
Abstract
Background
Ornithobacterium (O.) rhinotracheale is an emerging bacterial pathogen in poultry and not fully understood to date. Because of its importance particularly for the global turkey meat industry, reliable diagnostic and characterization methods are needed for early treatment and in future for better vaccine production. The host range of birds infected by O. rhinotracheale or carrying the bacterium in their respiratory tract has constantly increased raising important epidemiological and taxonomic questions for a better understanding of its diversity, ecology and transmission cycles. The purpose of this study was to introduce partial rpoB gene sequencing for O. rhinotracheale into routine diagnostics to differentiate strains isolated from poultry and more diverse avian hosts (i.e., birds of prey, corvids and pigeons) and to compare phylogenetic relationships with results from 16S rRNA gene analysis and multilocus sequence typing (MLST).
Results
Partial 16S rRNA gene analysis revealed a high level of homogeneity among the 65 investigated O. rhinotracheale sequences with similarity values ranging from 98.6 to 100% between sequences from non-galliform and poultry species. The corresponding rpoB gene sequences were heterogeneous and ranged in their similarity values from 85.1 to 100%. The structure of the rpoB tree was in strong correlation with previous MLST results revealing three main clusters A (poultry and birds of prey), B (poultry, birds of prey and corvids) and C (pigeons), which were clearly separated from each other.
Conclusions
By using partial sequences from a single gene, the rpoB gene analysis is in good agreement with MLST results with a slight decrease in resolution to distinguish more similar strains. The present results provide strong evidence that traditional phenotypic and genetic methods may not properly represent the heterogeneous group of bacteria classified as O. rhinotracheale. From housekeeping gene analyses, it is very likely that the genus Ornithobacterium includes additional species and partial rpoB gene sequencing can be recommended as fast, cost-effective and readily available method to identify strains and differentiate between O. rhinotracheale and Ornithobacterium-like bacteria.
Similar content being viewed by others

Background
Ornithobacterium (O.) rhinotracheale is a relatively novel, emerging bacterial pathogen in turkeys and chickens that causes high economic losses to the commercial poultry production. It was first recognized and taxonomically classified in the early 1990’s [1, 2] and has been isolated from poultry flocks worldwide [3, 4].
The genus Ornithobacterium belongs to the family of the Flavobacteriaceae [5], which - besides others - also includes the genus Riemerella with R. anatipestifer [6, 7] and the genus Coenonia with C. anatina [8]. Both bacterial species are also important poultry pathogens mainly of domestic ducks and geese. Besides a new species proposed as Candidatus Ornithobacterium hominis sp. nov. [9], O. rhinotracheale is the only bacterial species described within the genus Ornithobacterium but not fully understood to date. Because of its veterinary importance particularly for the global turkey meat industry [10] the need for reliable diagnostic and characterization methods is obvious for early treatment. Its cultural characteristics and fastidious requirements (i.e., small colony size, slow growth, enriched media and capnophilic incubation), however, may challenge bacterial isolations and reduce the detection rates [11, 12]. Therefore, molecular detection of O. rhinotracheale DNA from tissues or swabs targeting the 16S rRNA gene with specific primers is frequently used in routine diagnostics, but 16S sequences often lack the resolution to capture heterogeneity among the strains [13,14,15].
Over the past few decades, the host range of birds infected by O. rhinotracheale or carrying the bacterium in their respiratory tract has constantly increased [16]. The presence of O. rhinotracheale in apparently healthy, captive and free-ranging non-galliform species raises important epidemiological and taxonomic questions for a better understanding of its diversity, ecology and transmission cycles. Multilocus sequence typing (MLST) recently established by Thieme et al. [3, 16] not only revealed specific phylogenetic relationships in non-galliform birds such as pigeons and birds of prey, but also identified few strains from turkeys and chickens that clearly differed from the main poultry cluster. The aim of the present study was to introduce partial rpoB gene sequencing for O. rhinotracheale into routine diagnostics to differentiate strains isolated from poultry and more diverse avian hosts and to compare the results with those from 16S rRNA gene and MLST analyses. The rpoB gene has been proved very useful and powerful for bacterial identification and phylogenetic studies [17,18,19].
Methods
Bacterial strains
Sixty-five strains previously identified as O. rhinotracheale were used for genetic analyses (Table 1), including 51 strains that were already investigated by MLST and represent the 31 sequence types (ST) as described by Thieme et al. [3, 16], and six additional strains isolated from turkeys in Portugal (present study). Eight strains were identified only genetically from swab samples of different birds of prey and corvid species [20]. They were included in the rpoB gene analysis to investigate mixed bacterial DNA samples, when O. rhinotracheale cannot be obtained from bacterial cultures because of its fastidious growth requirements and concomitant fast-growing bacteria.
16S rRNA and rpoB gene analyses
Amplification of the O. rhinotracheale specific 16S rRNA gene fragment (784 bp) was performed according to Numee et al. [14] with primer sequences described by van Empel and Hafez [11]. The rpoB gene fragment (538 bp) was amplified using 1 μM of primers rpoBFla-f (5‘-TCAATTCGTTCTTTGGAAC- 3′) and rpoBFla-r (5‘-GCATCATGTTAGATCCCAT-3′) with cycling conditions as follows: 3 min denaturation at 94 °C, followed by 30 cycles at 94 °C for 30 s, 54 °C for 30 s and 72 °C for 45 s, and a final extension step at 72 °C for 5 min. rpoB primers were designed based on published genomic sequences within the family Flavobacteriaceae (including Riemerella strains NCTC 11014T and LMG 11607T, and O. rhinotracheale strain DSM 15997T) as described by Christensen and Bisgaard [19]. 16S rRNA and rpoB PCR products were gel purified (MinElute Gel Extraction Kit, Qiagen, Hilden, Germany) and Sanger sequenced in both directions at LGC Genomics, Berlin, Germany, using PeakTrace™ Basecaller and the PHRED 20 quality score. The identity of bacterial species was confirmed using BLAST search against the GenBank database.
Phylogenetic analyses of partial 16S rRNA (632 bp) and rpoB (538 bp) gene sequences were performed with MEGA6 [21] by using the Maximum Likelihood method based on the Jukes-Cantor model [22]. As several O. rhinotracheale strains were identical in their partial 16S rRNA and rpoB gene sequences, phylogenetic trees were built for 47 out of 65 strains and Riemerella anatipestifer DSM 15868T (accession number NC_017045) was included as outgroup. The O. rhinotracheale type strain DSM 15997T (accession number NC_018016) was included for sequence similarity comparisons (Table 2).
Results and discussion
Within the 16S rRNA gene analysis, the 65 O. rhinotracheale specific partial sequences showed a high level of homogeneity with similarity values ranging from 98.6 and 98.7% (ten strains from pigeons, GV37 and GV38) to 100% (strains from turkey or chicken and GV1) between non-galliform and poultry species. The structure of the phylogenetic tree (Fig. 1), however, was largely comparable to MLST results [16]. The majority of O. rhinotracheale strains were arranged in one cluster and separated from two subsets of three strains each isolated from poultry (family Phasianidae) or birds of prey (family Accipitridae), respectively. One of the O. rhinotracheale serotype reference strains (RefF) belonged to the aforementioned subset of the three strains from poultry (including GB 978/14/1 and GB 137/10/2) and formed a separate cluster in previous studies too [14, 15].

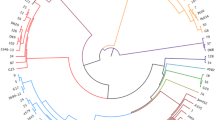
Phylogenetic tree based on partial 16S rRNA gene sequences (632 bp) and constructed in MEGA6 [21] by using the Maximum Likelihood method based on the Jukes-Cantor model [22]. The tree was built with 47 out of 65 O. rhinotracheale sequences (remaining identical sequences are indicated in grey) and Riemerella anatipestifer DSM 15868T (NC_017045) as outgroup. GenBank accession numbers are given in brackets. The percentage of replicate trees (> 50%) in which the associated taxa clustered together in the bootstrap analysis (100 replicates) is shown next to the branches. Initial tree(s) for the heuristic search were obtained by applying the Neighbor-Joining method to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site
The main 16S cluster was split into two subclusters with 47 strains collected from poultry, birds of prey and corvids, which are more closely related to each other and comprise a high diversity of bird families and species. In contrast, the second subcluster only involved 11 strains from pigeons and one strain from a white-tailed eagle (GV37, family Accipitridae). The latter grouped together with two strains isolated from feral pigeons, which were separated from strains collected from pigeons of a pigeon loft [16].
The 65 partial rpoB gene sequences were heterogeneous and ranged in their similarity values from 85.1 to 100%. The structure of the phylogenetic tree was in strong correlation with MLST results [16]. Three distinct clusters A (n = 6; poultry and birds of prey), B (n = 47; poultry, birds of prey and corvids) and C (n = 11; only pigeon) were identified (Fig. 2) that corresponded well to the MLST clusters [16]. Strain GV37 formed a distinct lineage and only shared 86.2 to 87.4% sequence similarity with strains of cluster A, 86.8 to 87.5% similarity with strains of cluster B and 85.1 to 85.5% similarity with strains of cluster C (Table 2). Within cluster A, three strains from turkeys and chicken (98.7 to 100% similarity) were clearly separated from three strains collected from birds of prey (99.4 to 100% similarity). Both groups shared only 94.2 to 94.8% sequence similarity. In contrast, rpoB gene sequence similarity values among strains from cluster B or cluster C ranged from 98.5 and 98.0% to 100%, respectively.

Phylogenetic tree based on partial rpoB gene sequences (538 bp) and constructed in MEGA6 [21] by using the Maximum Likelihood method based on the Jukes-Cantor model [22]. The tree was built with 47 out of 65 O. rhinotracheale sequences (remaining identical sequences are indicated in grey) and Riemerella anatipestifer DSM 15868T (NC_017045) as outgroup. GenBank accession numbers are given in brackets. The percentage of replicate trees (> 50%) in which the associated taxa clustered together in the bootstrap analysis (100 replicates) is shown next to the branches. Initial tree(s) for the heuristic search were obtained by applying the Neighbor-Joining method to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site
Sequence analysis of a specific rpoB gene fragment is widely used in addition to the 16S rRNA gene for more reliable bacterial identification and taxonomic classifications at genus and species level [17]. For members of the family Pasteurellaceae, rpoB similarity cut-offs of 85 and 95% were recommended for the description of new genera and species, respectively [23]. For the family Flavobacteriaceae, corresponding similarity cut-offs have not been defined so far [24]. Partial rpoB gene analyses, however, proved to be very useful to reveal clear phylogenetic relationships of Riemerella strains and related members of Flavobacteriaceae [19]. Likewise, O. rhinotracheale strains from this study show strong differences in similarity among their rpoB gene sequences. Strains of cluster A and cluster C as well as strain GV37 were clearly separated from cluster B, a genetically homogenous group that includes the majority of O. rhinotracheale strains and the O. rhinotracheale type strain DSM 15997T. Moreover, comparisons with O. rhinotracheale DSM 15997T or respective with strains from cluster B revealed sequence similarities of ≤90.0% but above 85.0% (Table 2), which would only support bacterial identification at genus level.
Conclusions
By using partial sequences from a single gene, the rpoB gene analysis is in good agreement with MLST results with a slight decrease in resolution to distinguish more similar strains. Eight rpoB gene sequences were received from swab samples (all non-galliform birds) [20] extending the avian host range and phylogenetic relationships investigated by MLST. On a bird family-level, the Columbidae specific cluster C and two different subclusters of three strains from Accipitridae species (namely 165–2/2015, GV8 and GV13) and seven strains from Falconidae species (namely GV9 to GV12, GV55, GV143 and GV149) were seen likewise to MLST [16]. At the same time, however, several O. rhinotracheale strains from different poultry species, birds of prey and corvid species have a close genetic relationship (similarity ≥98.5%) pointing towards the risk of interspecies transmission. Moreover, our results provide strong evidence that traditional phenotypic and genetic methods used for identification may not properly represent the heterogeneous group of bacteria classified as O. rhinotracheale. From housekeeping gene analyses, it is very likely that the genus Ornithobacterium includes additional species and partial rpoB gene sequencing can be recommended as fast, cost-effective and readily available method to identify strains and differentiate between O. rhinotracheale and Ornithobacterium-like bacteria.
Abbreviations
- MLST:
-
Multilocus sequence typing
- PCR:
-
Polymerase chain reaction
- ST:
-
Sequence type
References
Hafez HM, Kruse W, Emele J, Sting R. Eine Atemwegsinfektion bei Mastputen durch Pasteurella-ähnliche Erreger: Klinik, Diagnostik und Therapie. In: Proceedings of the international conference on poultry diseases. Potsdam: German Veterinary Medical Society (DVG); 1993. p. 105–12.
Vandamme P, Segers P, Vancanneyt M, van Hove K, Mutters R, Hommez J, et al. Ornithobacterium rhinotracheale gen. Nov., sp. nov., isolated from the avian respiratory tract. Int J Syst Bacteriol. 1994;44:24–37.
Thieme S, Mühldorfer K, Gad W, Lüschow D, Hafez MH. Molecular characterization of the recently emerged poultry pathogen Ornithobacterium rhinotracheale by multilocus sequence typing. PLoS One. 2016;11:e0148158.
Ha HJ, Christensen N, Humphrey S, Haydon T, Bernardi G, Rawdon T. The first detection of Ornithobacterium rhinotracheale in New Zealand. Avian Dis. 2016;60:856–9.
Hafez HM, Vandamme P, Ornithobacterium Vandamme GXXVI. Segers, Vancanneyt, Van Hove, Mutters, Hommez, Dewhirst, Paster, Kersters, Falsen, Devriese, Bisgaard, Hinz and Mannheim 1994, 35VP. In: Krieg NR, Stanley JT, Brown DR, Hedlund BJ, Paster NL, et al., editors. Bergey’s manual of systematic bacteriology. 2nd ed. New York: Springer; 2011. p. 250–314.
Segers P, Mannheim W, Vancanneyt M, De Brandt K, Hinz KH, Kersters K, et al. Riemerella anatipestifer gen. Nov., comb. nov., the causative agent of septicaemia anserum exsudativa, and its phylogentic affiliation within the Flavobacterium-Cytophaga rRNA homology group. Int J Syst Evol Microbiol. 1993;43:768–76.
Rubbenstroth D, Ryll M, Hotzel H, Christensen H, Knobloch JK-M, Rautenschlein S, et al. Description of Riemerella columbipharyngitis sp. nov., isolated from the pharynx of healthy domestic pigeons (Columba livia f. domestica), and emended descriptions of the genus Riemerella, Riemerella anatipestifer and Riemerella columbina. Int J Syst Evol Microbiol. 2013;63:280–7.
Vandamme P, Vancanneyt M, Segers P, Ryll M, Köhler B, Ludwig W, et al. Coenonia anatina gen. Nov., sp. nov., a novel bacterium associated with respiratory disease in ducks and geese. Int J Syst Bacteriol. 1999;49:867–74.
Salter SJ, Scott P, Page AJ, Tracey A, de Goffau MC, Ochoa-Montano B, et al. Candidatus Ornithobacterium hominis sp. nov.: insights gained from draft genomes obtained from nasopharyngeal swabs. bioRxiv. preprint. 2018. https://doi.org/10.1101/326074.
Sprenger SJ, Halvorson DA, Shaw DA, Nagaraja KV. Ornithobacterium rhinotracheale infection in turkeys: immunoprophylaxis studies. Avian Dis. 2000;44:549–55.
Van Empel PCM, Hafez MH. Ornithobacterium rhinotracheale: a review. Avian Pathol. 1999;28:217–27.
Zahra M, Ferreri M, Alkasir R, Yin J, Han B, Isolation SJ. Characterization of small-colony variants of Ornithobacterium rhinotracheale. J Appl Microbiol. 2013;51:3228–36.
Amonsin A, Wellehan JFX, Li L-L, Vandamme P, Lindeman C, Edman M, et al. Molecular epidemiology of Ornithobacterium rhinotracheale. J Clin Microbiol. 1997;35:2894–8.
Numee S, Hauck R, Hafez HM. Detection and typing of Ornithobacterium rhinotracheale from German poultry flocks. Avian Dis. 2012;56:654–8.
Montes de Oca-Jimenez R, Vega-Sanchez V, Morales-Erasto V, Salgado-Miranda C, Blackall PJ, Soriano-Vargas E. Phylogenetic relationship of Ornithobacterium rhinotracheale strains. J Vet Med Sci. 2018;80:869–73.
Thieme S, Hafez MH, Gutzer S, Warkentin N, Lüschow D, Mühldorfer K. Multilocus sequence typing of Ornithobacterium rhinotracheale isolated from pigeons and birds of prey revealed new insights into its population structure. Vet Anim Sci. 2016b;1:15–20.
Adékambi T, Drancourt M, Raoult D. The rpoB gene as a tool for clinical microbiologists. Trends Microbiol. 2009;17:37–45.
Korczak B, Christensen H, Emler S, Frey J, Kuhnert P. Phylogeny of the family Pasteurellaceae based on rpoB sequences. Int J Syst Evol Microbiol. 2004;54:1939.
Christensen H, Bisgaard M. Phylogenetic relationships of Riemerella anatipestifer serovars and related taxa and an evaluation of specific PCR tests reported for R. anatipestifer. J Appl Microbiol. 2009;108:1612–9.
Gutzer S, Lüschow D, Hafez HM. Wild birds as source of Ornithobacterium rhinotracheale infections. In: Hafez HM, editor. Proceedings of the 7th “Hafez” international symposium on Turkey production. Meeting of the working group 10 (Turkey) of World’s poultry science association. Berlin: Mensch & Buch Verlag; 2013. p. 193–5.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol and Evol. 2013;30:2725–9.
Jukes TH, Cantor CR. Evolution of protein molecules. In: Munro HN, editor. Mammalian Protein Metabolism. New York: Academic Press; 1969. p. 21–132.
Christensen H, Kuhnert P, Busse H-J, Frederiksen W, Bisgaard M. Proposed minimal standrads for the description of genera, species and subspecies of the Pasteurellaceae. Int J Syst Evol Microbiol. 2007;57:166–78.
Bernardet JF, Nakagawa Y, Holmes B. Subcommittee on the taxonomy of Flavobacterium and Cytophaga-like bacteria of the international committee on systematics of prokaryotes. Proposed minimal standards for describing new taxa of the family Flavobacteriaceae and emended description of the family. Int J Syst Evol Microbiol. 2002;52:1049–70.
Acknowledgements
The authors would like to thank Elke Dyrks and Gabriele Grotehenn for their excellent technical assistance, and Dr. Ana Martins from ALS Controlvet Portugal and Dr. Rita Silva from Desenvolvimento e Inovacão industrial Lda (DIN) for providing six O. rhinotracheale strains from turkeys in Portugal.
Funding
IMBV was funded by a postdoctoral Honors Fellowship 2013–2 from the Dahlem Research School (DRS), Freie Universität Berlin, Germany. The funding body had no role in any aspect of this study.
The publication of this article was funded in part by the Open Access Fund of the Leibniz Association.
Availability of data and materials
Newly obtained sequences of the different genes from the 47 representative strains used for phylogenetic analyses have been deposited in GenBank (www.ncbi.nlm.nih.gov/genbank) and are available under their accession numbers (16S rRNA accession numbers: MH746672 to MH746712; rpoB accession numbers: MH746625 to MH746671). 16S rRNA gene sequences of strains RefA to RefH, RefO and RefN are available under accession numbers KY809786 to KY809793 [15], U87102 and U87103 [13], respectively.
Author information
Authors and Affiliations
Contributions
IMBV and KM conceived and designed the experiments; IMBV, SG, DL performed the experiments; IMBV, SG, DL, HMH, KM contributed isolates/reagents/materials/analysis tools; KM analyzed the data and wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Samples and bacterial isolates used in the present study were obtained for diagnostic purposes and do not require ethical approval or owner consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Veiga, I.M.B., Lüschow, D., Gutzer, S. et al. Phylogenetic relationship of Ornithobacterium rhinotracheale isolated from poultry and diverse avian hosts based on 16S rRNA and rpoB gene analyses. BMC Microbiol 19, 31 (2019). https://doi.org/10.1186/s12866-019-1395-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-019-1395-9