularis and Ixodes pacificus (Acari: Ixodidae), enzootic transmission of Borrelia burgdorferi, and Lyme disease in North America. J Med Entomol. 2016;53(2):250–61.
Ahantarig A, Trinachartvanit W, Baimai V, Grubhoffer L. Hard ticks and their bacterial endosymbionts (or would be pathogens). Folia Microbiol. 2013;58(5):419–28.
Gall CA, Reif KE, Scoles GA, Mason KL, Mousel M, Noh SM, Brayton KA. The bacterial microbiome of Dermacentor andersoni ticks influences pathogen susceptibility. ISME J. 2016;10(8):1846–55.
Smith TA, Driscoll T, Gillespie JJ, Raghavan R. A Coxiella-like endosymbiont is a potential vitamin source for the lone star tick. Genome Biol Evol. 2015;7(3):831–8.
Zhong J, Jasinskas A, Barbour AG. Antibiotic treatment of the tick vector Amblyomma americanum reduced reproductive fitness. PLoS One. 2007;2(5):e405.
Swei A, Kwan JY. Tick microbiome and pathogen acquisition altered by host blood meal. ISME J. 2017;11(3):813–6.
Clow KM, Weese JS, Rousseau J, Jardine CM. Microbiota of field-collected Ixodes scapularis and Dermacentor variabilis from eastern and southern Ontario, Canada. Ticks Tick-Borne Dis. 2018;9(2):235–44.
Zolnik CP, Prill RJ, Falco RC, Daniels TJ, Kolokotronis SO. Microbiome changes through ontogeny of a tick pathogen vector. Mol Ecol. 2016;25(19):4963–77.
Hawlena H, Rynkiewicz E, Toh E, Alfred A, Durden LA, Hastriter MW, Nelson DE, Rong R, Munro D, Dong Q, et al. The arthropod, but not the vertebrate host or its environment, dictates bacterial community composition of fleas and ticks. ISME J. 2012;7:221.
Rynkiewicz EC, Hemmerich C, Rusch DB, Fuqua C, Clay K. Concordance of bacterial communities of two tick species and blood of their shared rodent host. Mol Ecol. 2015;24(10):2566–79.
Van Treuren W, Ponnusamy L, Brinkerhoff RJ, Gonzalez A, Parobek CM, Juliano JJ, Andreadis TG, Falco RC, Ziegler LB, Hathaway N, et al. Variation in the microbiota of Ixodes ticks with regard to geography, species, and sex. Appl Environ Microbiol. 2015;81(18):6200–9.
Thapa S, Zhang Y, Allen MS. Effects of temperature on bacterial microbiome composition in Ixodes scapularis ticks. MicrobiologyOpen. 2018;8(5):e00719.
Kurtti TJ, Felsheim RF, Burkhardt NY, Oliver JD, Heu CC, Munderloh UG. Rickettsia buchneri sp. nov., a rickettsial endosymbiont of the blacklegged tick Ixodes scapularis. Int J Syst Evol Microbiol. 2015;65(Pt 3):965–70.
Shade A. Diversity is the question, not the answer. ISME J. 2016;11(1):1–6.
Hunter DJ, Torkelson JL, Bodnar J, Mortazavi B, Laurent T, Deason J, Thephavongsa K, Zhong J. The Rickettsia endosymbiont of Ixodes pacificus contains all the genes of de novo folate biosynthesis. PLoS One. 2015;10(12):e0144552.
Azad AF, Beard CB. Rickettsial pathogens and their arthropod vectors. Emerg Infect Dis. 1998;4(2):179–86.
Paddock CD, Sumner JW, Comer JA, Zaki SR, Goldsmith CS, Goddard J, McLellan SL, Tamminga CL, Ohl CA. Rickettsia parkeri: a newly recognized cause of spotted fever rickettsiosis in the United States. Clin Infect Dis. 2004;38(6):805–11.
Noda H, Munderloh UG, Kurtti TJ. Endosymbionts of ticks and their relationship to Wolbachia spp. and tick-borne pathogens of humans and animals. Appl Environ Microbiol. 1997;63(10):3926–32.
Budachetri K, Browning RE, Adamson SW, Dowd SE, Chao C-C, Ching W-M, Karim S. An insight into the microbiome of the Amblyomma maculatum (Acari: Ixodidae). J Med Entomol. 2014;51(1):119–29.
Cape Cod deer tick project [https://www.cranberries.org/sites/default/files/uploads/pdf/Meeting_files/DeerTicks_LarryDapsis.pdf]. Accessed 3 Apr 2018.
Trout Fryxell RT, DeBruyn JM. The microbiome of Ehrlichia-infected and uninfected lone star ticks (Amblyomma americanum). PLoS One. 2016;11(1):e0146651.
Mousson L, Martin E, Zouache K, Madec Y, Mavingui P, Failloux AB. Wolbachia modulates chikungunya replication in Aedes albopictus. Mol Ecol. 2010;19(9):1953–64.
Bian G, Xu Y, Lu P, Xie Y, Xi Z. The endosymbiotic bacterium Wolbachia induces resistance to dengue virus in Aedes aegypti. PLoS Pathog. 2010;6(4):e1000833.
Duron O, Bouchon D, Boutin S, Bellamy L, Zhou L, Engelstadter J, Hurst GD. The diversity of reproductive parasites among arthropods: Wolbachia do not walk alone. BMC Biol. 2008;6(1):27.
Hartelt K, Oehme R, Frank H, Brockmann SO, Hassler D, Kimmig P. Pathogens and symbionts in ticks: prevalence of Anaplasma phagocytophilum (Ehrlichia sp.), Wolbachia sp., Rickettsia sp., and Babesia sp. in southern Germany. Int J Med Microbiol. 2004;293(Suppl 37):86–92.
Plantard O, Bouju-Albert A, Malard MA, Hermouet A, Capron G, Verheyden H. Detection of Wolbachia in the tick Ixodes ricinus is due to the presence of the hymenoptera endoparasitoid Ixodiphagus hookeri. PLoS One. 2012;7(1):e30692.
Benson MJ, Gawronski JD, Eveleigh DE, Benson DR. Intracellular symbionts and other bacteria associated with deer ticks (Ixodes scapularis) from Nantucket and Wellfleet, Cape Cod, Massachusetts. Appl Environ Microbiol. 2004;70(1):616–20.
Menchaca AC, Visi DK, Strey OF, Teel PD, Kalinowski K, Allen MS, Williamson PC. Preliminary assessment of microbiome changes following blood-feeding and survivorship in the Amblyomma americanum nymph-to-adult transition using semiconductor sequencing. PLoS One. 2013;8(6):e67129.
WCt B, Piesman J. Phylogeny of hard- and soft-tick taxa (Acari: Ixodida) based on mitochondrial 16S rDNA sequences. Proc Natl Acad Sci U S A. 1994;91(21):10034–8.
16S Illumina amplicon protocol [http://press.igsb.anl.gov/earthmicrobiome/protocols-and-standards/16s/]. Accessed 15 Apr 2017.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–41.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35(21):7188–96.
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–200.
Schloss PD, Westcott SL. Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Appl Environ Microbiol. 2011;77(10):3219–26.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069–72.
McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6(3):610–8.
Hill TC, Walsh KA, Harris JA, Moffett BF. Using ecological diversity measures with bacterial communities. FEMS Microbiol Ecol. 2003;43(1):1–11.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57(1):289–300.
Benjamini Y. Discovering the false discovery rate. J R Stat Soc. 2010;72(4):405–16.
Weiss S, Xu ZZ, Peddada S, Amir A, Bittinger K, Gonzalez A, Lozupone C, Zaneveld JR, Vázquez-Baeza Y, Birmingham A, et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017;5(1):27.
Acknowledgments
We thank Elizabeth Mitchell for laboratory and field assistance and helpful comments during the project, and Dr. Michael LaMontagne of the University of Houston-Clear Lake for assistance with collection of ticks in Cape Cod, MA, and helpful comments on the manuscript. We gratefully acknowledge Dr. Pete Teel (Texas A&M University) for many helpful discussions and the Texas DSHS Zoonosis Control Branch for providing tick samples from dogs.
Funding
Funding for this work was provided by the State of Texas and the University of North Texas Health Science Center. The funding entities had no role in the study design, data collection, interpretation, or publication of this study, and the results and conclusions expressed herein are solely those of the authors.
Author information
Authors and Affiliations
Contributions
ST and MSA conceived and designed the study. MSA supervised the study. ST prepared samples for sequencing, performed PCR and sequencing experiments. YZ processed the data using mothur. ST, YZ, and MSA analyzed data. ST performed statistical analyses with assistance from YZ. ST drafted the initial manuscript, and all authors provided feedback and insights into the manuscript. ST revised the manuscript, and all authors read, edited, and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval
Currently, there is no Institutional Animal Care and Use Committee (IACUC) protocol for handling, or processing of ticks. Tick samples collected from dogs were submitted by owners to the Zoonosis Control Branch of the Texas Department of State Health Services (DSHS), and subsequently forwarded to our lab as de-identified samples. No consent was obtained.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figure S1. Rarefaction curves of the number of OTUs observed in male and female I. scapularis. Figure S2. Relative abundance of bacterial phyla in I. scapularis ticks from Texas and Massachusetts, USA. Figure S3. Bacterial richness (ACE and Chao1 estimators) in I. scapularis ticks. Figure S4. PCoA plot of weighted UniFrac distance metrics in male and female I. scapularis ticks collected from Texas and Massachusetts, USA. Figure S5. Unweighted PCoA plot of Borrelia-positive and Borrelia-negative I. scapularis males and females collected from Massachusetts, USA. Figure S6. Comparison of the bacterial microbiomes in colony-reared and field-collected I. scapularis ticks. (DOCX 1633 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Thapa, S., Zhang, Y. & Allen, M.S. Bacterial microbiomes of Ixodes scapularis ticks collected from Massachusetts and Texas, USA. BMC Microbiol 19, 138 (2019). https://doi.org/10.1186/s12866-019-1514-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-019-1514-7