
Abstract
Background
For Aspergillus niger, a broad set of auxotrophic and dominant resistance markers is available. However, only few offer targeted modification of a gene of interest into or at a genomic locus of choice, which hampers functional genomics studies. We thus aimed to extend the available set by generating a histidine auxotrophic strain with a characterized hisB locus for targeted gene integration and deletion in A. niger.
Results
A histidine-auxotrophic strain was established via disruption of the A. niger hisB gene by using the counterselectable pyrG marker. After curing, a hisB - , pyrG - strain was obtained, which served as recipient strain for further studies. We show here that both hisB orthologs from A. nidulans and A. niger can be used to reestablish histidine prototrophy in this recipient strain. Whereas the hisB gene from A. nidulans was suitable for efficient gene targeting at different loci in A. niger, the hisB gene from A. niger allowed efficient integration of a Tet-on driven luciferase reporter construct at the endogenous non-functional hisB locus. Subsequent analysis of the luciferase activity revealed that the hisB locus is tight under non-inducing conditions and allows even higher luciferase expression levels compared to the pyrG integration locus.
Conclusion
Taken together, we provide here an alternative selection marker for A. niger, hisB, which allows efficient homologous integration rates as well as high expression levels which compare favorably to the well-established pyrG selection marker.
Similar content being viewed by others

Background
The filamentous fungus Aspergillus niger is an industrially exploited cell factory with a broad product portfolio including primary metabolites, proteins and enzymes [1]. Recent data proved that A. niger can also serve as a suitable host for secondary metabolite production [2, 3]. Additionaly, A. niger is a model system used to study fundamental molecular and cellular processes. Various selection systems are available for transformation of A. niger, including nutritional (pyrG, trpC, amdS, niaD, sC, agaA and argB) and antibiotic resistance (hph, ble) markers [4–11]. Recently this set was expanded by two new nutritional markers (nicB and adeA) which can be used for gene deletion in A. niger [12]. However, in order to study the function and interplay of several genes, or to construct/re-engineer a complete metabolic pathway in A. niger, it is of advantage having a range of selection markers at hand to choose the best one suited for a given approach. The number of nutritional markers for A. niger is limited to seven, as recently published [12] and although marker recycling using the Cre/loxP system has been established for Aspergilli [13, 14], they often suffer from poor recombination events ranging from 5 to 20%. So far, within the set of nutritional selection markers available for A. niger, only the pyrG and agaA loci [4, 15, 16] meet the need of a well characterized locus for efficient homologous integration of single copy expression cassettes. In fungi, a transcriptionally active, non-protein encoding locus that is targeted by exogenous expression cassettes at high frequency has been an important molecular technique underlying transformation, mutant complementation, and functional genomic approaches to study gene function [17]. Even the highly efficient CRISPR/Cas system which has been applied recently for filamentous fungi [17–21], depends on well characterized loci for genomic integration of the genes of interest.
In order to establish an alternative auxotrophic selection marker for A. niger, which can also be used for both gene targeting or insertion of an expression cassette into a well characterised locus, we chose the histidine biosynthesis pathway as a target. This pathway was intensively studied in Salmonella typhimurium, Escherichia coli and Corynebacterium glutamicum (for reviews see [22, 23]) and in A. nidulans [24]. It generates histidine in ten reaction steps catalysed by seven enzymes in a branched pathway out of phosphoribosyl pyrophosphate supplied via the pentose phosphate pathway. As it was shown that deletion of hisB in A. nidulans and A. fumigatus results in histidine auxotrophy [25, 26], we selected hisB, which catalyses the sixth step in the histidine biosynthesis pathway and disrupted it via a direct targeting approach in A. niger. Subsequent integration of the well-established Tet-on system [27] using luciferase as reporter gene enabled us to evaluate gene expression characteristics at the hisB locus in comparison to the widely used pyrG locus. We could furthermore demonstrate that genome editing using the hisB orthologue of A. nidulans is feasable.
Methods
Strains, growth conditions and molecular techniques
A. niger strains used in this study are listed in Table 1. The strains were grown at 30°C in minimal medium (MM) [28] or complete medium (CM), consisting of MM supplemented with 1% yeast extract and 0.5% casamino acids. 10 mM uridine or 10 mM histidine were added to the medium when required.
To obtain pyrG - strains via counterselection, 2 x 107 spores were plated on MM plates containing 75 mg/ml 5-Fluoroorotic acid (FOA), 10 mM uridine, 10 mM proline and 10 mM histidine. Plates were incubated at 30°C for 1-2 weeks until single colonies were visible. FOA-resistant mutants were purified on MM + FOA plates once and tested for their uridine auxothrophy on MM plates containing 10 mM histidine or 10 mM histidine and 10 mM uridine, respectively.
All molecular techniques were performed according to standard procedures [29] and the transformation, genomic DNA extraction and Southern blot were performed as described elsewhere [30].
Construction of a hisB disruption vector
To construct a hisB disruption plasmid we used an approach which was published recently [31]. In brief, 533 bp and 500 bp sequences of the hisB coding and 3’ sequence were amplified via PCR using primers listed in Additional file 1: Table S1. Both fragments were inserted via Gibson cloning into the BsrGI linearized plasmid pAO4-13 carrying the A. oryzae pyrG gene [32] giving rise to the counter-selectable hisB disruption plasmid pMF22.1.
Construction of an olvA deletion cassette
The plasmid pAW34 ([33], kindly provided by Arthur Ram) containing the AopyrG gene flanked by the 5’ and 3’ region of olvA was used as a backbone. The A. nidulans hisB (AN6536) gene was amplified using primers listed in Additional file 1: Table S1 and cloned into the XhoI/HindIII linearized pAW34 via Gibson cloning giving rise to plasmid pSE1.6.
Construction of luciferase reporter constructs
The A. niger pyrG* gene within Tet-on plasmids pVG2.2 (containing the empty Tet-on system, [27]) and pVG4.1 (containing a codon optimized version of the luciferase mluc under control of the Tet-on system, [27]) was replaced by a 2291 bp fragment amplified by fusion PCR, containing the full length hisB gene without a functional start codon flanked by 5’ and 3’ region of the hisB gene, giving rise to pTG1.2 and pTG2.15, respectively (Additional file 2: Figure S2).
Measurement of the luciferase activity
Ninety six well microtiter plate assays were performed as described earlier [27] with slight modifications. In brief, 5 x 104 spores were inoculated in 200 μl CM medium [30] supplemented 1.4 mM luciferin and 0, 5 (A) or 20 μg/ml (B) doxycycline in a microtiter plate and incubated at 30°C in a Victor3 (Perkin Elmer). OD and luminescence were measured every 30 min.
Results and discussion
In order to construct an alternative auxotrophic marker for A. niger, we choose the A. niger orthologue of the well characterised S. cerevisiae selection marker HIS3. The gene codes for an imidazole-glycerol-phosphate dehydratase which catalyses the sixth step of the histidine biosynthesis in S. cerevisiae, by specifically dehydrating imidazole-glycerol-3-phosphate, producing imidazole-acetole-phosphate (Fig. 1). Proteins Blasts of the HIS3 sequence against translated A. niger ORFs [34] revealed a single orthologue (An15g00610) for HIS3 in the genome of A. niger with a protein sequence identity of 57.6%, which was termed hisB. It is well known that gene expression depends on both position effects and on the availability of transcription factors [35]. We thus analysed whether gene expression levels of hisB and the widely used pyrG are comparable by scrutinzing an in-house database, which comprises genome-wide expression profiles of A. niger from 155 different cultivation conditions [36]. As depicted in Fig. 2, hisB is expressed under all conditions covered in the transcriptomic database in a range comparable to the pyrG gene (Fig. 2).


Expression profile of the A. niger pyrG and hisB genes. Our in-house transcriptomic database for A. niger covering 155 growth conditions was analyzed for hisB and pyrG expression levels. Gene expression levels were normalized against actin (actA) expression during the exponential growth phase of A. niger in maltose-based bioreactor cultivation. For improved visualization, data were assorted ascending for the pyrG expression level
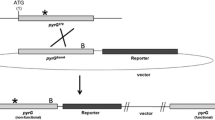
We used a gene disruption approach published earlier [31] to inactivate the hisB gene. A part of the open reading frame of the hisB gene was integrated together with the 3’ region of hisB into pOA4-13 which contains the Aspergillus oryzae pyrG gene as selection marker [32] giving rise to plasmid pMF22.1. Subsequent transformation of the vector into a pyrG - strain lacking a functional non-homologues-end-joining-pathway (NHEJ) [30], resulted in a single recombination event whereby the hisB gene became disrupted (Fig. 3). Counterselection on plates containing fluoroorotic acid (FOA) (see Methods) allowed recombination events between the duplicated parts of the hisB gene, thereby restoring gene functionality (Fig. 3). Alternatively, recombination between the 3’ regions resulted in a nonfunctional truncated hisB (hisB*) (Fig. 3). In total, 62 primary transformants were obtained from which 10 were randomly selected and tested for histidine auxotrophy. One of four transformants unable to grow on minimal medium (MM) lacking histidine (MF40.6) was counterselected as described in Material and Methods, resulting in 30 pyrG cured strains, which were purified and tested for their histidine auxotrophy. Only 10 strains out of 30 were found to be auxotrophic for histidine (Additional file 3: Figure S3, A), although the duplicated parts of the hisB gene and the 3’ regions did only differ in 33 bp in length. From the set of strains which were screened for histidine and uridine auxotrophy, strain MF41.3 (hisB - , pyrG -) was chosen for further analysis. The auxotrophy was stable under non-selective conditions, which was proven by several rounds of cultivation on complete medium supplemented with 10 mM histidine and uridine. After three rounds, MF41.3 still remained unable to grow on MM lacking uridine or histidine (data not shown), which is in good agreement with earlier work published for A. nidulans and A. fumigatus, which showed that deletion of hisB leads to auxotrophic strains, which can be rescued by the addition of 1-5 mM histidine to the medium, thereby restoring wildtype-like growth [25, 26]. The truncated hisB* locus in MF41.3 was subsequently sequenced and compared to the hisB locus of MA169.4, showing the expected DNA modifications (Fig. 3 and Additional file 4: Figure S4). In order to exclude polygenetic effects in subsequent phenotypic analysis caused by deletion of two essential auxotrophic gene markers, we restored the pyrG gene in MF41.3 by transforming this strain with linearized plasmid pAB4.1 [7], containing the full length A. niger pyrG gene, giving strain MF42.2. This strain was able to grow on plates lacking uridine and the correct integration of the linearized fragment was confirmed via Southern blot analysis (data not shown).

Schematic overview of the hisB disruption approach in A. niger and subsequent counterselection. The plasmid pMF22.1 was constructed via amplifying parts of the hisB gene and its 3’ region (numbers above the fragments indicate the DNA position from the start codon). Both fragments were inserted via Gibson cloning into the BsrGI linearized plasmid pAO4-13 carrying the Aspergillus oryzae pyrG [32] giving rise to the plasmid pMF22.1. The plasmid was transformed into the strain MA169.4 [15] thereby disrupting the hisB locus giving rise to MF40.6. Counterselection may either lead to a genomic rearrangement, thereby eliminating the plasmid via recombination in its hisB locus or the 5’ region, creating a truncated hisB* (MF41.3) or wildtype hisB loci, respectively
To evaluate whether the A. nidulans hisB gene (AN6536) could be used as selection marker to complement the histidine auxotrophy, the genomic and protein sequences were compared using BlastN and BlastP, revealing a high conservation (90.2%) on the protein level, whereas the nucleotide sequence conservation was considerably lower (74.1%), which was thought to be crucial for the correct integration of the deletion cassette into the region of choice instead of complementing A. niger hisB gene at its endogenous locus. For an easy read-out of the transformation and gene replacement efficiency, the olvA gene was chosen. This gene encodes a hydrolase involved in DHN-melanin synthesis in A. niger and its deletion results in an incomplete melanin biosynthesis and thus green spore formation [33].
In doing so, plasmids pSE1.6 (olvA::AnidhisB) and pAW34 (olvA::AopyrG) were linearized and transformed into MF42.2 (hisB -, pyrG +) or MA169.4 (pyrG -), respectively. Analysis of total transformants and spore color analysis revealed that deletion of olvA with the A. nidulans hisB gene leads to a deletion efficiency comparable to the pyrG marker (Table 2). Strains carrying the A. nidulans hisB gene at either the olvA locus or ectopically integrated into the genome were selected, purified, spotted on CM and compared to the olvA::AopyrG strain AW8.4 and MA169.4 with an ectopically integrated copy of pAW34. No macroscopic differences were detected between mutant or wild type strains except spore color formation (Additional file 5: Figure S5).
A luciferase based reporter system was used to test whether the truncated hisB* locus is suitable to allow gene expression of a gene of interest. In doing so, the luciferase reporter constructs pTG1.2 (containing the empty Tet-on system) and pTG2.15 (containing a codon optimized version of the luciferase mluc under control of the Tet-on system [27, 37]) were constructed. Both plasmids should integrate into the truncated hisB* locus via a single recombination event, because the A. niger hisB gene was used as selection marker, thereby restoring its functionality (Additional file 2: Figure S2). Transformation of MF42.2 resulted in 53 primary transformants for pTG1.2 and 22 primary transformants for pTG2.15, respectively. For both strains, 10 out of 20 transformants were proven by diagnostic PCR to harbor the expression constructs at hisB*, which was further confirmed via Southern analysis (Additional file 2: Figure S2). Transformants TG1.14 (carrying a single copy of Tet-on::hisB), TG2.3 (carrying a single copy of Tet-on::mluc::hisB) were selected and their luciferase activity determined and compared to the previously published strains VG7.2 and VG8.27 [21], harboring the Tet-on::pyrG* or Tet-on-mluc::pyrG* constructs at the pyrG locus, respectively (Fig. 4).

Luciferase activity assay of strains containing the Tet-on::mluc construct integrated into the hisB* or pyrG loci. The luciferase activity assay was performed as described earlier [27] using 5 μg/ml (panel a) or 20 μg/ml (panel b) doxycycline for induction. Strains TG1.14 and VG7.1 are negative-control strains (not expressing luciferase but harboring the empty Tet-on constructs), while strains TG2.3 and VG8.27 carry the Tet-on::mluc constructs integrated at the hisB or pyrG locus, respectively. Mean values of triplicate experiments are shown
While the luminescence activity of the strains carrying the reporter construct integrated into the pyrG or hisB locus are comparable (induction with 5 μg/ml doxycycline), the values of the TG2.3 are 3 times higher when using 20 μg/ml doxycycline, possibly reflecting a higher transcriptional activity at the hisB locus under the conditions used. It is notable that neither the vector control (TG1.14, 20 μg/ml doxycycline) nor the non-induced luciferase constructs (TG2.3 and VG8.27) showed any luciferase activity during the experiment, clearly demonstrating that the system is tight at the hisB locus in the absence of the inducer.
Conclusion
In summary, we report here a straight forward approach to rationally generate auxotrophic markers in the filamentous fungus A. niger which was employed to create a histidine auxotrophic strain which can be used as a recipient isolate for endogenous deletion of genes using the A. nidulans orthologue hisB. In addition, we characterized the hisB* locus for functionality to integrate expression constructs, which revealed an expression level for the luciferase reporter with a higher performance and tighter characteristics under non-induced conditions compared to the well-used pyrG locus. The tools described here significantly increase the tractability of A. niger at the molecular level and suggest hisB could be used for similar applications in other model or pathogenic filamentous fungi.
References
Meyer V, Fiedler M, Nitsche B, King R. The cell factory Aspergillus enters the big data era: opportunities and challenges for optimising product formation. Adv Biochem Eng Biotechnol. 2015;149:91–132.
Richter L, Wanka F, Boecker S, Storm D, Kurt T, Vural Ö, Süßmuth R, Meyer V. Engineering of Aspergillus niger for the production of secondary metabolites. Fungal Biol Biotechnol. 2014;1:4.
Süssmuth R, Zobel S, Boecker S, Kulke D, Heimbach D, Meyer V. Reprogramming the biosynthesis of cyclodepsipeptide synthetases rendering new to nature enniatins and beauvericins. Chembiochem. 2015;17:283–7.
Dave K, Ahuja M, Jayashri TN, Sirola RB, Punekar NS. A novel selectable marker based on Aspergillus niger arginase expression. Enzyme Microb Technol. 2012;51:53–8.
Unkles SE, Campbell EI, Carrez D, Grieve C, Contreras R, Fiers W, Van den Hondel CA, Kinghorn JR. Transformation of Aspergillus niger with the homologous nitrate reductase gene. Gene. 1989;78:157–66.
Buxton FP, Gwynne DI, Davies RW. Cloning of a new bidirectionally selectable marker for Aspergillus strains. Gene. 1989;84:329–34.
Van Hartingsveldt W, Mattern IE, van Zeijl CMJ, Pouwels PH, van den Hondel CA. Development of a homologous transformation system for Aspergillus niger based on the pyrG gene. Mol Gen Genet MGG. 1987;206:71–5.
Goosen T, van Engelenburg F, Debets F, Swart K, Bos K, van den Broek H. Tryptophan auxotrophic mutants in Aspergillus niger: inactivation of the trpC gene by cotransformation mutagenesis. Mol Gen Genet. 1989;219:282–8.
Lenouvel F, van de Vondervoort PJI, Visser J. Disruption of the Aspergillus niger argB gene: a tool for transformation. Curr Genet. 2002;41:425–32.
Punt PJ, van den Hondel CA. Transformation of filamentous fungi based on hygromycin B and phleomycin resistance markers. Methods Enzymol. 1992;216:447–57.
Kelly JM, Hynes MJ. Transformation of Aspergillus niger by the amdS gene of Aspergillus nidulans. EMBO J. 1985;4:475–9.
Niu J, Arentshorst M, Seelinger F, Ram AFJ, Ouedraogo JP. A set of isogenic auxotrophic strains for constructing multiple gene deletion mutants and parasexual crossings in Aspergillus niger. Arch Microbiol. 2016;198:861–8.
Forment JV, Ramón D, MacCabe AP. Consecutive gene deletions in Aspergillus nidulans: application of the Cre/loxP system. Curr Genet. 2006;50:217–24.
Krappmann S, Bayram O, Braus GH. Deletion and allelic exchange of the Aspergillus fumigatus veA locus via a novel recyclable marker module. Eukaryot Cell. 2005;4:1298–307.
Carvalho NDSP, Arentshorst M, Kwon MJ, Meyer V, Ram AFJ. Expanding the ku70 toolbox for filamentous fungi: establishment of complementation vectors and recipient strains for advanced gene analyses. Appl Microbiol Biotechnol. 2010;87:1463–73.
Arentshorst M, Lagendijk EL, Ram AF. A new vector for efficient gene targeting to the pyrG locus in Aspergillus niger. Fungal Biol Biotechnol. 2015;2:2.
Cairns TC, Studholme DJ, Talbot NJ, Haynes K. New and improved techniques for the study of pathogenic fungi. Trends Microbiol. 2016;24:35–50.
Krappmann S. CRISPR-Cas9, the new kid on the block of fungal molecular biology. Med Mycol. 2017;55:16–23.
Matsu-ura T, Baek M, Kwon J, Hong C. Efficient gene editing in Neurospora crassa with CRISPR technology. Fungal Biol Biotechnol. 2015;2:4.
Liu R, Chen L, Jiang Y, Zhou Z, Zou G. Efficient genome editing in filamentous fungus Trichoderma reesei using the CRISPR/Cas9 system. Cell Discov. 2015;1:15007.
Nødvig CS, Nielsen JB, Kogle ME, Mortensen UH. A CRISPR-Cas9 system for genetic engineering of filamentous fungi. PLoS ONE. 2015;10:e0133085.
Martin RG, Berberich MA, Ames BN, Davis WW, Goldberger RF, Yourno JD. Metabolism of Amino Acids and Amines Part B. In Methods in Enzymology. Volume 17. Elsevier Inc;1971. p. 3–44. [Methods in Enzymology].
Kulis-Horn RK, Persicke M, Kalinowski J. Histidine biosynthesis, its regulation and biotechnological application in Corynebacterium glutamicum. Microb Biotechnol. 2014;7:5–25.
Berlyn MB. Gene-enzyme relationships in histidine biosynthesis in Aspergillus nidulans. Genetics. 1967;57:561–70.
Busch S, Hoffmann B, Valerius O, Starke K, Düvel K, Braus GH. Regulation of the Aspergillus nidulans hisB gene by histidine starvation. Curr Genet. 2001;38:314–22.
Dietl A-M, Amich J, Leal S, Beckmann N, Binder U, Beilhack A, Pearlman E, Haas H. Histidine biosynthesis plays a crucial role in metal homeostasis and virulence of Aspergillus fumigatus. Virulence. 2016;7:465–76.
Meyer V, Wanka F, van Gent J, Arentshorst M, van den Hondel CAMJJ, Ram AFJ. Fungal gene expression on demand: an inducible, tunable, and metabolism-independent expression system for Aspergillus niger. Appl Environ Microbiol. 2011;77:2975–83.
Meyer V, Ram AFJ, Punt PJ. Genetics, genetic manipulation, and approaches to strain improvement of filamentous fungi. In: Manual of Industrial Microbiology and Biotechnology. Volume 1. 3rd ed. NY: Wiley; 2010. p. 318–29.
Green MR, Sambrook J. Molecular cloning : a laboratory manual. Volume 1-3. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2012.
Arentshorst M, Ram AFJ, Meyer V. Using non-homologous end-joining-deficient strains for functional gene analyses in filamentous fungi. Methods Mol Biol. 2012;835:133–50.
Delmas S, Llanos A, Parrou J-L, Kokolski M, Pullan ST, Shunburne L, Archer DB. Development of an unmarked gene deletion system for the filamentous fungi Aspergillus niger and Talaromyces versatilis. Appl Environ Microbiol. 2014;80:3484–7.
De Ruiter-Jacobs YM, Broekhuijsen M, Unkles SE, Campbell EI, Kinghorn JR, Contreras R, Pouwels PH, van den Hondel CA. A gene transfer system based on the homologous pyrG gene and efficient expression of bacterial genes in Aspergillus oryzae. Curr Genet. 1989;16:159–63.
Jørgensen TR, Park J, Arentshorst M, van Welzen AM, Lamers G, Vankuyk PA, Damveld RA, van den Hondel CAM, Nielsen KF, Frisvad JC, Ram AFJ. The molecular and genetic basis of conidial pigmentation in Aspergillus niger. Fungal Genet Biol. 2011;48:544–53.
Pel HJ, de Winde JH, Archer DB, Dyer PS, Hofmann G, Schaap PJ, Turner G, de Vries RP, Albang R, Albermann K, Andersen MR, Bendtsen JD, Benen JAE, van den Berg M, Breestraat S, Caddick MX, Contreras R, Cornell M, Coutinho PM, Danchin EGJ, Debets AJM, Dekker P, van Dijck PWM, van Dijk A, Dijkhuizen L, Driessen AJM, D’Enfert C, Geysens S, Goosen C, Groot GSP, et al. Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat Biotechnol. 2007;25:221–31.
Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–54.
Paege N, Jung S, Schäpe P, Müller-Hagen D, Ouedraogo J-P, Heiderich C, Jedamzick J, Nitsche BM, van den Hondel CA, Ram AF, Meyer V. A transcriptome meta-analysis proposes novel biological roles for the antifungal protein AnAFP in Aspergillus niger. PLoS ONE. 2016;11:e0165755.
Gooch VD, Mehra A, Larrondo LF, Fox J, Touroutoutoudis M, Loros JJ, Dunlap JC. Fully codon-optimized luciferase uncovers novel temperature characteristics of the Neurospora clock. Eukaryot Cell. 2008;7:28–37.
Bos CJ, Debets AJ, Swart K, Huybers A, Kobus G, Slakhorst SM. Genetic analysis and the construction of master strains for assignment of genes to six linkage groups in Aspergillus niger. Curr Genet. 1988;14:437–43.
Goto S, Bono H, Ogata H, Fujibuchi W, Nishioka T, Sato K, Kanehisa M. Organizing and computing metabolic pathway data in terms of binary relations. Pac Symp Biocomput 1997:175–86.
Arnaud MB, Chibucos MC, Costanzo MC, Crabtree J, Inglis DO, Lotia A, Orvis J, Shah P, Skrzypek MS, Binkley G, Miyasato SR, Wortman JR, Sherlock G. The Aspergillus genome database, a curated comparative genomics resource for gene, protein and sequence information for the Aspergillus research community. Nucleic Acids Res. 2010;38(Database issue):D420–7.
Christie KR, Weng S, Balakrishnan R, Costanzo MC, Dolinski K, Dwight SS, Engel SR, Feierbach B, Fisk DG, Hirschman JE, Hong EL, Issel-Tarver L, Nash R, Sethuraman A, Starr B, Theesfeld CL, Andrada R, Binkley G, Dong Q, Lane C, Schroeder M, Botstein D, Cherry JM. Saccharomyces genome database (SGD) provides tools to identify and analyze sequences from Saccharomyces cerevisiae and related sequences from other organisms. Nucleic Acids Res. 2004;32(Database issue):D311–4.
Acknowledgments
We are grateful to Susanne Engelhardt for her excellent technical assistance.
Funding
This work was financially supported by the Marie Curie Career Integration Grant (CIG303864) to VM.
Availability of data and materials
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
Authors’ contributions
MF and VM designed the study and drafted the manuscript. MF, TG and CK performed the experiments. All authors contributed to the writing and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Primers used in this study. (DOC 36 kb)
Additional file 2: Figure S2.
Schematic overview of the integration of the luciferase constructs into the hisB* locus and confirmation via Southern analysis. (TIFF 42235 kb)
Additional file 3: Figure S3.
Analysis of histidine auxotrophy of strains MF41.1-30. (TIFF 19712 kb)
Additional file 4: Figure S4.
Sequencing results of the hisB* locus. (TIFF 16195 kb)
Additional file 5: Figure S5.
Growth comparison of olvA - and olvA + mutants with AnidhisB or AopyrG background on CM plates. (TIFF 7346 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fiedler, M.R.M., Gensheimer, T., Kubisch, C. et al. HisB as novel selection marker for gene targeting approaches in Aspergillus niger . BMC Microbiol 17, 57 (2017). https://doi.org/10.1186/s12866-017-0960-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-017-0960-3