
Abstract
Background
Black spot disease in tree peony caused by the fungal necrotroph A. alternata, is a primary limiting factor in the production of the tree peony. The intricate molecular mechanisms underlying the tree peony resistance to A. alternata have not been thoroughly investigated.
Results
The present study utilized high-throughput RNA sequencing (RNA-seq) technology to conduct global expression profiling, revealing an intricate network of genes implicated in the interaction between tree peony and A. alternata. RNA-Seq libraries were constructed from leaf samples and high-throughput sequenced using the BGISEQ-500 sequencing platform. Six distinct libraries were characterized. M1, M2 and M3 were derived from leaves that had undergone mock inoculation, while I1, I2 and I3 originated from leaves that had been inoculated with the pathogen. A range of 10.22–11.80 gigabases (Gb) of clean bases were generated, comprising 68,131,232 − 78,633,602 clean bases and 56,677 − 68,996 Unigenes. A grand total of 99,721 Unigenes were acquired, boasting a mean length of 1,266 base pairs. All these 99,721 Unigenes were annotated in various databases, including NR (Non-Redundant, 61.99%), NT (Nucleotide, 45.50%), SwissProt (46.32%), KEGG (Kyoto Encyclopedia of Genes and Genomes, 49.33%), KOG (clusters of euKaryotic Orthologous Groups, 50.18%), Pfam (Protein family, 47.16%), and GO (Gene Ontology, 34.86%). In total, 66,641 (66.83%) Unigenes had matches in at least one database. By conducting a comparative transcriptome analysis of the mock- and A. alternata-infected sample libraries, we found differentially expressed genes (DEGs) that are related to phytohormone signalling, pathogen recognition, active oxygen generation, and circadian rhythm regulation. Furthermore, multiple different kinds of transcription factors were identified. The expression levels of 10 selected genes were validated employing qRT-PCR (quantitative real-time PCR) to confirm RNA-Seq data.
Conclusions
A multitude of transcriptome sequences have been generated, thus offering a valuable genetic repository for further scholarly exploration on the immune mechanisms underlying the tree peony infected by A. alternata. While the expression of most DEGs increased, a few DEGs showed decreased expression.
Similar content being viewed by others

Background
The tree peony (Paeonia suffruticosa Andrews) is a significant traditional ornamental flowering plant. It is renowned for its edible, medical, and ornamental values [1, 2], of which ‘Junyanhong’ is a tree peony variety with high ornamental value. A significant limitation to the yield of tree peony is the presence of black spot disease caused by the fungal necrotroph Alternaria alternata. August is the peak period for the onset of this disease, and severely affected plants will all wither and their leaves will fall off in the later stage, which will severely damage the ornamental and commercial worth of the tree peony, whether they are grown outdoors or in a greenhouse [3]. Therefore, this disease presents the greatest threat in warm and moist environments, creating a year-round challenge for greenhouse production [4]. Currently, there is a lack of foundational knowledge regarding the interaction between tree peony and black spot disease. However, in the jujube-A. alternata system, MYB gene, participating in lignin synthesis, was up-regulated [5]. The defense response to A. alternata in chrysanthemum was confirmed to be mediated by the signaling pathways of jasmonic acid (JA) and salicylic acid (SA) [6]. These findings are consistent with the research results of the tomato infected with A. alternata pathogen [7]. These pathways are essential for initiating plant defense mechanisms. For instance, in the tomato-Meloidogyne incognita system, the transcription of SA marker genes such as PR1 (pathogenesis-related gene 1) and PR5 were affected [8]. During the process of A. alternata infection in mint, there were significant proteomic changes observed as a result of the defensive action [9]. The pear has developed numerous mechanisms to mitigate the impact of biological stressors, primarily through PTI [PAMP (pathogen-associated molecular pattern)-triggered immunity] as well as ETI (effector-triggered immunity) [10]. A variety of defensive responses have been triggered in pear-A. alternata interaction, involving the generation of ROS (reactive oxygen species), activation of MAPKs (mitogen-activated protein kinases), and the biosyntheses of various phytohormones [10]. Amidst the invasion of a fungus, the chitin in fungi cell walls was degraded by plant chitinase. The released chitin oligosaccharides were recognized by plant receptors, thus triggering host immunity. This process is crucial in the defense mechanisms employed by plants to counter pathogenic attacks. Meanwhile, chitinase acting as an effector of immune response can disrupt the integrity of fungal cell wall, thereby resisting the of pathogens invasion [11]. The transcription of chitinase genes were induced in male Zizania latifolia when challenged by Ustilago esculenta [12]. Hence, it is exceedingly probable that homeotic genes and analogous signalling pathways are elicited in various other plants. However, the phenomenon has not been comprehensively recorded at the transcriptome level in tree peony.
RNA-Seq technique, which can generate tens of millions of reads, is deemed to a more precise detection and analytical approach at the level of transcription [13], because the count of reads that align to a specific area of the genome may directly influence the transcription level in that area [14]. When applied to Macrobrachium rosenbergii hemocytes infected with Staphylococcus aureus, immune-related genes were found to play a crucial role in antibacterial function and exhibited particular effects on gram-positive bacteria [13]. Similarly, the chrysanthemum-Alternaria tenuissima interaction has been shown that the DEGs excavated were primarily associated with immune recognition, hormone signalling, cell wall reinforcement, ROS production, and photosynthesis. Additionally, abundant TFs were also uncovered [6]. Finally, 15 JAZ (jasmonate ZIM domain) genes were all down-regulated in wheat after inoculation with Fusarium spp., leading to a strong activation of the JA signaling pathway [15]. Here, the response induced by A. alternata in tree peony was elucidated through a comprehensive RNA-seq analysis. A transcriptomic comparison was carried out between mock- and pathogen-inoculated leaves of tree peony, and the DEGs were identified, sorted, and subjected to in-depth analysis. The DEGs were annotated using seven public protein databases, indicating their potential involvement in immune recognition, reactive oxygen species generation, plant hormone signaling, and a range of TFs from different families were validated. This experiment has revealed new insights into the genes that are activated or suppressed during the defense response. Validation of the transcription patterns of 10 selected DEGs was conducted using qRT-PCR, thereby confirming the accuracy and reliability of the transcriptome sequencing data. Our research findings will enhance the understanding of the tree peony transcriptomic response to black spot disease.
Results
Colony and morphological characteristics of pathogen that caused black spot in Tree peony
The isolated and purified fungal colonies were nearly round and covered in thick white aerial mycelium. At the beginning, the front of the colony is grayish white, and then gradually changes to dark to bluish brown (Fig. 1B); the back of the colony appears dark brown to dark brown (Fig. 1A). After 5 days of culture at 25℃, black and brown spots appeared within the colony and on the mycelium surface, forming conidial discs. The fungus grew rapidly on the PDA medium, resulting in dark brown, villous colonies (Fig. 1A, B).
The mycelium is colorless and has no diaphragm, about 2.0 ~ 3.1 μm in diameter, conidiophore are clumped, branched, erect or curved (Fig. 1D). The conidia are sepia, obovate, pear-shaped or inverted stick shape, sepia, with 3 to 8 Transverse septum and 1 to 4 longitudinal septum, the part of division narrows slightly, with a short columnar beak, with a size of 18.3 to 32.3 μm×3.4 to 7.1 μm (Fig. 1C). The pathogen of black spot in tree peony was preliminatively identified as Alternaria alternata according to its morphological characteristics.

Characteristics of the colony and morphology of the pathogen responsible for black spot on PDA medium. A: back of colony; B: front of colony; C: conidia; D: mycelia and conidiophore
Pathogen ribosomal DNA-ITS sequence analysis
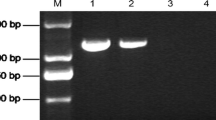
The genomic DNA of the isolated strain was amplified using the fungal universal primer ITS1/ITS4, and a fragment of 534 bp was amplified. The PCR products were purified and sequenced, and the sequencing results were submitted to GenBank. NCBI Blast analysis of the 534 bp fragment revealed that it had 100% homology with the majority of Alternaria alternata strains, such as MW009024.1 (Fig. 2) and MW008981.1 (Fig. 3) in GenBank. According to the results of morphological characteristics, the pathogen of black spot in tree peony was identified as Alternaria alternata.

Sequence alignment of the tree peony pathogen and Alternaria alternata (MW009024.1)

Sequence alignment of the tree peony pathogen and Alternaria alternata (MW008981.1)
Symptoms of tree peony inoculated with A. alternata causing black spot disease
The disease of healthy tree peony leaves began to develop 24 h after infected with A. alternata. Black circular lesions, approximately 1.5 mm in diameter, were formed. By 72 h later, the diameter of the lesions had expanded to about 8 mm, with villous black-green mycelium clusters on the surface and obvious black spots on the back of the leaves. In contrast, there was no significant change in the mock-infected group.
Transcriptome sequencing and de novo assembly
The main features of the six cDNA libraries (Fig. 4) are outlined in Tables 1 and Additional file 1: Figure S1. The amount of raw reads each library varied from ~ 75 to ~ 88 M, and the overall count of clean bases sequenced ranged from 10.22 to 11.8 Gb (Table 1). (Accession ID: library M1 SAMN40497892, M2 SAMN40497893, M3 SAMN40497894; I1 SAMN40497889, I2 SAMN40497890, I3 SAMN40497891). After eliminating reads containing adaptor sequences, and those with a proportion of unknown bases“N” ≥ 5%, as well as reads where low quality value bases (< 10) accounted for over 20% of the total reads. A total of, respectively 73.70, 78.28, 78.63, 78.19, 68.13 and 78.41 M clean reads were acquired, equivalent to 11.06, 11.74, 11.80, 11.73, 10.22 and 11.76 Gb clean bases (Table 1). The percentage of clean reads per library was found to be more than 89.36% (Additional file 1: Figure S1).
Read mapping
Using the Bowtie2 software, 83.42% of the clean reads were successfully mapped to Unigenes, with 29.69% of those being unique reads. The Q20% was over 97.08%, and the Q30% was over 88.98% (Table 1). In total, 99,721 Unigenes were generated (total length of 126,288,636 bp) with an average length of 1266 bp, a N50 length of 1935 bp, a N70 length of 1326 bp and a N90 length of 610 bp by assembly with Trinity Software (Table 1). In total, 16,234 Unigenes (16.28%) were ranging from 200 to 300 nt in length, while 8212 Unigenes (8.23%) had a length ranging from 300 to 400 nt. Additionally, there were 3721 Unigenes (3.73%) that fell within the range of 900 to 1000 nt in length, and finally, there were also7516 Unigenes (7.54%) which exceeded a length of 3000 nt (Fig. 5).

The cDNA libraries were obtained from mock- and A. alternata-infected ‘Junyanhong’ tree peony. M: mock infection, I: infection with a spore suspension of A. alternata. Leaves were collected at 0, 6, 24, 48, and 72 h post-infection (hpi). Three mock-infected samples (M1, M2, and M3), and three A. alternata-infected samples (I1, I2, and I3)

Length distribution analysis of de novo assembled Unigenes in tree peony
Functional annotation
Transcriptome annotation was performed using Trinotate v3.2.2 [16], which utilized BLAST for sequence homology searches against the SwissProt database, hmmscan [17] for Pfam database analysis, and associated with GO, NR, NT, KOG, KEGG databases.
Functional annotation was done using BLAST against the database of NR, NT, Pfam, KOG, KEGG, GO and SwissProt (E value < 105). Our results showed that out of 99,721 unigenes 66,641 (66.83%) were annotated in at least one database, and 18,138 genes (18.19%) were annotated in all seven databases. The success rate of Unigene annotation was 61,818 in NR (61.99%), 45,369 in NT (45.50%), 46,190 in SwissProt (46.32%), 49,194 in KEGG (49.33%), 50,041 in KOG (50.18%), 47,029 in Pfam (47.16%), and 34,759 in GO (34.86%) (Additional file 2: Table S1). On the basis of NR annotation findings, we counted the proportion of various species in the annotation. The species distribution for the best match of each sequence showed that 26.51% of the genes matched Vitis vinifera, followed by 6.34% for Juglans regia, 3.43% for Hevea brasiliensis, 3.34% for Nelumbo nucifera, 3.09% for Theobroma cacao and 57.28% for other species (Additional file 3: Figure S2).
GO classification of DEGs
A comparison of the transcriptome in mock- and A. alternata-infected leaves yielded 14,693 DEGs (Fig. 6). Out of these DEGs, 9,858 Unigenes showed significant up-regulation and 4,835 Unigenes displayed down-regulation (Fig. 6). The MYB and bHLH transcription factor families encompassed over 200 and 150 genes, respectively (Fig. 7), while the AP2-EREBP and C3H families each contained more than 100 genes. The WRKY, NAC, G2-like, FAR1, and C2H2 families each included nearly 100 genes. Additionally, the mTERF, Trihelix, Tify, SBP, GRAS, C2C2-GATA, C2C2-Dof, ARF, and ABI3VP1 families each comprised about 50 genes (Fig. 7).
In total, 6,453 DEGs were GO annotated and classified into three major groups: “biological process”, “cellular component”, and “molecular function” (Additional file 4: Table S2 and Fig. 8). Among them, 3679 Unigenes were categorized under “biological process”, 3835 Unigenes under “cellular components”, and 5465 Unigenes under “molecular function”. The identified Unigenes encompassed a wide array of functional GO categories. Both “metabolic process” and “cellular process” exhibited a higher number of DEGs compared to others in the “biological process” category. The groups that were most abundant were “organelle”, “cell”, “cell part”, “membrane part” and “membrane” in the “cellular component” category. In the category of “cellular component”, the most abundant groups were identified as “organelle”, “cell”, “cell part”, “membrane part” and “membrane”. Conversely, the least abundant groups were found to be “virion” and “virion part”. The Unigenes were predominantly found in the classes of “binding” and “catalytic activity” in the “molecular function” category. Contrasting M and I, a slight amount of DEGs belonged to the categories ‘molecular transducer activity’, ‘carbon utilization’ and ‘growth’ (Fig. 8).

The quantity of DEGs identified in MOCK-vs-Inoculation library

Classification of transcription factor families to which genes belong

GO functional classifications of DEGs. The x-axis displays DEGs number, while the y-axis depicts annotation of gene function in Gene Ontology categories. The findings are categorized into three primary groups: biological processes, cellular components and molecular functions

The pathway assignment of DEGs based on KEGG
We further conducted analysis of the Unigenes using the KEGG database. Among the 14,693 DEGs identified, a total of 4958 Unigenes were categorized into five primary groups encompassing 19 various pathways (Fig. 9). Within these five primary categories, the largest category was “metabolism”, which encompassed 6574 Unigenes. This was followed by the categories of “genetic information processing”, “environmental information processing”, “cellular processes” and “organismal systems”, encompassing 1680, 532, 392, and 275 Unigenes, respectively. Various defense pathways regulated by the main phytohormones JA, SA, and ET were also triggered in defense response. Many transcription factors, pathogenesis-related genes, and metabolism-related genes all exhibited differential expression. Additionally, infection with A. alternata in tree peony led to the down-regulation of genes involved in photosynthesis.
Transcription factors and pathogenesis-related protein genes were mediated through A. alternata infection
The findings from the expression analysis showed that A. alternata inoculation mediated the transcription of numerous transcription factors and genes related to pathogenesis. The expression of the WRKY transcription factor family was found to be significantly higher in A. alternata-infected leaves compared to the mock-infected leaves.
For validating RNA-Seq data, ten Unigenes were further analyzed using qRT-PCR (Table 2; Fig. 10). These Unigenes included six transcription factors and four pathogenesis-related protein genes (CL6209.Contig3_All, CL7367.Contig1_All, CL11001.Contig7_All, and Unigene5195_All). Six transcription factors contain AP2-EREBP (Unigene27620_All), WRKY (CL3108.Contig2_All), MYB (Unigene30574_All), bHLH (CL6244.Contig1_All), NAC (CL11763.Contig4_All), and C2H2-type zinc finger transcription factor (Unigene26849_All). These genes in the pathogen-inoculated leaves showed significant up- or down-regulation. The consequences of qRT-PCR assay demonstrated that our findings were in concordance with those obtained employing transcriptome sequencing methodology.
The expression of AP2-EREBP, WRKY, MYB, bHLH, C2H2-type zinc finger, NAC, PR5-like receptor kinase, Chitinase and PR10 homologues exhibited a significant increase in response to A. alternata inoculation (Fig. 10A-I), while Lipoxygenase was down-regulated (Fig. 10J). The transcript abundance of AP2-EREBP, WRKY, MYB, bHLH, C2H2-hype zinc finger, NAC and PR10 homologues increased to a marked maximum after 48 h of the presence of A. alternata inoculum, however, it subsequently declined (Fig. 10A-F, I). Particularly, the expression of WRKY and AP2-EREBP were 85 and 44 fold higher, respectively, than the mock-infected tree peonies at 48 hpi (Fig. 10A, B). The expression of MYB, NAC, C2H2-type zinc finger, PR10 homologues and bHLH were respectively 20, 17, 10, 4 and 3 times higher than those of the mock-infected tree peonies at 48 hpi (Fig. 10C-F, I). The transcript levels of PR5-like receptor kinase and Chitinase genes was significantly upregulated upon inoculation with A. alternata, reaching peak transcript abundance at 24 hpi (Fig. 10G, H). Significantly, at 24 hpi, the transcript level of PR5-like receptor kinase gene was 978 fold higher than the mock-infected plants (Fig. 10G), and Chitinase gene was induced by about 21 fold (Fig. 10H). The findings from Table 2; Fig. 10 supported the reliability and stability of RNA-Seq results. They also suggested that genes implicated in plant-pathogen interaction and transcription factors are interconnected, enabling the host to adjust its defense response effectively.
These genes exhibited significant up- or down-regulation (P value < 0.05). The FPKM data were acquired through transcriptome sequencing analysis, and the relative transcript abundance was determined using qRT-PCR analysis.

Differential gene expression changes in the leaves of‘Junyanhong’following mock inoculation (MOCK) and A. alternata-inoculation (Inoculation). The RNA of plant leaves at 0, 6, 24, 48 and 72 hpi was extracted for qRT-PCR analysis. The tree peony GAPDH gene served as the internal control gene. The values depicted in the figure above represent the mean of three replicates and standard error. Asterisks (*) indicate significant differences as compared to the MOCK based on one-way analysis of variance (**P < 0.01 and *P < 0.05)
Discussion
Global transcriptional responses to A. alternata infection
The defense mechanism of tree peony against A. alternata inoculation is not well understood at present. Advances in next-generation sequencing technology have now allowed us to identify genes implicated in the response to A. alternata infection and investigate the molecular mechanism underlying this process. Approximately 80% of reads in each RNA-Seq library could be mapped to known transcripts (Table 1), a higher proportion than the chrysanthemum-A. tenuissima system [6], likely due to the more comprehensive tree peony transcriptome. The 20% of unmappable reads are likely linked to unidentified transcripts. The transcripts adjusted by A. alternata infection were validated using qPCR, demonstrating that RNA-Seq methodology is appropriate for analyzing induced transcription in the tree peony as part of its defensive response (Table 2). We identified 14,693 DEGs in the tree peony after inoculation with A. alternata (Fig. 6). Comparing lettuce-Botrytis cinerea, Arabidopsis thaliana-B. cinerea and Chrysanthemum-A. tenuissima transcriptomic studies [18,19,20], we identified two commonly induced DEGs in the aforesaid three systems: Lsa004290.1, At1g74360 and Unigene7965_All (Leucine-rich repeat protein kinase gene) and Lsa016859.1, At4g17500, Unigene17395_All (ERF1). In the present research, we also excavated two types of DEGs. Specifically, the expression of twenty-five Leucine-rich repeat protein kinase genes was induced, while seven gene expression was inhibited (Additional file 5: Table S3). All four ethylene-responsive transcription factor (ERF) genes were up-regulated, including three ERF1 and one ERF2 (Additional file 6: Table S4). At the initial stages of lettuce-B. cinerea, A. thaliana-B. cinerea and chrysanthemum-A. tenuissima interactions, genes engaged in the ET pathway play a pivotal role in defensive reaction. This discovery was also verified in the interaction between tree peony and A. alternata. The analysis of RNA-seq dataset revealed that a multitude of genes were significantly related to the plant response within 6–72 hpi, including those associated with SA, JA and ET pathways (Fig. 11). A more detailed discussion on selected genes within these pathways will follow.
Genes related to pathogen recognition affected by A. alternata inoculation
Plant innate immunity depends upon the recognition of conserved microbe-specific molecules, known as PAMPs/MAMPs (Pathogen- or Microbe-Associated Molecular Patterns), by PRRs (pattern recognition receptors) situated on the cell surface [21]. PRRs can be either RLPs (receptor-like proteins) or RLKs (receptor-like kinases), playing critical roles in perceiving pathogen invasion [22].

Summary of tree peony defense gene expression patterns after inoculation with A. alternata. Genes associated with signaling pathways for defense mechanisms SA, JA and ET are shown as colored boxes. Arrow red up and arrow purple down represents separately up- and down-regulation of gene expression. Gray indicates no change in expression
It has been well documented experimentally that several RLKs are involved in certain host-microbe interactions [6, 20, 22]. Our RNA-seq data confirmed the differential expression of multiple RLKs in the chrysanthemum-A. tenuissima, lettuce-B. cinerea interaction, consistent with previous findings [6, 20]. The LRR-RLKs (leucine-rich repeat RLKs), a type of PRR, exert a crucial and indispensable influence in various immune responses in plants [6, 23].
In A. thaliana, BRI1 (Brassinosteroid Insensitive 1) encoding an LRR-RLK recruits BAK1 (BRI1-associated receptor kinase 1), which is another LRR-RLK, and they form a heterodimer [24]. BAK1 functions as a negative regulatory factor in the induction of cell death by microbial infection [25]. Bak1 mutants increased susceptibility after inoculation with B. cinerea, because programmed cell death was triggered. The transcription of ten BRI1 genes (CL1230.Contig4_All, CL1230.Contig11_All, Unigene31363_All, Unigene27001_All, CL1230.Contig9_All, Unigene27896_All, CL4790.Contig1_All, CL4790.Contig2_All, Unigene27106_All, CL1230.Contig2_All), two BAK1 genes (CL11629.Contig5_All and CL11629.Contig4_All) was modulated by A. alternata infection in the DEGs (Additional file 7: Table S5 and Fig. 11). In the chrysanthemum-A. tenuissima interaction system, four BRI1 genes (Unigene18133_All, Unigene15368_All, Unigene55939_All, Unigene29292_All) and six BAK1 genes (Unigene14705_All, Unigene16709_All, Unigene27008_All, Unigene36228_All, Unigene37501_All and Unigene16958_All) were detected in the mock- vs. pathogen-inoculation contrast [6]. Moreover, it was observed that the BRI-like gene Lsa034184.1 was up-regulated, while no presence of BAK1 genes was detected in the interaction of lettuce with B. cinerea [20].
Previous research showed that SERK1 (Somatic Embryogenesis Receptor Kinase 1), one additional LRR-RLKs, positively regulates host defense response to fungal infection [26]. SERK1 was found during Magnaporthe grisea inoculation and exposure to different defense signaling molecules [26]. Two SERK (Somatic Embryogenesis Receptor Kinase) genes, Unigene14416_All and Unigene22508_All, were detected in the chrysanthemum infected by A. tenuissima [6]. Previous research showed that one additional LRR-RLKs, we detected five SERKs (CL7324.Contig2_All, CL1080.Contig3_All, CL1080.Contig2_All, Unigene21635_All, CL1080.Contig6_All) during RNA-seq analysis of A. alternata inoculated tree peony. All these five genes were down-regulated except for CL1080.Contig6_All (Additional file 5: Table S3 and Fig. 11). Collectively, these data suggested that LRR-RLKs played an important role in conferring resistance to A. alternata inoculation in tree peony. The rapid activation of these genes could be how tree peonies recognize pathogens and initiate an appropriate defense response.
ROS genes regulated by A. alternata infection
MAMPs can trigger defense responses and an oxidative burst, leading to rapid generation of ROS. These are largely produced from plasma membrane anchored NADPH (nicotinamide adenine dinucleotide phosphate) oxidases, known as rboh (respiratory burst oxidase homolog) in plants [6, 27, 28]. It has been determined that rbohD and rbohF play an essential role in the accumulation of ROS in A. thaliana, demonstrating their importance in plant defense response [29]. In the chrysanthemum-A. tenuissima system, rbohD (Unigene 300_All) and rbohF (Unigene45792_All) were all detected [6]. The lettuce rbohD (Lsa002796.1) expression was induced 48 hpi with B. cinerea, while rbohF (Lsa018309.1) remained unaffected [20]. Four tree peony rboh genes were all upregulated: Unigene12350_All (rbohD), Unigene28605_All (rbohF), Unigene2268_All (homologue of rbohF) and Unigene20126_All (rbohD), in the MOCK- vs. pathogen-inoculation contrast. (Additional file 8: Table S6 and Fig. 11). Similar gene up-regulation has been documented in lettuce [20] and chrysanthemum [6] in response to pathogen infection.
Genes related to photosynthesis were mainly suppressed by A. alternata infection
Twenty-seven out of 34 detected DEGs involved in photosynthesis were downregulated, and seven genes (CL603.Contig28_All, CL1718.Contig2_All, CL11724.Contig12_All, Unigene4054_All, Unigene29717_All, CL517.Contig23_All, CL6952.Contig2_All) were upregulated (Additional file 9: Table S7). The response is similar to findings in other plant-pathogen interactions, such as chrysanthemum-A. tenuissima [6], lettuce-B. cinerea [20] and lettuce-Verticillium dahlia [29] interactions, as well as in other documented interactions between plants and pathogens [30,31,32,33,34]. According to the GO analyses, another process down-regulated in the tree peony is the circadian rhythm. The Unigenes we identified related to circadian rhythm (Additional file 10: Table S8) may play vital roles in activating defense genes during fungal infection. For example, CL11161.Contig12_All, involving in Circadian rhythm-plant interaction, was down-regulated in DEGs (Additional file 10: Table S8). Similar gene suppression by pathogen infection has also been observed within other species, for instance, lettuce [20], A. thaliana [35] and chrysanthemum [8]. In the A. thaliana-B. cinerea interaction system, the oscillating expression of core clock components was moderated, potentially indicating manipulation of plant defense response by pathogens. This is because certain defense-related genes are influenced by circadian rhythm [36].
Genes linked to JA, SA and ET signaling pathways affected by A. alternata infection
Our RNA-seq data revealed the presence of a collection of genes associated with the JA, SA and ET signaling pathways, JAZ, MYC2 (transcription factor MYC2); TGA (transcription factor TGA), PR1; ETR (ethylene receptor), CTR1 (Raf-like protein kinase constitutive triple response 1), EIN3 (ethylene-insensitive protein 3), ERF1 and ERF2 (Additional file 6: Table S4 and Fig. 11), with varying levels of transcripts after A. alternata inoculation. JAZ proteins repress the JA-signaling pathway by inhibiting the expression of JA-responsive genes through suppressing transcriptional regulators such as MYC2. This role plays a crucial part in the plant resistance to both biotic and abiotic stresses. The degradation of JAZ repressors eliminates the inhibition of JA-signalling mediated by JAZ, thereby facilitating the transcription of JA response genes [36]. JA and SA are key phytohormones which are essential for protecting plants from pathogen infections [37]. Here, many DEGs involed in JA signalling pathway, for example the expression of JAR1 (Unigene10364_All) and MYC2 (Unigene28661_All, Unigene18397_All, CL6096.Contig1_All, CL1547.Contig1_All, CL1547.Contig2_All, CL11641.Contig14_All and CL11641.Contig13_All) were all obviously induced, but the expression of JAZ (Unigene42074_ All) was suppressed. In A. thaliana, TGAs are important regulators of gene expression in SA-mediated plant immunity, PR1 are SA-responsive genes [38]. We detected TGA (CL966.Contig19_All) and PR-1 (Unigene21511_All, Unigene21510_All, Unigene14741_All and Unigene14213_All) were all up-regulated. Interestingly, the transcription of NPR1 remained unchanged significantly. The ETRs in A. thaliana, along with CTR1, negatively modulate ET signal transduction [39]. When ethylene is present, ETRs and consequently CTR1 are deactivated, leading to reduced phosphorylation and increased accumulation of EIN2, which undergoes proteolytic cleavage, resulting in the release of EIN2-CEND (the C-terminal domain). Then EIN2-CEND moves to the nucleus, where it indirectly triggers the activation of essential transcription factors, EIN3 and EIN3-like (EIL), playing a crucial role in ethylene signaling [40]. Subsequently, this process leads to the activation of ethylene responsive genes. In this study, many DEGs related to ethylene, such as ETR (CL804.Contig3_All), EIN3 (CL10811.Contig1_All), ERF1 (CL2975.Contig1_All) and ERF2 (Unigene27620_All) were all up-regulated, while CTR1 (CL2204.Contig3_All and CL10777.Contig7_All) were all down-regulated. Signaling of key defense phytohormones (JA, SA, and ET) was also notably stimulated in lettuce following B. cinerea inoculation [20]. In conclusion, a large amount of genes involved in the JA/SA/ET signaling pathways are crucial in the defense of tree peonies after A. alternata inoculation.
Transcription factors responding to A. alternata infection
Transcription factors play a central role in activating defense response genes. They can specifically combine cis-acting regulatory elements in defence gene promoters to regulate the transcription of target genes, playing an important part in the defense of plants against pathogenic invaders [6, 41]. In this study, we identified six transcription factor classes (AP2-EREBP, WRKY, MYB, bHLH, C2H2-type zinc finger and NAC) among the DEGs that responded to A. alternata infection. In A. thaliana, AtWRKY6, AtWRKY40, AtWRKY41, AtWRKY46, AtWRKY70 and AtWRKY53 are all involved in defending against pathogen attacks [42,43,44,45,46]. Specifically, AtWRKY46, AtWRKY70 and AtWRKY53 have overlapping and synergistic roles in the basic defense of plants [34]. The coexpression of AtWRKY40 and AtWRKY18 increased the sensitivity of A. thaliana to both pathogens (P. syringae and B. cinerea) [47]. Our data detected a single WRKY copy (CL3108.Contig3_All), identified as a homologue of AtWRKY40, which was transcribed prominently began 6 h post-inoculation with A. alternata, especially at 48 hpi, the induced expression reached the highest (Additional file 11: Table S9 and Fig. 10). Another WRKY (CL8806.Contig3_All, homologous to AtWRKY33) (Additional file 11: Table S9 and Fig. 11) was also up-regulated by A. alternata infection (Fig. 10). In A. thaliana, the transcription of AtWRKY33 is pivotal for defense toward Botrytis cinerea [48]. Therefore, it was speculated that WRKYs (CL3108.Contig3_All and CL8806.Contig3_All) might be involved in the defence response against A. alternata. Transcriptional activator AtMYB15 is capable of activating the transcription of genes for plant defense [49]. In this study, it was observed that one MYB copy (Unigene30574_All), which is homologous to AtMYB15, showed up-regulation in response to A. alternata inoculation (Additional file 12: Table S10 and Fig. 10). AtERF2 has been associated with the induction of defense genes in numerous research studies [50,51,52,53]. The expression level of one ERF copy (Unigene27620_All, homologous to AtERF2) increased significantly at 48 h after A. alternata inoculation (Fig. 10). Transcription factors belonging to the bHLH, C2H2-type zinc finger, and NAC families were found to be highly expressed in tree peony leaves after A. alternata inoculation. Many studies have emphasized the importance of five families of transcription factors (AP2-EREBP, WRKY, MYB, bHLH, and NAC proteins) in their role in plant responses to pathogens [54,55,56,57,58,59,60,61,62]. In short, clearly, a large variety of genes was induced after the tree peony leaf was infected with A. alternata. The response involved interactions among transcription factors, pathogenesis-related genes, and genes within the JA/SA/ET signaling pathways. It is speculated that the tree peony has evolved effective strategies to defend themselves against pathogen invasion in their struggle for life. Understanding the characteristics of these genes would help clarify the molecular mechanism behind tree peony defense response toward A. alternata infection. Consequently, this could help identify potential candidate genes for targeted genetic modification of tree peony.
Virion and virion part responding to A. alternata infection
The GO classification indicates categories like “virion” and “virion part” (Fig. 5). When A. alternata infects plants, it can produce virus-like particles [63, 64], which may be involved in the infectious mechanism of the disease. A. alternata, specifically the Japanese pear pathotype, is known to produce a host-specific virus called A. alternata chrysovirus 1 (AaCV1). This novel mycovirus has dual effects: it hampers the growth of the host fungus while making it ‘hypervirulent’ to the plant [63]. Currently, effective and environmentally friendly control measures for tree peony black spot disease are lacking. Mycovirus-mediated attenuated strains represent an important biological control method. Numerous fungal viruses have been discovered and identified in various plant pathogenic fungi [65,66,67,68,69]. These findings may offer a new approach for biologically controlling tree peony black spot using low-virulence fungal viruses.
Materials and methods
Experimental plant
Tree peony ‘Junyanhong’ was acquired from Shenzhou Peony Garden (Luoyang, China). Potting was conducted using 25 cm diameter plastic flower pots enriched with a mixture of peat soil, vermiculite and perlite in a 2:1:1 volume ratio. The plants were cultivated at a temperature of 25 °C during the day and 15 °C at night, with an 14 h/10 h day/night cycle, and 60% relative humidity.
Isolation of pathogenic fungi
By using the conventional tissue block isolation method, a pathogenic strain was isolated from the tree peony leaves with naturally occurring black spot disease. The diseased leaves collected from the field were rinsed with tap water. A small piece of tissue at the junction of diseased and healthy position on the leaf was cut, and immersed in 70% alcohol for 10 s, and then transferred to 0.1% mercuric chloride (HgCl2) solution for 3 min to perform disinfection. The tissue pieces were then violently shaken in sterile water for 5 min, repeated 3–4 times, and placed on sterile filter paper. After draining the excess water, they were inoculated into the pre-prepared PDA medium (generally a 9 cm petri dish), placed at a constant temperature of 25 ± 1℃ for 2–10 days, and their growth was regularly observed. When a black mold layer appeared in the colony, it indicated that spores had been produced. A small amount of spores was selected in the ultra-clean workbench and a freehand section was made, which was observed under an optical microscope. After determining the spore morphology of the fungus, the spores were washed with sterile water and a single spore was selected and purified under the microscope. The purified strains were stored on PDA bevels and kept in refrigeration at 4℃ for future use.
Traditional morphological identification of pathogen
The purified strains were inoculated on a PDA plate and incubated in an incubator at a temperature of 25℃, with a relative humidity of 70% and alternating light and dark cycles (12 h/12 h) for 5 days. The morphology and color of the colonies were observed and recorded. The mycelia were selected with an inoculating needle to prepare slides. The morphology of conidium and conidiophore was observed under an optical microscope, and the size of spores and diameter of mycelia were measured.
Ribosomal DNA-ITS sequence analysis of pathogen
The above strain will be inoculated onto PDA medium (25℃, dark) and allowed to grow for 3 days. Subsequently, the mycelia will be gently scraped off using a sterile blade. The collected mycelia will then undergo genomic DNA extraction using the “E.Z.N.A.TM Fungal DNA Mini Kit” (OMEGA). ITS region of rDNA was amplified and sequenced using primers ITS1 (5′-TCCGTAGGTGAACCTGCGG-3′) / ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) [70]. The optimal reaction system had a volume of 50 µL, including DNA template 2 µL, 2×ES Taq Master Mix 25 µL, primer 2 µL, ddH2O 19 µL. The PCR conditions included an initial denaturation at 94 °C for 1 min, followed by 35 cycles of denaturation at 94 °C for 30 s, primer annealing at 58 °C for 30 s, and extension at 72 °C for 1 min, with a final extension at 72 °C for 5 min.
Amplified PCR products were detected using 1% agarose gel electrophoresis, and the specific band of expression was excised, purified, and sent to Beijing Qingke Biotechnology Co., Ltd. for sequencing. The sequencing results were analyzed online using the BLAST tool in NCBI to search for known sequences with high homology.
Inoculation
A spore solution containing 106 spores per ml was prepared by adding 0.1% Triton X-100 serves as a surfactant. The plant leaves were pierced with a sterile needle, and a drop of spore solution (10 µl) was applied to the wound [6]. MOCK-inoculation of the control was performed in a similar manner, but with 10 µl of sterilized distilled water droplets instead of the fungal suspension. The inoculated plants were placed in a dark environment with 100% relative atmospheric humidity at 25 °C for 24 h, followed by exposure to cool-white fluorescent lamps at a light intensity of 130 µmol m− 2 s− 1 for 14 h per day. Leaves from three plants per treatment were collected at different time points [0, 6, 24, 48, and 72 hpi (hours post-infection)] and pooled for RNA-seq analysis.
RNA extraction
Six individual libraries (M1, M2, and M3; I1, I2, and I3) were obtained. Leaves from plants were collected and divided into libraries M (mock-inoculated) and I (pathogen-inoculated). Total RNA was extracted using the modified CTAB method as recommended by the manufacturer [71]. RNA quality was assessed utilizing the 2100 Bioanalyzer (Agilent Technologies) with criteria of RIN (RNA Integrity Number) ≥ 6.5 and 28 S:18 S ≥ 1.0. RNA concentration was measured with the NanoDrop spectrophotometer (Thermo Fisher Scientific Inc., Wilmington, DE, USA). The OD260/OD280 and OD260/OD230 were within the range of 1.8-2.0 and not less than 1.8, respectively. Total RNA extracted from each of the three sampled plants was mixed together in equimolar amounts (at least 10 µg) [6].
Construction of cDNA library for sequencing
The total RNA was treated with DNase I (RNase-free; TaKaRa, Dalian, China) to remove any residual genomic DNA. Messenger RNA was captured using oligo(dT) beads, subsequently cleaved into 200-bp sizes with a fragmentation buffer. cDNA was initially synthesized by random hexameric primers. Subsequently, it was further synthesized with the assistance of RNase H and DNA polymerase I. The synthesized double-stranded cDNA underwent an end-filling process, followed by phosphorylation at the 5’ ends and addition of a single A (adenine) nucleotide to the 3’ ends. Subsequently, it was ligated to the bulbous connector with a protruding “T” at the 3’ end. Transcriptome sequencing was conducted utilizing the BGISEQ-500 sequencing platform (BGI, China).
Filtering and assembling high-quality reads from raw data
Raw reads were cleaned via removing adaptor sequences, reads with more than 5% unknown bases (N), and those with low quality value bases (< 10) accounting for over 20% of the read. The filtered “Clean Reads” from each library were then assembled de novo utilizing Trinity software [72] to generate Unigenes, which were saved in FASTQ format. Unigenes were derived from the clustering and redundancy elimination of assembled clean read segments (also called transcripts) using the TGICL (TGI Clustering Tools) [73]. To examine multiple samples, the preceding step should be repeated using TGICL for further Unigene analysis. All acquired Unigenes were collectively called “All-Unigenes”. They were divided into two types: the first type consisted of clusters, resulted from further elimination of redundancy, the same cluster contained a quantity of Unigenes exhibiting a high similarity (over 70%) (beginning with “CL”, representing a cluster, followed by their respective gene family ID). Another type comprised singletons (beginning with “Unigene”), referring to unclustered, separate Unigenes. The clean reads were mapped to the assembled unique gene utilising Bowtie2 for comparison [74, 75]. The FPKM value (Fragments Per Kilobase of transcript per Million mapped fragments) is the quantitative index for gene expression level [76]. Differential gene expression between pathogen- and mock-inoculated plants was determined by performing a calculation on the log2 ratio of the two FPKM values.
Identification of DEGs
DEGs were detected using the DEGseq, DEseq2 methods [77, 78]. P-values were adjusted to q-values using two methods outlined by Benjamini Y et al. [79] and Storey J et al. [80]. P-values were corrected by FDR (False Discovery Rate). In this research, we identified DEGs based on the criteria of q-value ≤ 0.05, FDR ≤ 0.05, and |log2 Ratio|≥1.00 between pathogen- and mock-inoculated samples. Transcriptomes in mock- and A. alternata-infected leaves were de novo assembled and annotated using Trinity and Trinotate, respectively. Unigenes of tree peony compared with the following seven databases, inclusive of NR, SwissProt, KEGG, GO, KOG, Pfam and NT. All Unigenes were annotated by comparing them with genes annotated in these databases. For plant transcription factors, we first identify the open reading frame (ORF) of Unigene using getorf. Next, we compare the ORF to the transcription factor protein domain using hmmsearch (data from TF). Finally, we utilize the characterization of the transcription factor family described by PlantTFDB to enable identification of Unigene capabilities. Enrichment analysis was conducted based on GO and KEGG pathway gene function annotations, utilizing the phyper function in R software (https://www.r-project.org).
qRT-PCR verification
The transcription of 10 selected genes was verified using qRT-PCR. The pooled and unpooled samples collected at various times were analyzed separately by qRT-PCR. First-strand cDNA was synthesized utilizing the PrimeScript™ RT reagent Kit with gDNA Eraser (Code No. RR047A, TaKaRa, Japan). The primers (given in Table 3) were designed using Primer5.0 software. qRT-PCR was conducted utilizing the TB Green® Premix Ex Taq™ II Tli RNaseH Plus (TaKaRa, Code: No. RR820A) on CFX96 Real-Time System (Bio-Rad). The tree peony GAPDH gene served as the internal reference for data normalization [81]. The GAPDH primers were as below: F: 5′-GGTTGATCTCACTGTTAGGC-3′; R: 5′-TCAGACTCCTCCCTACAAG-3′ [70]. Each 20 µl reaction included 10 µL of TB Green (2×), 0.3 µM of gene-specific primers, and 8 ng cDNA. The qRT-PCR reactions were conducted under the following conditions: 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s, 55 °C for 30 s, and 72 °C for 30 s. The transcription abundance is represented by the mean ± standard error (SE) of three replicated measurements. Relative transcription level of each gene was calculated utilizing the 2−ΔΔCt method [6].
Statistical analysis
The obtained data was analyzed with one-way analysis of variance using IBM SPSS version 19.0 (SPSS Inc., Chicago, IL, USA) to determine any significant differences. Graphs were drawn using SigmaPlot 14.0 (Systat Software Inc.) and Microsoft Excel 2021.
Conclusions
In the present study, we performed the first transcriptome analysis of tree peony leaf and presented a comparison of DEGs related to the interplay between tree peony and A. alternata. The research findings significantly contribute to the current sequence databases of tree peony, providing a solid foundation for further exploration and characterization of gene transcription patterns in the dynamic interaction between tree peony and A. alternata. Most DEGs detected were found to participate in recognition of pathogens, ROS detoxification, and the signalling of phytohormones. Furthermore, transcription factors belonging to five different families were also discovered, this will help us understand more about the immune mechanisms involved in the defense response of tree peony to A. alternata.
Data availability
Sequence data that support the findings of this study have been deposited in the sequence read archive (SRA) of NCBI (Additional file 13: Table S11) (BioProject ID: PRJNA1088763) (https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA1088763).
Abbreviations
- A. alternata :
-
Alternaria alternata
- RNA-seq:
-
High-throughput RNA sequencing
- NR:
-
Non-Redundant Protein Sequence
- NT:
-
Nucleotide Sequence
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- KOG:
-
Clusters of euKaryotic Orthologous Groups
- Pfam:
-
Protein family
- GO:
-
Gene Ontology
- TGICL:
-
TGI Clustering Tools
- qRT-PCR:
-
Quantitative real-time PCR
- SA:
-
Salicylic Acid
- JA:
-
Jasmonate Acid
- ET:
-
Ethylene
- ERF:
-
Ethylene response factor
- TF:
-
Transcription Factor
- EIN3:
-
Ethylene-insensitive protein 3
- ERF1:
-
Ethylene-responsive transcription factor 1
- ERF2:
-
Ethylene-responsive transcription factor 2
- TGA:
-
Transcription factor TGA
- PR1:
-
Pathogenesis-Related Protein 1
- PR5:
-
Pathogenesis-related protein 5
- PTI:
-
Pattern-Triggered Immunity
- ETI:
-
Effector-Triggered Immunity
- MAPKs:
-
Mitogen-Activated Protein Kinases
- ROS:
-
Reactive Oxygen Species
- PAMP:
-
Pathogen-Associated Molecular Pattern
- A. tenuissima :
-
Alternaria tenuissima
- JAZ:
-
Jasmonate ZIM-Domain
- DEGs:
-
Differentially Expressed Genes
- FPKM:
-
Fragments Per Kilobase per Million mapped fragments
- A. thaliana :
-
Arabidopsis thaliana
- B. cinerea :
-
Botrytis cinerea
- PRRs:
-
Pattern Recognition Receptors
- RLKs:
-
Receptor-Like Kinases
- RLPs:
-
Receptor-Like Proteins
- LRR-RLKs:
-
Leucine-Rich Repeat RLKs
- BRI1:
-
Brassinosteroid Insensitive 1
- BAK1:
-
BRI1-associated receptor kinase 1
- SERK1:
-
Somatic Embryogenesis Receptor Kinase 1
- rboh:
-
Respiratory burst oxidase homolog
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- ETR:
-
Ethylene Receptor
- CTR1:
-
Raf-like protein kinase constitutive triple response 1
- ITS:
-
The internal transcribed spacer
References
Guo L, Li Y, Lei Yang, Gao J, Song C, Guo Dalong, et al. Genome-scale investigation and identification of variations associated with early flowering based on whole genome resequencing and transcriptome integrated analysis in tree peony. Sci Hortic-Amsterdam. 2023;310:111695.
Zhang X, Li Y, Wang X, Peng L, Liu Z, Shu Q, et al. Overexpression of a novel F-box protein PsFFL1 from tree peony (Paeonia suffruticosa) confers drought tolerance in tobacco. Plant Growth Regul. 2023;101(1):131–43.
Duan Y. Study on the identification and biological characteristics of pathogenic fungi of Peony leaf spot. Luoyang: Henan University of Science and Technology. 2009;1–73 (in Chinese).
Tian Y, Che Z, Sun D, Yang Y, Lin X, Liu S. Resistance identification of tree peony cultivars of different flowering time to gray mold pathogen Botrytis Cinerea. Hortic Sci. 2019;54(2):328–30.
Liu Y, Lei X, Deng B, Chen O, Deng L, Zeng K. Methionine enhances disease resistance of jujube fruit against postharvest black spot rot by activating lignin biosynthesis. Postharvest Biol Tec. 2022;190:111935–44.
Li H, Chen S, Song A, Wang H, Fang W, Guan Z, et al. RNA-Seq derived identification of differential transcription in the chrysanthemum leaf following inoculation with Alternaria tenuissima. BMC Genomics. 2014;15:9–22.
Jia C, Zhang L, Liu L, Wang J, Li C, Wang Q. Multiple phytohormone signalling pathways modulate susceptibility of tomato plants to Alternaria alternata f. sp. Lycopersici J Exp Bot. 2013;64(2):637–50.
Kamaruzzaman M, Zhao LF, Zhang JA, Zhu LT, Li Y, Deng XD, et al. MiPDCD6 effector suppresses host PAMP-triggered immunity to facilitate Meloidogyne incognita parasitism in tomato. Plant Pathol. 2023;72:195–206.
Sinha R, Chattopadhyay S. Changes in the leaf proteome profile of Mentha arvensis in response to Alternaria alternata infection. J Proteom. 2011;74(3):327–36.
Xing C, Chen Q, Qiao Q, Gu S, Cheng X, Dong H, et al. PbrWRKY70 increases pear (Pyrus Bretschneideri Rehd) black spot disease tolerance by negatively regulating ethylene synthesis via PbrERF1B-2. Plant Science. Int J Experimental Plant Biology. 2023;334:1–14.
Li L, Zhao D. Ectopic overexpression of EuCHIT30.7 improves Nicotiana tabacum resistance to powdery mildew. Phyton-Int J Exp Bot. 2023;92(11):3043–61.
Xu W, Zhou NN, Guo P, Zhou H, Xu S, He X, et al. Physiological and chitinase gene expression responses of male zizania latifolia to Ustilago esculenta infection. J Am Soc Hortic Sci. 2023;148(2):74–82.
Zhan F, Zhou S, Shi F, Li Q, Lin L, Qin Z. Transcriptome analysis of Macrobrachium rosenbergii hemocytes in response to Staphylococcus aureus infection. Fish Shellfish Immun. 2023;139:1–17.
Hu T, Chitnis N, Monos D, Dinh A. Next-generation sequencing technologies: an overview. Hum Immunol. 2021;82(11):801–11.
Duan S, Jin J, Gao Y, Jin C, Mu J, Zhen W, et al. Integrated transcriptome and metabolite profiling highlights the role of benzoxazinoids in wheat resistance against Fusarium crown rot. Crop J. 2022;10:407–17.
Bryant DM, Johnson K, DiTommaso T, Tickle T, Couger MB, Payzin-Dogru D, et al. A tissue-mapped Axolotl De Novo Transcriptome enables identification of limb regeneration factors. Cell Rep. 2017;18:762–76.
Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:29–37.
Ferrari S, Galletti R, Denoux C, De LG, Ausubel FM, Dewdney J. Resistance to Botrytis cinerea induced in Arabidopsis by elicitors is independent of salicylic acid, ethylene, or jasmonate signalling but requires PHYTOALEXIN DEFICIENT3. Plant Physiol. 2007;144:367–79.
Mulema JMK, Denby KJ. Spatial and temporal transcriptomic analysis of the Arabidopsis thaliana-Botrytis cinerea interaction. Mol Biol Rep. 2012;39:4039–49.
De Cremer K, Mathys J, Vos C, Froenicke L, Michelmore RW, Cammue B, et al. RNAseq-based transcriptome analysis of Lactuca sativa infected by the fungal necrotroph Botrytis Cinerea. Plant Cell Environ. 2013;36:1992–2007.
Yang C, Dolatabadian A, Fernando WGD. The wonderful world of intrinsic and intricate immunity responses in plants against pathogens. Can J Plant Pathology: Revue Canadienne De Phytopathologie. 2022;1:44.
Yang C, Wang E, Liu J. CERK1, more than a co-receptor in plant-microbe interactions. New Phytol. 2022;234(5):1606–13.
Xiong W, Berke L, Michelmore R, Dirk-Jan M, Workum, Becker Frank FM, Schijlen E, et al. The genome of Lactuca saligna, a wild relative of lettuce, provides insight into non-host resistance to the downy mildew Bremia Lactucae. Plant J. 2023;115(1):108–26.
Zhang Y, Tian H, Chen D, Zhang H, Sun M, Chen S, et al. Cysteine-rich receptor-like protein kinases: emerging regulators of plant stress responses. Trends Plant Sci. 2023;28(7):776–94.
Kemmerling B, Schwedt A, Rodriguez P, Mazzotta S, Frank M, Qamar SA, et al. The BRI1-associated kinase 1, BAK1, has a brassinolide-independent role in plant cell-death control. Curr Biol. 2007;17(13):1116–22.
Hu H, Xiong L, Yang Y, Rice. SERK1 gene positively regulates somatic embryogenesis of cultured cell and host defense response against fungal infection. Planta. 2005;222(1):107–17.
Arnaud D, Deeks MJ, Smirnoff N. Organelle-targeted biosensors reveal distinct oxidative events during pattern-triggered immune responses. Plant Physiol. 2023;191:2551–69.
Galeou A, Stefanatou C, Prombona A. Circadian clock-dependent and-independent response of Phaseolus vulgaris to Pseudomonas syringae. Physiol Mol Plant Pathol. 2023;124:101944.
Torres MA, Dangl JL, Jones JD. Arabidopsis gp91phox homologues AtrbohD and AtrbohF are required for accumulation of reactive oxygen intermediates in the plant defense response. Proc Natl Acad Sci USA. 2002;99(1):517–22.
Milli A, Cecconi D, Bortesi L, Persi A, Rinalducci S, Zamboni A, et al. Proteomic analysis of the compatible interaction between Vitis vinifera and Plasmopara Viticola. J Proteom. 2012;75(4):1284–302.
Gyetvai G, Sonderkaer M, Goebel U, Basekow R, Ballvora A, Imhoff M, et al. The transcriptome of compatible and incompatible interactions of potato (Solanum tuberosum) with Phytophthora infestans revealed by DeepSAGE analysis. PLoS ONE. 2012;7(2):e31526.
Bonfig KB, Gabler A, Simon UK, Luschin-Ebengreuth N, Hatz M, Berger S, et al. Post-translational derepression of invertase activity in source leaves via down-regulation of invertase inhibitor expression is part of the plant defense response. Mol Plant. 2010;3(6):1037–48.
Swarbrick PJ, Schulze-Lefert P, Scholes JD. Metabolic consequences of susceptibility and resistance (race-specific and broad-spectrum) in barley leaves challenged with powdery mildew. Plant Cell Environ. 2006;29(6):1061–76.
Scharte J, Schon H, Weis E. Photosynthesis and carbohydrate metabolism in tobacco leaves during an incompatible interaction with Phytophthora nicotianae. Plant Cell Environ. 2005;28(11):1421–35.
Windram O, Madhou P, McHattie S, Hill C, Hickman R, Cooke E, et al. Arabidopsis defense against Botrytis Cinerea: chronology and regulation deciphered by high-resolution temporal transcriptomic analysis. Plant Cell. 2012;24(9):3530–57.
Ye L, Cao L, Zhao X, Guo X, Ye K, Jiao S, et al. Investigation of the JASMONATE ZIM-DOMAIN Gene Family reveals the canonical JA-Signaling pathway in Pineapple. Biology. 2022;11:445.
Zhong Q, Hu H, Fan B, Zhu C, Chen Z. Biosynthesis and roles of salicylic acid in balancing stress response and growth in plants. Int J Mol Sci. 2021;22:11672.
Li J, Luo C, Yang X, Peng L, Lu T, Yang J, et al. Genome-wide identification of the mango pathogenesis-related 1 (PR1) gene family and functional analysis of MiPR1A genes in transgenic Arabidopsis. Sci Hortic-Amsterdam. 2023;321:112254.
Shakeel SN, Gao Z, Amir M, Chen YF, Schaller GE, Rai MI, et al. Ethylene regulates levels of ethylene receptor/CTR1 signaling complexes in Arabidopsis thaliana. J Biol Chem. 2015;290(19):12415–24.
Park HL, Seo DH, Lee HY, Bakshi A, Park C, Chien YC, et al. Ethylene-triggered subcellular trafficking of CTR1 enhances the response to ethylene gas. Nat Commun. 2023;14(365):1–15.
Xu M, Gao C, Ji L, Zhu L, Gao Y, Feng H, et al. A fungal microRNA-like RNA regulated effector promotes pathogen infection by targeting a host defense-related transcription factor. Plant J. 2023;115(3):803–19.
Robatzek S, Somssich IE. A new member of the Arabidopsis WRKY transcription factor family, AtWRKY6, is associated with both senescence-and defence-related processes. Plant J. 2001;28(2):123–33.
Xu X, Chen C, Fan B, Chen Z. Physical and functional interactions between pathogen-induced Arabidopsis WRKY18, WRKY40, and WRKY60 transcription factors. Plant Cell. 2006;18:1310–26.
Higashi K, Ishiga Y, Inagaki Y, Toyoda K, Shiraishi T, Ichinose Y. Modulation of defense signal transduction by flagellin-induced WRKY41 transcription factor in Arabidopsis thaliana. Mol Genet Genomics. 2008;279:303–12.
Knoth C, Ringler J, Dangl JL, Eulgem T. Arabidopsis WRKY70 is required for full RPP4-mediated disease resistance and basal defense against Hyaloperonospora Parasitica. Mol Plant Microbe Interact. 2007;20(2):120–8.
Hu Y, Dong Q, Yu D. Arabidopsis WRKY46 coordinates with WRKY70 and WRKY53 in basal resistance against pathogen Pseudomonas syringae. Plant Sci. 2012;185:288–97.
Xu X, Chen C, Fan B, Chen Z. Physical and functional interactions between Pathogen-Induced Arabidopsis WRKY18, WRKY40, and WRKY60 transcription factors. Plant Cell. 2006;18(5):1310–26.
Birkenbihl RP, Diezel C, Somssich IE. Arabidopsis WRKY33 is a key transcriptional regulator of hormonal and metabolic responses toward Botrytis cinerea infection. Plant Physiol. 2012;159(1):266–85.
Shi H. Function analysis of transcription factor AtMYB44 and AtMYB15 during defense response in Arabidopsis. Nan jing: Nanjing Agricultural University; 2010.
Solano R, Stepanova A, Chao Q, Ecker JR. Nuclear events in ethylene signaling: a transcriptional cascade mediated by ETHYLENE-INSENSITIVE3 and ETHYLENE-RESPONSE-FACTOR1. Genes Dev. 1998;12:3703–14.
Berrocal-Lobo M, Molina A, Solano R. Constitutive expression of ETHYLENE-RESPONSE-FACTOR1 in Arabidopsis confers resistance to several necrotrophic fungi. Plant J. 2002;29:23–32.
McGrath KC, Dombrecht B, Manners JM, Schenk PM, Edgar CI, Maclean DJ, et al. Repressor- and activator-type ethylene response factors functioning in jasmonate signaling and disease resistance identified via a genome-wide screen of Arabidopsis transcription factor gene expression. Plant Physiol. 2005;139:949–59.
Onate-Sanchez L, Anderson JP, Young J, Singh KB. AtERF14, a Member of the ERF family of transcription factors, plays a nonredundant role in Plant Defense. Plant Physiol. 2007;143:400–9.
Su XF, Lu GQ, Guo HM, Zhang KX, Li XK, Cheng HM. The dynamic transcriptome and metabolomics profiling in Verticillium Dahliae inoculated Arabidopsis thaliana. Sci Rep-UK. 2018;8:15404.
Zheng Z, Qamar SA, Chen Z, Mengiste T. Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant J. 2006;48(4):592–605.
Wang W, Li T, Chen J, Zhang X, Wei L, Yao S, et al. A self-regulated transcription factor CsWRKY33 enhances resistance of citrus fruit to penicillium digitatum. Postharvest Biol Tec. 2023;198:112267.
Singh KB, Foley RC, Oñate-Sánchez L. Transcription factors in plant defense and stress responses. Curr Opin Plant Biol. 2002;5(5):430–6.
Dubos C, Stracke R, Grotewold E, Weisshaar B, Martin C, Lepiniec L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010;15(10):573–81.
Du H, Yang SS, Feng BR, Tang YX, Huang YB, Liang Z. Genome-wide analysis of the MYB transcription factor superfamily in soybean. BMC Plant Biol. 2012;12(1):106–27.
Wang FT, Lin RM, Feng J, Qiu DW, Chen WQ, Xu SC. Wheat bHLH transcription factor gene, TabHLH060, enhances susceptibility of transgenic Arabidopsis thaliana to Pseudomonas syringae. Physiol Mol Plant P. 2015;90:123–30.
Lin R, Zhao W, Meng X, Wang M, Peng Y. Rice gene OsNAC19 encodes a novel NAC-domain transcription factor and responds to infection by Magnaporthe Grisea. Plant Sci. 2007;172(1):120–30.
Xia N, Zhang G, Liu XY, Deng L, Cai GL, Zhang Y, et al. Characterization of a novel wheat NAC transcription factor gene involved in defense response against stripe rust pathogen infection and abiotic stresses. Mol Biol Rep. 2010;37(8):3703–12.
Okada R, Ichinose S, Takeshita K, Urayama SI, Fukuhara T, Komatsu K, et al. Molecular characterization of a novel mycovirus in Alternaria alternata manifesting two-sided effects: down-regulation of host growth and up-regulation of host plant pathogenicity. Virology. 2018;519:23–32.
Shamsi W, Sato Y, Jamal A, Shahi S, Kondo H, Suzuki N, et al. Molecular and biological characterization of a novel botybirnavirus identified from a Pakistani isolate of Alternaria alternata. Virus Res. 2019;263:119–28.
Nuss DL. Hypovirulence: mycoviruses at the fungal-plant interface. Nat Rev Microbiol. 2005;3(8):632–42.
Yu X, Li B, Fu YP, Xie JT, Cheng JS, Ghabrial S, et al. Extracellular transmission of a DNA mycovirus and its use as a natural fungicide. P Natl Acad Sci Usa. 2013;110(4):1452–7.
Wang LP, Jiang JJ, Wang YF, Hong N, Zhang F, Xu WX, Wang GP. Hypovirulence of the phytopathogenic fungus Botryosphaeria Dothidea: association with a coinfecting chrysovirus and a partitivirus. J Virol. 2014;88(13):7517–27.
Li H, Bian R, Liu Q, Yang L, Pang T, Salaipeth L, et al. Identification of a novel hypovirulence-inducing hypovirus from Alternaria alternata. Front Microbiol. 2019;10:1076.
Das T, Prasad A, Dey A. Mycoviral gene-incorporating phytopathogenic fungi: a biocontrol agent. Trends Plant Sci. 2023;8:28.
Ciordia M, Loureiro MD, González AJ. First report of Neofusicoccum parvum causing canker on Castanea sativa in Spain. Plant dis. 2022;106:1299.
Zhang Y, Cheng Y, Ya H, Xu S, Han J. Transcriptome sequencing of purple petal spot region in tree peony reveals differentially expressed anthocyanin structural genes. Front Plant Sci. 2015;6:964–72.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52.
Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, et al. TIGR gene indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinf (Oxford England). 2003;19(5):651–2.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Li B, Dewey CN. Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323.
Tsai CC, Wu KM, Chiang TY, Huang CY, Chou CH, Li SJ, et al. Comparative transcriptome analysis of Gastrodia elata (Orchidaceae) in response to fungus symbiosis to identify gastrodin biosynthesis-related genes. BMC Genomics. 2016;17:212.
Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26(1):136–8.
Love MI, Huber W, Anders S. Moderated estimation of Fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B. 1995;57(1):289–300.
Storey JD, Tibshirani R. Statistical methods for identifying differentially expressed genes in DNA microarrays. Methods Mol Biol. 2003;224:149–57.
Wang YJ, Dong L, Zhang C, Wang XQ. Reference gene selection for real-time quantitative PCR normalization in tree peony (Paeonia suffruticosa Andr). J Agric Biotechnol. 2012;20(5):521–8.
Acknowledgements
None.
Funding
This work was supported by the Henan Province Science and Technology Breakthrough Project (192102110035).
Author information
Authors and Affiliations
Contributions
H.L. and Y.Z. conceived and supervised the study. H.L., Y.L., Q.R., Z.Y.L., S.L., R.R., F.W. and Y.L. performed the experiments. H.L., Y.Z., Y.L., Z.X.L., Q.R., Y.L. and Z.Y.L. participated in data analysis and experiment materials preparation. H.L. wrote the original manuscript of this paper. H.L. and Y.Z. revised the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The collection of the tree peony variety ‘Junyanhong’ used in our study complied with local guidelines. ‘Junyanhong’ is a cultivated and open variety rather than a wild one. The Shenzhou Peony Garden in Luoyang, China, clarified the permissions granted for collecting plant materials of the tree peony variety ‘Junyanhong’ for research in this article.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
12864_2024_10784_MOESM1_ESM.docx
Additional file 1: Figure S1. Composition of the raw reads in the six sample libraries. “Clean” reads are those remaining after removal of adaptor sequences, reads in which the proportion of unknown bases “N” was ≥ 5% and reads in which low quality (<15) bases represented >20% of the reads. The numbers in parentheses indicate the number and percentage of each type of read present.
12864_2024_10784_MOESM3_ESM.docx
Additional file 3: Figure S2. Species distribution of the BLASTX results. This figure shows the species distribution of Unigene BLASTX results against the nr protein database with a cutoff E-value<10-5 and the proportions of each species. Different colors represent different species. Species with proportions of more than 3% are shown.
12864_2024_10784_MOESM5_ESM.xlsx
Additional file 5: Table S3. The differential transcription of LRR receptor-like serine/threonine-protein kinase (FLS2) genes in the contrast MOCK vs Inoculation. The criteria applied for assigning significance were: q-value ≤ 0.05 and estimated absolute |log2Ratio(Inoculation/MOCK)| ≥ 1. FPKM: Fragments Per Kilobase per Million mapped fragments.
12864_2024_10784_MOESM6_ESM.xlsx
Additional file 6: Table S4. The differential transcription of SA, JA and ET signalling pathway-related genes in the contrast MOCK vs Inoculation. The criteria applied for assigning significance were: q-value ≤ 0.05 and estimated absolute |Ratio(Inoculation/MOCK)| ≥ 1. FPKM: Fragments Per Kilobase per Million mapped fragments.
12864_2024_10784_MOESM7_ESM.xlsx
Additional file 7: Table S5. The differential transcription of brassinosteroid insensitive 1 (BRI1), brassinosteroid insensitive 1-associated receptor kinase 1 (BAK1), somatic embryogenesis receptor kinase (SERK), and chitin elicitor receptor kinase 1 (CERK1) genes in the contrast MOCK vs Inoculation. The criteria applied for assigning significance were: q-value ≤ 0.05 and estimated absolute |Ratio(Inoculation/MOCK)| ≥ 1. FPKM: Fragments Per Kilobase per Million mapped fragments.
12864_2024_10784_MOESM8_ESM.xlsx
Additional file 8: Table S6. The differential transcription of respiratory burst oxidase genes in the contrast MOCK vs Inoculation. The criteria applied for assigning significance were: q-value ≤ 0.05 and estimated absolute |Ratio(Inoculation/MOCK)| ≥ 1. FPKM: Fragments Per Kilobase per Million mapped fragments.
12864_2024_10784_MOESM9_ESM.xlsx
Additional file 9: Table S7. The differential transcription of photosynthesis-related genes in the contrast MOCK vs Inoculation. The criteria applied for assigning significance were: q-value≤ 0.05 and estimated absolute |Ratio(Inoculation/MOCK)| ≥ 1. FPKM: Fragments Per Kilobase per Million mapped fragments.
12864_2024_10784_MOESM10_ESM.xlsx
Additional file 10: Table S8. The differential transcription of circadian rhythm-related genes in the contrast MOCK vs Inoculation. The criteria applied for assigning significance were: q-value ≤ 0.05 and estimated absolute |Ratio(Inoculation/MOCK)| ≥ 1. FPKM: Fragments Per Kilobase per Million mapped fragments.
12864_2024_10784_MOESM11_ESM.xlsx
Additional file 11: Table S9. The differential transcription of WRKY transcription factors in the contrast MOCK vs Inoculation. The criteria applied for assigning significance were: q-value ≤ 0.05 and estimated absolute |Ratio(Inoculation/MOCK)| ≥ 1. FPKM: Fragments Per Kilobase per Million mapped fragments.
12864_2024_10784_MOESM12_ESM.xlsx
Additional file 12: Table S10. The differential transcription of MYB transcription factors in the contrast MOCK vs Inoculation. The criteria applied for assigning significance were: q-value ≤ 0.05 and estimated absolute |Ratio(Inoculation/MOCK)| ≥ 1. FPKM: Fragments Per Kilobase per Million mapped fragments.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, H., Lu, Y., Liu, Z. et al. Transcriptomic analysis unveils alterations in the genetic expression profile of tree peony (Paeonia suffruticosa Andrews) infected by Alternaria alternata. BMC Genomics 25, 861 (2024). https://doi.org/10.1186/s12864-024-10784-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10784-3