
Abstract
Biological networks serve a crucial role in elucidating intricate biological processes. While interspecies environmental interactions have been extensively studied, the exploration of gene interactions within species, particularly among individual microorganisms, is less developed. The increasing amount of microbiome genomic data necessitates a more nuanced analysis of microbial genome structures and functions. In this context, we introduce a complex structure using higher-order network theory, “Solid Motif Structures (SMS)”, via a hierarchical biological network analysis of genomes within the same genus, effectively linking microbial genome structure with its function. Leveraging 162 high-quality genomes of Microcystis, a key freshwater cyanobacterium within microbial ecosystems, we established a genome structure network. Employing deep learning techniques, such as adaptive graph encoder, we uncovered 27 critical functional subnetworks and their associated SMSs. Incorporating metagenomic data from seven geographically distinct lakes, we conducted an investigation into Microcystis’ functional stability under varying environmental conditions, unveiling unique functional interaction models for each lake. Our work compiles these insights into an extensive resource repository, providing novel perspectives on the functional dynamics within Microcystis. This research offers a hierarchical network analysis framework for understanding interactions between microbial genome structures and functions within the same genus.
Similar content being viewed by others


Background
Biological networks [1,2,3] serve as effective frameworks that elucidate the multifaceted relationships among biological entities. They are instrumental in advancing the understanding of a wide array of biological processes, including but not limited to cell differentiation, pharmacological interactions with biological pathways and the discovery of disease pathways [4,5,6]. These processes’ architectural structures and interactions can be accurately depicted as graphs (networks), where nodes signify biological units and edges depict various forms of connections or relationships between them. Utilizing networks allows for these complex biological processes to be visually and conceptually simplified. Analytical methods such as graph theory, machine learning and deep learning are leveraged to model and elucidate their complex molecular mechanisms, thereby facilitating a comprehensive exploration and understanding of biological phenomena across multiple dimensions and scales [7]. Despite considerable progress in the field of biological networks, the majority of existing research has been confined to interactions within communities and species (ecological networks [8, 9]) and between functional genes (metabolic networks [10,11,12]). Although studies have explored functional interactions between genes within a species from the perspective of pan-genomics [13,14,15], studying the patterns of functional gene interactions within a microbial group at the genus level from the perspective of biological networks remains an underexplored area of the field.
Owing to the rapid accumulation of microbiome genomic data, there is an imperative requirement for the integration and representation of the vast and intricate microbial communities [16, 17]. Employing microbial network analysis as a tool to discern community states and ecological niches has gained significant traction in the study of microbial community structures [18, 19]. Studies suggest that community structures are influenced by both internal and external functional interactions [20, 21]. These interactions afford a comprehensive perspective on microbial communities and enhance the understanding of functional distributions within them [22, 23]. While current microbial network analyses have yielded valuable insights, the emphasis has largely been on interactions among microbial communities in diverse ecosystems [24, 25]. In contrast, the functional interactions for the same genus have been comparatively neglected.
For the exploration of gene function interactions within individual prokaryotic microorganisms, this research has chosen Microcystis as the focal organism. As a ubiquitous freshwater cyanobacterium with toxigenic capabilities, Microcystis thrives in a diverse array of ecological niches [26,27,28,29]. Its role in shaping aquatic microbial ecosystems under scenarios of global change is escalating in significance. From an ecological standpoint, Microcystis, via its extracellular polysaccharides, serves as a nutrient-rich substrate for a plethora of other bacteria, while also providing them with a physical shield against predation [30,31,32]. The volume of publicly published Microcystis genomes has exponentially increased. Within the Microcystis genus, traits such as nutrient affinity, absorption rates, cell quotas, nitrogen metabolism, and toxin production manifest remarkable heterogeneity [28, 33, 34]. Although Microcystis genomes demonstrate a high degree of sequence conservation and a relatively consistent core gene set [28, 29, 35], they exhibit considerable variation in both genome size and gene count [29, 36]. This marked genomic variability is largely attributable to the genome’s inherent plasticity and the occurrence of horizontal gene transfer (HGT), factors that are pivotal in the evolutionary trajectory and environmental adaptability of Microcystis genomes [37,38,39,40,41]. Therefore, given the genome with both conserved and open characteristics, extensive environmental resilience, and intricate microenvironments, Microcystis represents an ideal model for studying genome structures with specialized functions. It is crucial to investigate the functional gene interaction patterns and environmental stability within Microcystis communities from a genome structure perspective, further solidifying Microcystis as an exemplary research subject.
Higher-order relationships refer to interactions beyond two nodes, are observed to be prevalent in a biological network such that they almost comprise the entire network structure [42]. For example, in protein-protein interaction networks, there are a large number of more than two protein interactions. In the field of neuroscience, shifting from considering pairwise interactions to capturing higher-order relationships provides a more nuanced analysis of information interaction and functional response mechanisms within brain networks [43, 44]. Under the limitations of methodology, most extraction of high-order structures in biological networks is based on pairwise relationships, that is, when there is a triadic relationship between three nodes, it is considered that there is a high-order relationship between these three nodes [45,46,47]. This study pioneers the concept of “solid motif structures (SMS)”, thereby laying the foundation for a new hierarchical framework within network biology. SMS is the complex structure extracted from the Microcystis functional network using higher-order network theory. This type of structure represents stable functional interaction among three or more entities in the network. From a topological perspective, this type of higher-order relationship is more meaningful than the interaction between multiple nodes filtered through pairwise relationships [48, 49]. It dissects the interaction patterns between functional genes within the microbial genome from the same genus, with the ultimate goal of achieving a nuanced understanding of the complex interactions between functional genes in genome for the same genus. In summary, this study constructed a hierarchical analysis framework for functional microbial genome from a network perspective and explored the functional interaction patterns of Microcystis at different network scales. Moreover, metagenomic data from seven globally distributed lakes were chosen for in-depth analysis of the localized characteristics of Microcystis genome structures in distinct ecological settings and to model the functional interaction patterns. As an additional contribution, we have compiled a comprehensive resource repository of Microcystis functional interactions, serving as an invaluable repository for future research endeavors.
Results
Precision-guided elucidation of local structures in the microcystis genome network
Efficient construction of the microcystis genome structure network
This study assembled a genome structure network based on 162 high-quality publicly available Microcystis whole genomes. These genomes encompassed a total of 718,579 protein sequences, yielding a network with 39,094 nodes (see Sect. Microcystis genome structure network construction). The compression ratio of the Microcystis network nodes to sequences is 18.4, dramatically condensing the genomic information. Of the 5,594 nodes (representing 296,626 genes) annotated to functions (functional orthologs level, see Sect. Metagenome analysis for microcystis genomes), 5,255 nodes (94%) were associated with a unique K number (from KEGG Orthology Database [50], A K number represents a functional ortholog), attesting to the high fidelity of the Microcystis genome structure network. An analysis of species number distribution within nodes (Fig. 1a) showcased a concentration at both extremities of the species distribution. This pattern indicates the coexistence of both conserved and distinct nodes within the network, reflecting the intricate balance between the openness and conservation inherent in the Microcystis genome.
Harnessing ‘uniform’ topological units to ensure information integrity
To study the topological distribution of genome structural networks, this study utilized an optimal community detection scheme (see Sect. Community detection scheme) based on deep learning to isolate structures to elucidate the local topological features of the genome structure network, identifying 18,069 communities. Each community represents a topologically meaningful cluster, referred as a topological unit. In terms of community scale (defined as the number of nodes contained in the community) and thickness (defined as the average number of genes contained in all nodes in the community), the community detection scheme effectively partitioned the Microcystis genome network into ‘uniform’ topological units (Fig. 1b). This means that the Microcystis genome structure network has a clear topological pattern distribution, rather than a random network.
To assess whether this ‘uniform’ community partitioning would compromise the network’s information structure and to determine if the network’s information could be reconstructed from the partitioned units, this study clustered species based on their co-occurrence in communities. CVTree constructs whole-genome based phylogenetic trees by using a Composition Vector (CV) approach to infer evolutionary relatedness of microbial organisms [51, 52]. The clustering results were aligned with the CVTree constructed from the 162 Microcystis genomes to verify the scheme’s fidelity (Fig. 1c). Comparison of the optimal scheme against five other community detection methods, including two classic traditional methods (Deepwalk [53] and Louvain [54]) and three cutting-edge deep learning methods (GAE [55], ARGA [56], and GALA [57]). These methods are often used as benchmark for community detection schemes. The greater the number of consistent clustering pairs, the shorter the distance between trees and the larger the consistent subgraph, which means the higher the consistency between the two trees. Based on four evaluation metrics (Table 1) revealed that the optimal scheme was closest to the CVTree and exhibited the most consistent clustering relationships. This consistency manifested as two large subgraphs, whereas other methods yielded fragmented clusters. These findings corroborate the optimal scheme’s superior capability for information restoration and its evolutionary relevance in genome structure.
Exploration of functionally associated components across multiple dimensions
In order to explore whether these topological clustering results are functionally meaningful after the genome structure network is divided into several topological units through the community detection scheme, this study isolated three tiers of functional association components based on three dimensions in KEGG database (functional orthologs(K), functional pathways(ko), functional modules(M), [50]) and refined by co-occurrence correlations observed within topological units (Supplementary Fig. 1a-c). At the K dimension, 309 functional association components were identified, incorporating 1,154 functional orthologs, which constitute 59.7% of all genes annotated with K numbers. At the ko dimension, 26 functional association components were discovered, including 182 functional pathways, making up 63.2% of all genes annotated with ko numbers. At the M dimension, 47 functional association components were uncovered, comprising 346 functional modules, accounting for 91.3% of all genes annotated with M numbers. A complete list of functional association components is available in Supplementary Tables 1–3. The results showed that Microcystis exhibited obvious non-random functional clustering phenomenon, forming functional association components, indicating that the clustering of topological features in genome structure and functional clustering are correlated, which laid the foundation for further mapping topological units onto the PPI network to divide functional subnetworks.
Regional specificity and conservation in Microcystis genome
Further explorations were conducted to ascertain the presence of region-specific topological units within the genome structure network. The 162 Microcystis strains were categorized into six groups based on geographic (based on continent) and climatic (based on climatic zone) criteria (North America temperate, South America tropical, South America temperate, Asia temperate, Europe temperate, Africa temperate). A substantial number of region-specific communities were identified (440 in the Asia temperate, 424 in the North America temperate, 110 in the South America tropical, 102 in the Europe temperate, 7 in the South America temperate, and 2 in the Africa temperate, Fig. 2a, red in figure). Further analysis of the abundance disparities of 27 key Microcystis functions between region-specific and shared communities (Fig. 2b) revealed that functions such as xenobiotics metabolism (XM), restriction modification system (RM), methane metabolism (MM), and carbon fixation (CF) displayed significant regional specificity. Conversely, other key functions appeared to be relatively conserved. This suggests that while the Microcystis genome structure is influenced by environmental variables like climate and geography, certain key functions remain relatively stable across different regions.

Evaluating the Efficacy of the Optimal Community Detection Scheme. (a) Species Distribution in the Genome Structure Network: This subplot depicts the distribution of species counts within nodes of the Microcystis genome structure network. There’s a concentration of nodes at both ends of the distribution, highlighting the presence of both conserved and unique nodes. The x-axis represents the count of species and the y-axis details the count of nodes. (b) Distribution of Community Dimensions: This subplot illustrates the distribution of community scale and thickness within the Microcystis genome structure network. The community detection approach effectively segmented the Microcystis genome network into topologically consistent units. The x-axis community scale is defined as the number of nodes contained in the community. The y-axis community thickness is defined as the average number of genes contained in all nodes in the community. (c) Comparative Analysis with CVtree Species Clustering: This part contrasts the optimal community detection scheme with the CVtree species clustering method. The results show that there are a large number of corresponding clusters between the two trees, which reflects the reliability and precision of the scheme

In-depth Analysis of Functional Associations in the Microcystis Genome Structure Network. a. Interactions Among Regional Communities: The upper bar chart quantifies the number of communities associated with each type of regional interaction. The dot matrix beneath delineates the specific regional interactions. The left-side bar chart enumerates communities per regional categorization. Red represents topological units unique to a single group. Key regional abbreviations include AF_TE (African temperate), SA_TE (South American temperate), SA_TR (South American tropical), EU_TE (European temperate), NA_TE (North American temperate), and AS_TE (Asian temperate). b. Regional Distribution of Key Functions: This segment delves into the distinct and shared abundance of pivotal Microcystis functions across varied climatic and geographical regions, illuminating the environmental impact on functional prevalence. The x-axis represents the function types and the y-axis represents the proportion of function abundances in different regions. ‘Conservative’ indicates the number of functional labels that are present in all regions. Note that the sum of the proportion in the existing regional groups for almost all functions is less than 100%, which is caused by the lack of geographical information in 11 of the 162 Microcystis genomes (see Sect. Data Acquisition)
Overcoming structural constraints to construct microcystis key functional subnetworks
Constructing key functional subnetworks in microcystis
To transcend the limitations imposed by the location structure of the genome and to enable a more comprehensive analysis of local functional linkages in the Microcystis genome, topological units (or communities), were mapped onto the protein-protein interaction network, resulting in the construction of 27 Microcystis key functional subnetworks (Supplementary Figs. 2,3). Detailed network information is cataloged in Supplementary Table 4. The criterion for partitioning these functional subnetworks was to include all nodes within the community where a specific functional node is located (see Sect. Partitioning functional subnetworks). Consequently, each key functional subnetwork comprises nodes associated with other key functions as well as unknown functions.
The existing functional annotation rates across these functional subnetworks exhibited variability (Fig. 3a). The annotation rate of nodes generally ranged between 0.5 and 0.7, suggesting that the proportion of proteins with identified functions remained relatively consistent across different networks. However, the annotation rate for edges fluctuated considerably, ranging from 0.2 to 0.8, which highlighted significant disparities in protein interaction types among various functions. For functions such as response to oxidative stress (ROS), exopolysaccharide synthesis (EPS), restriction modification system (RM), and antibiotic-related functions (KKS), existing research offered some insights into many key proteins. Nevertheless, the comprehension of the intricate interactions governing these functions remains somewhat limited. Within our current knowledge paradigm, the 27 Microcystis functions showcased a unique association clustering pattern (Fig. 3b), highlighting the possible functional interaction patterns between these key functions.
Distinct topological characteristics across key functional subnetworks
The topological attributes of each key functional subnetwork display variability (Supplementary Table 5). Ten key functions, namely TCA cycle (TCA), response to oxidative stress (ROS), gas vesicle (GVP), amino acid transporters (AAT), carbon dioxide concentration mechanism (CCM), nitrogen metabolism (N), arginine, and proline metabolism (AP), exopolysaccharide synthesis (EPS), xenobiotics metabolism (XM), CRISPR-Cas systems (CAS), manifested distinct scale-free properties. This indicated the existence of highly significant proteins, often referred to as hub proteins, within these functional subnetworks. Conversely, the remaining 17 key functions displayed small-world properties, signifying that nodes within these subnetworks exhibited pronounced functional and structural clustering. Notably, no functional network manifested random network properties, affirming that evolutionary selection pressure has guided the Microcystis key functional subnetworks towards a specific, non-random functional or stability state.
Conserved topological patterns across functional subnetworks
Based on the motif count distribution pattern depicted in Fig. 3c, it was evident that despite the varying scale and complexity of the Microcystis functional subnetworks, they generally adhered to a specific topological distribution pattern and exhibited a preference for particular topological shapes. This suggested that Microcystis maintains consistent local structural patterns across its different functional subsystems. Such recurring patterns could represent fundamental, conserved functional units and indicate that local structures serve as a viable starting point for deciphering the functional interaction patterns within Microcystis.

Topological Characteristics of the 27 Principal Microcystis Functional Subnetworks. a. Node and Edge Annotation Rates: The annotation rate for nodes is defined as the ratio of nodes annotated to specific functions to all nodes in the network. The annotation rate for edges is defined as the ratio of edges annotated to functional interaction types to all edges in the PPI network. Across the 27 functional subnetworks, the proportion of proteins with discerned functions remains relatively stable. However, the edge annotation rate exhibits significant variability. b. Interaction Clustering Heatmap: This clustering heatmap underscores functional cohesion among the 27 key functions based on euclidean distance, laying the groundwork for deeper insights into their interdependencies. The horizontal axis (on the left) gauges the interaction intensity between functional subnetworks, while the vertical axis measures the co-occurrence correlation of various functions within these subnetworks. Red represents functions consistent with the scale-free model and black represents functions consistent with the small-world model. The color in figure indicates the degree of correlation (top left figure), the brighter the color, the higher the degree of correlation. c. Distribution of Motif Counts: Almost all Microcystis functional subnetworks follow specific topological distribution patterns, showing a preference for certain topological configurations. The x-axis enumerates motif types within groups of 3–5 nodes, amounting to 29 categories (3 nodes: 2 types, 4 nodes: 6 types, 5 nodes: 21 types)
Local topological patterns in the microcystis genome functional network
Classification of SMS types reveals functional complexity
Solid Motif Structure (SMS) is a type of higher-order interaction structure that takes into account the meaning of network topology. Since this study calculates this high-order structure on a PPI network, SMS express high-order interaction information at the functional level. SMS as unique local configurations within the functional network, functioning as specialized functional modules. The composition of these modules highlights the critical roles that various functions play within subnetworks during specific biological processes or signaling pathways (Fig. 4a). SMS can be classified into three types based on their functional composition and the proportion of unknown functions: unknown-function SMS (comprising over 50% of nodes with unknown functions), pure SMS (with over 50% of nodes sharing the same known function), and complex SMS (with over 50% of nodes have different known functions). A total of 11,736 SMSs were identified within the 27 key functional subnetworks of Microcystis, including 2,977 Pure SMSs (24.9%), 4,935 Complex SMSs (43.1%), and 3,824 unknown-function SMSs (32.0%). In terms of sheer numbers, the distribution across these three SMS types appears relatively balanced.
The scale of pure SMSs within the Microcystis key functional network was notably extensive. For crucial functionalities such as such as Gas vesicle (GVP), CRISPR-Cas systems (CAS), Response to Oxidative Stress (ROS), and Amino acid transporters (AAT), the prevalence of pure SMSs is pronounced. This suggested a high level of functional modularization, reflecting the specificity and differentiation of functions within these networks. All these functions align with the scale-free model. Conversely, most functional networks, such as Methane metabolism (MM), Alanine, aspartate and glutamate metabolism (AAG), and Carbon Dioxide Concentration Mechanism (CCM), exhibited a relatively low proportion of pure SMSs (below 20%). This indicated a high degree of functional overlap within these networks. The proportion of unknown-function SMSs served as an indicator of the current understanding of the functional network. Almost all subnetworks had a proportion of unknown-function SMSs higher than 20%, signifying that considerable gaps remain in our understanding of Microcystis key functional interactions.
Scale and distribution of SMSs indicate functional flexibility and robustness
The scale and distribution of the SMSs served as indicators of the association strength between various functions within the functional subnetwork (Fig. 4b). Network with many small SMSs suggested a multitude of protein interactions of similar importance. In the Microcystis functional subnetworks, functions like response to oxidative stress (ROS), gas vesicle (GVP), and xenobiotics metabolism (XM) predominantly followed this pattern. Most functional subnetworks featured SMSs of various scales, indicating that most Microcystis key functional networks prefer a diversified and flexible mode of functional interaction. This diversity conferred greater robustness and adaptability to the network, enabling it to modulate various functional interactions in response to environmental changes.
Structural roles of nodes reveal functional association
To delve deeper into the significance of each node, a comprehensive analysis was conducted based on the structural roles of nodes. The diversity of roles occupied by different functional nodes illuminated the various mechanisms by which proteins participate within network. Functions that assume multiple structural roles demonstrate strong evolutionary adaptability, capable of fulfilling diverse roles in different biological processes. All key functions occupy multiple roles in their respective functional subnetworks and assume 1–2 roles in other functional subnetworks. This suggested that these key proteins exhibited a high degree of functional diversity and adaptability within their own networks while maintaining specificity in other networks. Rich role interactions were particularly evident in the carbon fixation (CF), restriction modification system (RM), and metabolism of trace elements and vitamins (TEVIT) subnetworks (Fig. 4c). These functions appeared to serve as hub functions in the interaction of Microcystis key functions, playing a crucial role in maintaining the physiological processes and association of Microcystis. These hub functions likely coordinated and integrated multiple biological processes, potentially serving as the origin of functional complexity and diversity in biological processes.

Local topological features of Microcystis functional subnetworks. (a) SMS Composition Distribution: Across the 27 functional subnetworks, the functional composition of these SMSs elucidates the critical roles that various functions play within subnetworks. (b) Kernel Density Distribution of SMS Scales: SMS scale refers to the number of nodes that make up the SMSs, describing the scale of this high-order relationship. A subplot represents a functional subnetwork, the x-axis represents the SMS scale, and the y-axis represents the density of the kernel density distribution. The distribution of SMS scales indicates diverse functional modules and interaction patterns among proteins within the subnetworks. (c) Structural Role Differentiation: Take the three functional subnetworks of carbon fixation (CF), restriction modification system (RM), and metabolism of trace elements and vitamins (TEVIT) as examples. The x-axis represents the functional subnetwork, the y-axis represents the functional type, and each pie chart represents the richness of the node role corresponding to the functional node(y-axis) in the functional subnetwork (x-axis). The colors in the pie chart represent different role types, each color represents a topological grouping. The more colors in the pie chart, the higher the role richness
Advantages of the hierarchical network perspective in identifying key functional interactions of microcystis in different environments
New perspective reveals functional interaction differences across diverse environments
To assess the efficacy of the proposed new perspective in analyzing functional interactions in real environments, this study employed metagenomic data from seven globally distributed lakes to construct functional subnetworks. While the abundance distribution of key functional genes in these seven lakes was largely consistent (Fig. 5a), there were significant differences in the topological structures of their functional subnetworks (Fig. 5b). This demonstrated the capability of the network-based approach to identify nuanced functional interaction differences, thereby revealing that the manifestation and network construction of Microcystis key functions vary depending on the environments.
The SMS scale distribution pattern under different environments
In order to explore the differences in functional interaction patterns within Microcystis communities under different environments, the distribution of SMS scale in the functional subnetwork was used as an observation indicator. Functions (Fig. 5c) like alanine, aspartate and glutamate metabolism (AAG), fatty acid metabolism (FAM), amino sugar and nucleotide sugar metabolism (SUM), photosynthesis (PH), restriction modification system (RM), and metabolism of trace elements and vitamins (TEVIT) exhibited consistent SMS scale distributions across the seven lakes. This indicates that these functions preserve a relatively invariant role and expression within Microcystis. Functions like bacterial secretion system (BBS), carbon fixation (CF), and nitrogen metabolism (N) displayed marked variances in SMS scales. This observation underscores their potential criticality as Microcystis adapts to diverse ecological settings.
Functional interaction models reveal environment-stable and environment-specific flows
To delve deeper into the functional interaction pattern differences across various lakes, this study constructed functional interaction models for the seven lakes based on the importance roles of nodes in their functional subnetworks (Fig. 5d, Supplementary Fig. 4, see Sect. Metagenome analysis for microcystis genomes). These models revealed three environment-stable functional flows that were extremely consistent across all seven lakes, as well as numerous environment-specific functional interaction pattern s. Notably, certain information flows, those between xenobiotics metabolism (XM) and oxidative phosphorylation (OP), and between glycolysis (GLY) and pentose phosphate pathway (PPP), were exclusive to Lake FP23, underscoring its distinct ecological dynamics. Most other functional interaction paradigms manifested in at least three lakes, with the information flows coalescing into a connected graph.
Functions like fatty acid metabolism (FAM), antibiotic-related functions (KKS), xenobiotics metabolism (XM), and secondary metabolism (SM, [58]) appeared only in information flows in specific lakes, hinting at their environment-dependent interactions. Functions like gas vesicle (GVP, [59]), CRISPR-Cas systems (CAS, [60]), bacterial secretion system (BBS), exopolysaccharide synthesis (EPS, [61]), amino acid transporters (AAT), restriction modification system (RM, [62]), and phosphorus metabolism (P) remained absent from the functional interaction models, suggesting their low interaction with other Microcystis key functions. The bulk of the functional interactions exhibited conservation across the seven lakes. This layered exploration offers a new perspective to probe the intricate interplay between key microbial functions and their surrounding ecosystems.

Elucidating Microcystis Key Functional Interactions Across Diverse Environments Using a Hierarchical Perspective. a. Distribution of Functional Genes Across Lakes: The abundance distribution of pivotal functional genes across seven lakes demonstrates a significant level of consistency. b. Topological Variability of Key Functional Subnetworks: Distinct environments manifested significant differences in the topological structures of their functional subnetworks, underscoring the framework’s ability to discern subtle functional interaction disparities. Metrics from top left to bottom right include node count, edge count, average degree, and clustering coefficient. c. SMS Scale Distribution in Functional Subnetworks: This visualization delves deeper into the correlation between the abundance of distinct functional SMSs and lakes. Each subplot represents a functional subnetwork, the x-axis represents the SMS scale and the y-axis quantifies the proportion of SMSs based on 162 Microcystis samples from each lake. d. Functional Interaction Models: This model visuals the conservation and specificity in key functional interactions within the Microcystis functional framework across diverse environmental contexts. The left segment (in blue) showcases environment-stable model, representing interaction patterns consistent across all seven lakes. The right segment (in red) highlights environment-specific model, pinpointing interaction patterns unique to specific lakes
Discussion
Hierarchical dissection of the microcystis functional genome: bridging current research and unveiling ecosystems complexity
This study employed a hierarchical deconstruction approach, ranging from macro to micro (network-subnetwork-SMS-node), to illuminate the interaction patterns of key function in Microcystis. The diversity of these interaction patterns, which aligns with existing research, highlights the inherent complexity and dynamism inherent in microbial ecosystems.
At the macro level, the study identified association patterns among 27 key functions based on co-occurrence patterns within functional subnetworks. Strong associations between nitrogen and phosphorus metabolic modes were confirmed, corroborating findings by Downing et al. [63]. This adds to the understanding that the regulation of Microcystis toxins may be indirectly influenced by these metabolic modes. Additionally, the study found a close association between heavy metal genes and antibiotic resistance genes, which aligned with existing evidence linking antibiotic resistance gene abundance to environmental heavy metal pollution [64]. The relative independence of the gas vesicle function opened up new avenues for research into its regulatory mechanisms [65].
A deeper examination of the SMSs composition revealed intricate patterns of key functional combinations and interactions. Within the photosynthesis subnetwork, eight SMSs associated with the response to oxidative stress were identified, aligning with existing research on the impact of light intensity and hydrogen peroxide on Microcystis [66, 67]. The photosynthesis network displayed a highly modularized structure, encompassing five subprocesses (Photosystem I, Photosystem II, Cytochrome b6f complex, F-type ATPase, and Photosynthetic electron transport) and showed extensive interactions with phosphorus metabolism, thereby confirming its regulatory role in the synthesis of Microcystis secondary metabolites secondary metabolites.
At the micro level, the study focused on role composition within functional subnetworks. Functions like CRISPR-Cas systems, exopolysaccharide synthesis, response to oxidative stress, and secondary metabolism each occupy multiple roles within their respective subnetworks, corroborating their abundance studies in Microcystis. Despite the extensive connections of phosphorus metabolism-related functional genes within the nitrogen metabolism subnetwork, phosphorus metabolism assumed a singular role. This suggested a unique specificity of phosphorus metabolism-related genes in nitrogen metabolism, potentially contributing to the stability of the nitrogen metabolism network.
Through the hierarchical network analysis framework, this research not only corroborated established findings but also unveiled previously uncharted facets of microbial ecosystem complexity and adaptability.
Future extensions for multi-species and spatiotemporal scales: expanding the horizon of microbial genome
This study, focusing on the microbial functional genome for the same genus, employed a network-based approach to dissect the interaction patterns of key functions. This methodology offers remarkable scalability, opening up new avenues for future research that extends to multi-species relationships and broader spatiotemporal scales.
In the context of multi-species research, the network-based perspective can provide a nuanced understanding of biological evolutionary relationships and functional interaction patterns. By comparing these SMSs across diverse species, we can identify both shared and unique biological processes. This has the potential to illuminate the underlying patterns and drivers of evolutionary change, thereby enriching the understanding of species diversity, common ancestry and ecosystem interactions.
The network-based methodology also offers considerable scalability across spatial and temporal scales. On the spatial front, comparing Microcystis SMSs across various geographical settings can yield insights into how environment factors shape the functional characteristics and survival strategies of Microcystis. On the temporal scale, monitoring dynamic changes in these SMSs can unveil how Microcystis adapts to environmental fluctuations and seasonal shifts. Such insights would contribute to the biological adaptation mechanisms and evolutionary trajectories.
Conclusion
In this research, we introduce the concept of Solid Motif Structures (SMS) and employ a hierarchical network analysis framework to scrutinize the Microcystis functional genome. This approach enhances our understanding of the intricate dynamics between genome structural and functional interconnections within microorganisms of the same genus. Our findings illuminate the specificity and conservation inherent in microbial functional genomes. Furthermore, our research offers a novel perspective on the adaptive mechanisms by which microbial functional genomes respond to environmental stress.
Materials and methods
Data acquisition
Microcystis genome repository: A total of 162 publicly available Microcystis genomes (Supplementary Table 7) were sourced from the NCBI database (https://www.ncbi.nlm.nih.gov/). Geospatial data were collected for 151 of these strains, with the majority originating from the United States (51 strains), Canada (26 strains), Japan (25 strains), Brazil (24 strains), and China (13 strains). (Supplementary Fig. 5a)
Functional gene information:
-
Information pertaining to antibiotic-related functions (KKS) were procured from the CARD database (https://card.mcmaster.ca/).
-
Information on heavy metal (HM) related functional genes was sourced from the BacMet database (http://bacmet.biomedicine.gu.se/).
-
Information on restriction modification system-related functional genes was sourced from the REBASE database (http://rebase.neb.com/rebase/rebase.seqs.html).
-
Additional functional genes related to gas vesicle, CRISPR-Cas systems and secondary metabolism were extracted from Microcystis protein FASTA files.
-
All remaining functional gene data were retrieved from the KEGG database through functional annotation (https://www.ncbi.nlm.nih.gov/).
Protein-protein interaction data: Interaction datasets were obtained from the STRING database (https://string-db.org/).
Lake metagenome data: Metagenomic datasets for seven distinct lakes were sourced from the open-access Project PRJNA575023. (Supplementary Fig. 5b, Supplementary Table 8)
Hierarchical network analysis framework for functional microbial genome
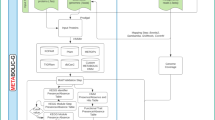
This study introduces a hierarchical framework for the functional genomic analysis of Microcystis from a network perspective.
Step 1: Utilizing the protein sequence information and coding sequence information (CDS) of the Microcystis genome, a genome structural network (Fig. 6a, see Sect. Microcystis genome structure network construction) is constructed. This network integrates extensive genome information from the same genus, illustrating gene positional relationships within the same genus.
Step 2: Employing a developed scheme (optimal community detection scheme), the genome structure network is segmented into several topologically meaningful clusters, referred to as topological units (Fig. 6b, see Sect. Community detection scheme).
Step 3: To correlate the topological aggregation with functional interactions, topological units are mapped onto the Microcystis protein-protein interaction (PPI) network. The PPI network is then divided into 27 subnetworks based on key functions (the selection of key functions is based on the current focus within Microcystis research) (Fig. 6c, see Sect. Partitioning functional subnetworks). These functional subnetworks retain dual information on gene positional relationships and functional interactions within the Microcystis genome.
Step 4: To further refine the study of functional interactions in the Microcystis genome from a network perspective, network analysis theories are utilized to extract higher-order topological structures, Solid Motif Structures (SMS), and to focus on the importance of each node within the networks (Fig. 6d-e, see Sect. Microcystis genome structure network construction).
Step 5: Considering real environment, the deconstructed functional interaction relationships are applied to metagenomic data from seven global lakes to model functional interaction patterns within the Microcystis in actual environments (Fig. 6f, see Sect. Metagenome analysis for microcystis genomes).
Step 6: The functional interaction relationships extracted at various levels of the framework (network level, subnetwork level, SMS level, and node level) are organized to construct a comprehensive resource repository (Fig. 6g).

Hierarchical Network Analysis Framework for Functional Microbial Genome. This figure delineates a top-down hierarchical framework tailored for the intricate analysis of the functional microbial genome. (a) Drawing from 162 publicly accessible Microcystis genomes, the Microcystis genome structure network was constructed (see Sect. Microcystis genome structure network construction) to represent the location relationship between genome structures and functions for the same genus. (b) A community detection scheme (see Sect. Community detection scheme) based on deep learning was harnessed to identify and extract topological units from the network, ensuring optimal information fidelity. (c) These discerned topological units were seamlessly integrated into the overarching protein-protein interaction network, leading to the derivation of 27 pivotal Microcystis functional interaction subnetworks, thereby facilitating a nuanced exploration of key functional interplays across structural paradigms. (d) Building upon these functional interaction subnetworks, the study embarked on the identification of SMSs (see Sect. Partitioning functional subnetworks), aiming to provide a granular deconstruction of interaction patterns within these subnetworks. (e) A comprehensive analysis was undertaken, with a focal emphasis on the structural roles of individual nodes (see Sect. Microcystis genome structure network construction), underscoring the significance of each node within the network. (f) Taking into account real environment, functional interaction models were constructed to amalgamate both conserved and unique functional interaction patterns based on metagenomic data (see Sect. Metagenome analysis for microcystis genomes). (g) Synthesizing the insights, a comprehensive resource repository of Microcystis functional interactions was curated, serving as an indispensable reference for subsequent research endeavors in the realm of microbial genomes
Microcystis genome structure network construction
The genome structure network is a network model that uses a large number of genomes to reflect the gene position relationship within the same genus [68]. Each node represents a cluster of genes with high sequence similarity, while each edge represents an upstream or downstream positional relationship between two nodes based on gene location.
Node construction: Sequence similarity among all genes within the Microcystis genome was accessed using BLAST. To efficiently identify similar genes, this study employed a bidirectional best hits approach with an e-value threshold of 1e-6, aggregating sequences that exhibited both percent identity and coverage rate surpassing 80% into a single node [68].
Edge construction: Edges within the genome structure network symbolize the positional relationship between gene pairs within a given genome using the data of coding DNA sequence (CDS). Initially, positional relationship pairs between genes from each genome were established (Fig. 7). Subsequently, sequence similarity for all genes was evaluated and identical node IDs were assigned to genes demonstrating high similarity and coverage. Finally, these gene positional relationship pairs were integrated to form a genome structure network. Edge weight is defined as the number of relationship pairs between nodes.

Schematic diagram of edge construction. This figure is quoted from our previously published results [68]. The figure shows the process of constructing the edges of the genome structure network. First, the position relationship pairs between two genes in each genome are obtained, and then the genome structure network is constructed based on the nodes. The edge weight represents the number of relationship pairs
Community detection scheme
Louvain community detection
The Louvain algorithm [54] serves as an optimized community detection algorithm commonly employed for identifying community structures in large-scale network graphs. The algorithm’s fundamental objective is to maximize modularity within the network graph, where modularity is a quantitative index assessing the quality of community structures. A higher value signifies more cohesive intra-community connections and fewer inter-community links. The algorithm operates in a two-phase cycle, iteratively executed until modularity ceases to improve (Fig. 8a). The calculation of modularity is as follows:
Where \(\:\varDelta\:M\) is the modularity change caused by moving node i into community C, where \(\:\sum\:in\:\)is the sum of edge weights inside community C; \(\:\sum\:tot\) is the sum of edge weights of all nodes in C; \(\:{k}_{i}\) is the sum of weights that edges connected to node i; \(\:{k}_{i,\text{i}\text{n}}\) is the sum of the weights that edges connecting node i to the internal nodes of C; W is the sum of the weights of all edges in the network.
Consistency clustering
Given that the Louvain algorithm is a greedy algorithm sensitive to random seeds and initial conditions, it may yield varying community partitions for the same network. To address this, the study employs a robust community consistency clustering approach, outlined as follows (Fig. 8b):
-
Step 1: Execute the Louvain algorithm on the genome structure network n times, yielding n sets of community partitions.
-
Step 2: Aggregate all community detection outcomes. Edge weights are defined as the probability Dij that two nodes are grouped within the same community across all runs.
-
Step 3: Implement a threshold τ to filter edges and derive consistent community partitions.
AGE algorithm
Unlike the Louvain algorithm, which focuses solely on topological node similarity, the Adaptive Graph Encoder (AGE) is a graph embedding framework utilizing graph convolution networks to offer a more nuanced representation of graphs, thereby enriching the community partitioning of attribute-rich graphs [69]. The AGE algorithm employs an efficient graph filter for Laplacian smoothing and uses an adaptive learning strategy for node feature training.
Optimal community detection scheme
By synergistically integrating the consistency and AGE community detection methods, this study introduces a novel community detection combination scheme. Specifically, the node community co-occurrence matrix D obtained through consistency clustering and the community clustering matrix S derived from the AGE algorithm are point-multiplied to generate the combined community clustering matrix C. This matrix is subsequently clustered to yield the final community detection outcome. (Fig. 8c). Two adjustable parameters are involved: τ, the consistency clustering threshold, and n, the number of communities partitioned by the AGE algorithm. The optimal settings were determined to be τ = 0.7, n = 100. (Supplementary Table 6).
Quantitative evaluation metrics for tree consistency
In this research, a species clustering tree was constructed based on the community-species co-occurrence relationships derived from the community detection scheme. Using CVtree as a reference point, we calculated the tree consistency between the CVtree and the species clustering tree generated by each scheme. This approach aimed to evaluate the biological validity and scientific rationale of the community detection scheme. To achieve a comprehensive and quantitative assessment of tree consistency, we employed four distinct metrics:
-
Distance between trees: The distance between two nodes on a tree is quantified by the number of root nodes separating them. The difference in distance between two nodes across two trees is calculated as the disparity in their respective distance on each tree. This metric is the cumulative sum of the distance differences for all node pairs across two trees, serving as an index for the divergence in clustering outcomes between the trees.
-
Number of consistent clustering pairs: Node pairs maintaining identical distances across two trees are considered to exhibit a consistent clustering relationship. This metric counts the node pairs that exhibit consistent clustering across two trees, thereby quantifying the degree of clustering consistency.
-
Number of consistent subgraphs and the largest consistent subgraph: This metric involves linking the node pairs with consistent clustering relationships across two trees and enumerating the resultant subgraphs and their maximum sizes, thereby providing a measure of the overall clustering consistency.

Comprehensive Community Detection Scheme. (a) Louvain Algorithm Overview: The Louvain algorithm operates in two iterative stages until modularity plateaus. Stage one: Each node is considered an autonomous community. Nodes then strive to align with neighboring communities that maximize modularity, culminating in the formation of small communities. Stage two: Each emergent community is recast as a “super-node” and interconnections between these super-nodes are established to preserve original inter-community connections. The graph is then simplified into a new network where nodes symbolize communities and edges denote inter-community relationships. (b) Consistency Clustering Overview: Stage one: The Louvain algorithm is applied to the genome structure network n times, generating n sets of community partitions. Stage two: These partitions are amalgamated and edge weights are defined as the probability Dij that two nodes are grouped within the same community across all runs. Stage three: A threshold τ is applied to filter edges and derive consistent community partitions. (c) Scheme Synthesis: The node community co-occurrence matrix D obtained from consistent clustering and the community clustering matrix S derived from the AGE algorithm are point-multiplied to produce the combined community clustering matrix C. This matrix is subsequently clustered to yield the final community detection outcome
Partitioning functional subnetworks
The construction of functional subnetworks is achieved by slicing the genome structure network communities (i.e., topological units) after mapping them onto the protein-protein interaction network. The criterion for slicing the functional subnetworks is to include all nodes within the communities where specific functional nodes reside. For example, to construct the functional subnetwork for function FA, first, all nodes labeled with function FA (i.e., N1, N2) are extracted. Knowing that N1 belongs to community C1, with other nodes in the same community being N3, N4, N5; and N2 belongs to community C2, with other nodes in the same community being N6, N7. These two nodes’ communities (C1, C2) are then extracted, along with all nodes within them (N1-N7), and mapped onto the PPI network. Thus, these 7 nodes form the interaction subnetwork based on function FA.
Complex network analysis techniques
All the following methods are implemented through python using networkx, Rolx and graph_tool packages.
Degree distribution
In network theory, a node’s degree signifies the count of unique nodes to which it is directly linked. Degree distribution serves as a statistical measure of the degrees across all nodes in a network and is commonly employed to characterize the network’s topological properties.
Clustering coefficient
The clustering coefficient of a node quantifies the ratio of actual connections among its neighbors to the maximum potential connections between them. The network overall clustering coefficient is the arithmetic mean of the clustering coefficients of all individual nodes, offering insights into the network’s local sub attributes.
Motif counting
A motif represents a recurring local structural configuration within a network, usually comprising a limited set of nodes and their interconnections. Motif counting involves enumerating the occurrences of specific motifs within the network, thereby aiding in the understanding of both its structural and functional dynamics.
SMS inference
The solid motif structure (SMS) is characterized as a tightly interconnected local modules within the functional network, indicative of robust functional correlations. This study employs a Bayesian approach to infer high-order interactions from low-order network configurations, thereby identifying SMSs within the network [70]. Part of the algorithm code has been open source and can be called through the python package graph_tool.
Node importance
This metric evaluates a node’s relative significance within the network based on four primary criteria: degree centrality, closeness centrality, betweenness centrality, and eigenvector centrality.
Node role discovery
The objective of role discovery is to ascertain and scrutinize the functional or positional roles of nodes within a network, extending beyond mere connectivity patterns [71].
Small world model and scale-free model
The scale-free model is a network generation paradigm where the network’s degree distribution adheres to a power-law distribution. This study assesses a network’s scale-free nature based on whether the variance of the network’s degree distribution exceeds its average degree. The small-world model is another network generation paradigm characterized by a minimal average path length between any pair of nodes and a high clustering coefficient.
Metagenome analysis for microcystis genomes
Data acquisition
Raw metagenomic data corresponding to water bloom events were sourced from the publicly accessible project PRJNA575023.
Quality control
Trimmomatic (v0.39) was employed for quality assurance of the raw reads, eliminating adapters sequences, low-quality reads and reads shorter than 50 base pairs(bp).
Assembly and binning
Clean reads were assembled into contigs using MEGAHIT (v1.2.9). Contigs exceeding 1500 bp in length were subjected to binning processes using MaxBin (2.2.6) and MetaBAT (v2.12.1). Bins were refined using MetaWRAP (v1.2.2) and their completeness and contamination levels were evaluated using CheckM (v1.0.12). Bins exhibiting completeness above 85% and contamination below 5% were retained for further analysis.
Functional annotation
Annotation of functional elements within the bins was conducted using KofamKOALA (v2023-04-01).
Functional interaction model construction
The construction of the functional interaction model is based on the importance of the functional nodes in each functional subnetwork. The importance of each function node in the functional subnetwork is quantified by calculating the four node centralities of all nodes (see Sect. Microcystis genome structure network construction). For example, if function A has a high level of node importance in the subnetwork of function B, a directed edge (information flow) from function A to function B is defined in the directed graph of the functional interaction model, which means that function A has important interactions in function B.
Data availability
All public databases used have been detailed with open access links in Sect. Data Acquisition Data Acquisition. The comprehensive resource library, which includes detailed insights into the interaction patterns of key functions in the Microcystis genome, can be accessed via the github: https://github.com/laxlyt/Microcystis-resource-library. This library encompasses three levels of functional association pattern information (K, ko, M) in the Microcystis network, detailed information on the 27 key functional subnetworks, Solid Motif Structures (SMS) data and node role information for each subnetwork. Researchers and enthusiasts are encouraged to explore and utilize this data for further studies and insights.
References
Alm E, Arkin AP. Biological networks[J]. Curr Opin Struct Biol. 2003;13(2):193–202.
Pavlopoulos GA, Secrier M, Moschopoulos CN, et al. Using graph theory to analyze biological networks[J]. BioData Min. 2011;4:1–27.
Koutrouli M, Karatzas E, Paez-Espino D, et al. A guide to conquer the biological network era using graph theory[J]. Front Bioeng Biotechnol. 2020;8:34.
Reuter JA, Spacek D, Snyder MP. High-throughput sequencing technologies. Mol Cell. 2015;58(4):586–97.
Hu T, Chitnis N, Monos D, et al. Next-generation sequencing technologies: an overview[J]. Hum Immunol. 2021;82(11):801–11.
Zhou XG, Ren LF, Li YT, et al. The next-generation sequencing technology: a technology review and future perspective[J]. Sci China Life Sci. 2010;53:44–57.
Muzio G, O’Bray L, Borgwardt K. Biological network analysis with deep learning. Brief Bioinform. 2021;22(2):1515–30.
Montoya JM, Pimm SL, Solé RV. Ecological networks and their fragility[J]. Nature. 2006;442(7100):259–64.
Guimaraes PR Jr. The structure of ecological networks across levels of organization[J]. Annu Rev Ecol Evol Syst. 2020;51:433–60.
Lacroix V, Cottret L, Thébault P, et al. An introduction to metabolic networks and their structural analysis[J]. IEEE/ACM Trans Comput Biol Bioinf. 2008;5(4):594–617.
Jeong H, Tombor B, Albert R, et al. The large-scale organization of metabolic networks[J]. Nature. 2000;407(6804):651–4.
Cottret L, Frainay C, Chazalviel M, et al. MetExplore: collaborative edition and exploration of metabolic networks[J]. Nucleic Acids Res. 2018;46(W1):W495–502.
Ma B, France M, Ravel J. Meta-pangenome: at the crossroad of pangenomics and metagenomics[J]. Pangenome, 2020: 205.
Rosconi F, Rudmann E, Li J, et al. A bacterial pan-genome makes gene essentiality strain-dependent and evolvable[J]. Nat Microbiol. 2022;7(10):1580–92.
Vallenet D, Calteau A, Dubois M, et al. MicroScope: an integrated platform for the annotation and exploration of microbial gene functions through genomic, pangenomic and metabolic comparative analysis[J]. Nucleic Acids Res. 2020;48(D1):D579–89.
Durán P, Thiergart T, Garrido-Oter R, et al. Microbial interkingdom interactions in roots promote Arabidopsis survival. Cell. 2018;175(4):973–83. e14.
Röttjers L, Faust K. From hairballs to hypotheses–biological insights from microbial networks. FEMS Microbiol Rev. 2018;42(6):761–80.
Kumar M, Ji B, Zengler K, et al. Modelling approaches for studying the microbiome. Nat Microbiol. 2019;4(8):1253–67.
Xiao Y, T M, Angulo, Friedman J, et al. Mapping the ecological networks of microbial communities. Nat Commun. 2017;8(1):2042.
Ellegaard KM, Engel P. Beyond 16S rRNA community profiling: intra-species diversity in the gut microbiota. Front Microbiol. 2016;7:1475.
de Vries FT, Griffiths RI, Bailey M, et al. Soil bacterial networks are less stable under drought than fungal networks. Nat Commun. 2018;9(1):3033.
Raman AS, Gehrig JL, Venkatesh S, et al. A sparse covarying unit that describes healthy and impaired human gut microbiota development. Science. 2019;365(6449):eaau4735.
Surana NK, Kasper DL. Moving beyond microbiome-wide associations to causal microbe identification. Nature. 2017;552(7684):244–7.
Banerjee S, Schlaeppi K, van der Heijden. M G A. Keystone taxa as drivers of microbiome structure and functioning[J]. Nat Rev Microbiol. 2018;16(9):567–76.
Matchado MS, Lauber M, Reitmeier S, et al. Network analysis methods for studying microbial communities: a mini review[J]. Comput Struct Biotechnol J. 2021;19:2687–98.
van Gremberghe I, Leliaert F, Mergeay J, et al. Lack of phylogeographic structure in the freshwater cyanobacterium Microcystis aeruginosa suggests global dispersal. PLoS ONE. 2011;6(5):e19561.
Cook KV, Li C, Cai H, et al. The global Microcystis interactome. Limnol Oceanogr. 2020;65:S194–207.
Dick GJ, Duhaime MB, Evans JT, et al. The genetic and ecophysiological diversity of Microcystis. Environ Microbiol. 2021;23(12):7278–313.
Harke MJ, Steffen MM, Gobler CJ, et al. A review of the global ecology, genomics, and biogeography of the toxic cyanobacterium, Microcystis spp. Harmful Algae. 2016;54:4–20.
HW P. Growth and reproductive strategies of freshwater blue-green algae (cyanobacteria). Growth and reproductive strategies of freshwater phytoplankton. 1988; 261–315.
Worm J, Søndergaard M. Dynamics of heterotrophic bacteria attached to Microcystis spp.(Cyanobacteria). Aquat Microb Ecol. 1998;14(1):19–28.
Brunberg AK. Contribution of bacteria in the mucilage of Microcystis spp. (Cyanobacteria) to benthic and pelagic bacterial production in a hypereutrophic lake. FEMS Microbiol Ecol. 1999;29(1):13–22.
Shen H, Song L. Comparative studies on physiological responses to phosphorus in two phenotypes of bloom-forming Microcystis. Hydrobiologia. 2007;592:475–86.
Tan X, Gu H, Ruan Y, et al. Effects of nitrogen on interspecific competition between two cell-size cyanobacteria: Microcystis aeruginosa and Synechococcus Sp. Harmful Algae. 2019;89:101661.
Lepère C, Wilmotte A, Meyer B. Molecular diversity of Microcystis strains (Cyanophyceae, Chroococcales) based on 16S rDNA sequences. Syst Geogr Plants. 2000;275–83.
Otsuka S, Suda S, Li R, et al. Morphological variability of colonies of Microcystis morphospecies in culture. J Gen Appl Microbiol. 2000;46(1):39–50.
Frangeul L, Quillardet P, Castets AM, et al. Highly plastic genome of Microcystis aeruginosa PCC 7806, a ubiquitous toxic freshwater cyanobacterium. BMC Genomics. 2008;9:1–20.
Meyer KA, Davis TW, Watson SB, et al. Genome sequences of lower great lakes Microcystis sp. reveal strain-specific genes that are present and expressed in western Lake Erie blooms. PLoS ONE. 2017;12(10):e0183859.
Pérez-Carrascal OM, Terrat Y, Giani A, et al. Coherence of Microcystis species revealed through population genomics. ISME J. 2019;13(12):2887–900.
Humbert JF, Barbe V, Latifi A, et al. A tribute to disorder in the genome of the bloom-forming freshwater cyanobacterium Microcystis aeruginosa. PLoS ONE. 2013;8(8):e70747.
Willis A, Woodhouse JN. Defining cyanobacterial species: diversity and description through genomics. CRC Crit Rev Plant Sci. 2020;39(2):101–24.
Shoval O, Alon U. SnapShot: network motifs[J]. Cell. 2010;143(2):326–326. e1.
Guo G, Zhao Y, Liu C, et al. Method for persistent topological features extraction of schizophrenia patients’ electroencephalography signal based on persistent homology. Front Comput Neurosci. 2022;16:1024205.
Sizemore AE, Giusti C, Kahn A, et al. Cliques and cavities in the human connectome. J Comput Neurosci. 2018;44(1):115–45.
Hu L, Zhang J, Pan X, et al. HiSCF: leveraging higher-order structures for clustering analysis in biological networks[J]. Bioinformatics. 2021;37(4):542–50.
Murgas KA, Saucan E, Sandhu R. Hypergraph geometry reflects higher-order dynamics in protein interaction networks[J]. Sci Rep. 2022;12(1):20879.
He Y, Yang Y, Su X, et al. Incorporating higher order network structures to improve miRNA–disease association prediction based on functional modularity[J]. Brief Bioinform. 2023;24(1):bbac562.
Torres L, Blevins AS, Bassett D, et al. The why, how, and when of representations for complex systems[J]. SIAM Rev. 2021;63(3):435–85.
Benson AR, Gleich DF, Higham DJ. Higher-order network analysis takes off, fueled by classical ideas and new data[J]. arXiv preprint arXiv:2103.05031, 2021.
Kanehisa M, Sato Y, Kawashima M, et al. KEGG as a reference resource for gene and protein annotation[J]. Nucleic Acids Res. 2016;44(D1):D457–62.
Qi J, Wang B, Hao BI. Whole proteome prokaryote phylogeny without sequence alignment: AK-string composition approach[J]. J Mol Evol. 2004;58:1–11.
Zuo G, Hao B. CVTree3 web server for whole-genome-based and alignment-free prokaryotic phylogeny and taxonomy[J]. Genomics Proteom Bioinf. 2015;13(5):321–31.
Perozzi B, Al-Rfou R, Skiena S, Deepwalk. Online learning of social representations[C]//Proceedings of the 20th ACM SIGKDD international conference on Knowledge discovery and data mining. 2014: 701–710.
De Meo P, Ferrara E, Fiumara G et al. Generalized louvain method for community detection in large networks[C]//2011 11th international conference on intelligent systems design and applications. IEEE, 2011: 88–93.
Kipf TN, Welling M. Variational graph auto-encoders[J]. arXiv preprint arXiv:1611.07308, 2016.
Pan S, Hu R, Long G et al. Adversarially regularized graph autoencoder for graph embedding[J]. arXiv preprint arXiv:1802.04407, 2018.
Park J, Lee M, Chang HJ et al. Symmetric graph convolutional autoencoder for unsupervised graph representation learning[C]//Proceedings of the IEEE/CVF international conference on computer vision. 2019: 6519–6528.
Cao H, Xu D, Zhang T, et al. Comprehensive and functional analyses reveal the genomic diversity and potential toxicity of Microcystis[J]. Harmful Algae. 2022;113:102186.
Dick GJ, Duhaime MB, Evans JT, et al. The genetic and ecophysiological diversity of Microcystis[J]. Environ Microbiol. 2021;23(12):7278–313.
Pattharaprachayakul N, Lee M, Incharoensakdi A, et al. Current understanding of the cyanobacterial CRISPR-Cas systems and development of the synthetic CRISPR-Cas systems for cyanobacteria[J]. Enzym Microb Technol. 2020;140:109619.
Van Le V, Srivastava A, Ko SR, et al. Microcystis colony formation: extracellular polymeric substance, associated microorganisms, and its application[J]. Bioresour Technol. 2022;360:127610.
Papoulis SE, Wilhelm SW, Talmy D, et al. Nutrient loading and viral memory drive accumulation of restriction modification systems in bloom-forming cyanobacteria[J]. Mbio. 2021;12(3). https://doi.org/10.1128/mbio. 00873 – 21.
Downing TG, Meyer C, Gehringer MM, et al. Microcystin content of Microcystis aeruginosa is modulated by nitrogen uptake rate relative to specific growth rate or carbon fixation rate. Environ Toxicology: Int J. 2005;20(3):257–62.
De la Iglesia R, Valenzuela-Heredia D, Pavissich JP, et al. Novel polymerase chain reaction primers for the specific detection of bacterial copper P‐type ATPases gene sequences in environmental isolates and metagenomic DNA. Lett Appl Microbiol. 2010;50(6):552–62.
Xiao M, Li M, Reynolds CS. Colony formation in the cyanobacterium Microcystis. Biol Rev. 2018;93(3):1399–420.
Morris JJ, Johnson ZI, Szul MJ, et al. Dependence of the cyanobacterium Prochlorococcus on hydrogen peroxide scavenging microbes for growth at the ocean’s surface. PLoS ONE. 2011;6(2):e16805.
Piel T, Sandrini G, White E, et al. Suppressing cyanobacteria with hydrogen peroxide is more effective at high light intensities. Toxins. 2019;12(1):18.
Lu Y, Li Q, Li T, PPA-GCN:. A efficient GCN Framework for Prokaryotic pathways assignment. Front Genet. 2022;13:839453.
Cui G, Zhou J, Yang C et al. Adaptive graph encoder for attributed graph embedding. Proceedings of the 26th ACM SIGKDD international conference on knowledge discovery & data mining. 2020;976–985.
Young JG, Petri G, Peixoto TP. Hypergraph reconstruction from network data. Commun Phys. 2021;4(1):135.
Henderson K, Gallagher B, Eliassi-Rad T et al. Rolx: structural role extraction & mining in large graphs. Proceedings of the 18th ACM SIGKDD international conference on Knowledge discovery and data mining. 2012;1231–1239.
Acknowledgements
The authors would like to thank the support from Yan Lin.
Funding
This research was supported by the National Key Research and Development Program of China (Grant No. 2020YFA0907402 and No. 2018YFA0903100), and the National Natural Science Foundation of China (Grant No. 92251304).
Author information
Authors and Affiliations
Contributions
YL wrote the main manuscript text and prepared figures. TL conceived and supervised the project. QL and TL reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.





Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Lu, Y., Li, Q. & Li, T. A novel hierarchical network-based approach to unveil the complexity of functional microbial genome. BMC Genomics 25, 786 (2024). https://doi.org/10.1186/s12864-024-10692-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10692-6