
Abstract
Background
Gibberella ear rot (GER) is one of the most devastating diseases in maize growing areas, which directly reduces grain yield and quality. However, the underlying defense response of maize to pathogens infection is largely unknown.
Results
To gain a comprehensive understanding of the defense response in GER resistance, two contrasting inbred lines ‘Nov-82’ and ‘H10’ were used to explore transcriptomic profiles and defense-related phytohormonal alterations during Fusarium graminearum infection. Transcriptomic analysis revealed 4,417 and 4,313 differentially expressed genes (DEGs) from the Nov-82 and H10, respectively, and 647 common DEGs between the two lines. More DEGs were obviously enriched in phenylpropanoid biosynthesis, secondary metabolites biosynthesis, metabolic process and defense-related pathways. In addition, the concentration of the defense-related phytohormones, jasmonates (JAs) and salicylates (SAs), was greatly induced after the pathogen infection. The level of JAs in H10 was more higher than in Nov-82, whereas an opposite pattern for the SA between the both lines. Integrated analysis of the DEGs and the phytohormones revealed five vital modules based on co-expression network analysis according to their correlation. A total of 12 hub genes encoding fatty acid desaturase, subtilisin-like protease, ethylene-responsive transcription factor, 1-aminocyclopropane-1-carboxylate oxidase, and sugar transport protein were captured from the key modules, indicating that these genes might play unique roles in response to pathogen infection,
Conclusions
Overall, our results indicate that large number DEGs related to plant disease resistance and different alteration of defensive phytohormones were activated during F. graminearum infection, providing new insight into the defense response against pathogen invasion, in addition to the identified hub genes that can be further investigated for enhancing maize GER resistance.
Similar content being viewed by others


Introduction
Gibberella ear rot (GER) caused by Fusarium graminearum (F. graminearum) is one of the most prevalent diseases in maize production field [1, 2]. The spores of F. graminearum can naturally infect maize kernels upon conveyance through wind, rain splash or insect infestation. Infected cob is observed with a reddish-pink mold starting at the tip of a rotten ear [3]. GER not only reduces yield but also contains diverse mycotoxins from the fungal, including deoxynivalenol (DON) and zearalenone (ZEN), which threatening human and livestock health [4, 5]. Agronomic and chemical practices controlling the disease are not very effective when climatic conditions are favorable for the pathogen [6]. In Southwest China, severe occurrence of GER leads to serious economic losses for maize production [7]. The preferred method for controlling GER is to breed and cultivate resistant genotypes.
When plants suffer from pathogens attack, defense responses of host towards biotic stresses are activated to orchestrate the establishment of different defensive barriers, including regulation of defense genes expression, production of reactive oxygen species (ROS) burst, induction of programmed cell death, changes of phytohormones and signaling pathways [8, 9]. The phytohormones are small molecules within plants that governing diverse physiological processes [10]. Jasmonate (JA) and salicylic acid (SA) are major defense-related phytohormones tightly associated with plant defense [11, 12]. The signaling networks of these defensive phytohormones can mediate and induce a local or systematic defense response in plant immune systems [13]. Accumulating evidence suggests that JA and SA generally involved in the activation of defense against necrotrophic and biotrophic pathogens, respectively [14]. In maize ear rot, a previous study reported the JA and SA were directly involved in resistance to Fusarium verticillioides-induced Fusarium ear rot (FER) [15]. Moreover, Christensen et al. demonstrated that the JA-mediated process was a major pathway involved in defence against the pathogen in maize FER disease [16].
With the rapid development of multi-omics techniques such as transcriptomics and metabolomics, a massive data have been generated, which are possible to explore the genetic mechanism of plant resistance by integrating the multi-perspective data. In recent years, substantial progress on the characterization of the complexity inherent in maize ear rot has been made. For example, Wang et al. characterized the transcriptome of FER disease by using RNA sequencing (RNA-seq) and provided many differentially expressed genes (DEGs) involved in SA and JA pathways related to defense responses against Fusarium verticillioides [15]. Maschietto et al. identified the DEGs lypoxygenase, heat shock proteins and transcription factors associated with FER resistance [17]. In another study, a series of metabolites such as sugars, polyols, aminoacids, acids, and polyamins were found specially involved in resistance to the pathogen in maize FER disease [18]. In addition, many previous studies described that the effect of fatty acid composition can also mediate of maize FER defense [19, 20]. In maize GER disease, Yuan et al. characterized the transcriptional profiles conferring resistance to F. graminearum infection and found 195 specific DEGs related to defense activities [21]. In the research, the DEGs associated with pathogenesis-related proteins and SA pathways were significantly enriched, suggesting the DEGs were crucial for GER defense responses. To our knowledge, combination of transcriptomics and metabolomics is less used in maize GER. Therefore, it is necessary to integrate the two perspective data for further exploring the specific defense activation mechanism in response to F. graminearum infection.
Weighted gene co-expression network analysis (WGCNA) is one of the most effective methods for analyzing the expression patterns of multiple genes associated with defined phenotypic characteristics [22]. In the approach, highly connected genes (hub genes) are centrally defined in different particular modules with certain biological processes [23]. Recently, the WGCNA method has been become a powerful approach to identify key modules or hub genes according to gene-to-metabolite correlations for characterizing the desired traits in numerous crops [22, 24,25,26,27]. The multi-omics networks could provide a great opportunity for understanding comprehensively defense mechanisms against pathogens in plant diseases.
In this study, to clarify the transcriptional profiles and hormone-dependent crosstalk conferring maize GER resistance, a combined analysis of transcriptional responses and defense-related phytohormonal alterations in two contrasting inbred lines was conducted to identify the defence mechanisms in response to F. graminearum infection. The intergrating view of transcriptional analysis and phytohormonal changes will enhance the knowledge of host-specificity mechanisms of maize GER and pave the way for genetic improvement of the disease resistance.
Materials and methods
Plant materials and field inoculation
Based on our previous work, an association panel consisting of 316 maize diverse inbred lines have been evaluated for GER resistance across multiple environments [28]. Among them, two inbred lines, Nov-82 and H10, were evaluated with high resistance and susceptibility to GER, respectively. So, the two lines with contrasting GER severity were selected and planted at the Agricultural Experimental Station of Sichuan Agricultural University (30°43′N, 103°52′E). An aggressive isolate of Fusarium graminearum (F. graminearum, strain Fg12002) was used for further phenotypic evaluation via artificially inoculated in field. For inoculation, the spore suspension of the isolate was adjusted to a final concentration of approximately 1.0 × 106 spores mL− 1 based on microscopic count. Inoculation was performed approximately 10–12 days after maize pollination. The middle-upper of the ear was pierced through the husk by the needle of syringe, piercing the kernels, but not reaching the cob, and then 2 ml of conidial suspension was injected. GER severity assessment was performed at maturity and the inoculated ear was scored using a rating scale from 1 to 7, where 1 represented no visible disease symptoms and grades 2 to 7 describe visible symptoms according to the percentage of infected ear areas (1 = 0%, 2 = 1–3%, 3 = 4–10%, 4 = 11–25%, 5 = 26–50%, 6 = 51–75%, and 7 = 76–100%, respectively) [1].
DAB and trypan blue staining
To further characterize the GER resistance of the Nov-82 and H10, the leaves from the two lines at the V5 stage were inoculated with the isolate F. graminearum using a needle syringe. ROS accumulation was detected using DAB (3, 3′-Diaminobenzidine) for the inoculated leaves at 72 h post-inoculation (hpi). Inoculated leaves of the two lines were dipped in freshly prepared 100 lg/mL DAB solution, containing 1 mg/mL of DAB-HCl, pH 5.0 for 4 h at room temperature. Stained leaves were placed in boiling ethanol for 10 min to remove chlorophyll and then transferred to destaining solution prior to photography [29]. Meanwhile, dead cells were detected using trypan blue staining for the inoculated leaves at 72 hpi. The inoculated leaves were soaked in staining stock solution (0.1% w/v trypan blue in a solvent consisting of equal volumes of water-saturated phenol, lactic acid, glycerol, and water), boiled for 2 min, and stained overnight. The stained leaves were destained in chloral hydrate solution (2.25 g/ml chloralhydrate in water) and then observed under microscope. Each sample contained at least three independent maize leaf.
Transcriptome analysis
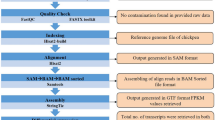
In the current study, we performed a transcriptomic analysis of Nov-82 and H10 upon F. graminearum inoculation. The two inbred lines were artificially inoculated with the isolate under controlled conditions. Husk tissues around the inoculated spots of the two lines were collected at 0, 12, 24, 48, 72 and 96 hpi after inoculation with F. graminearum, respectively, and immediately frozen in liquid nitrogen before maintained at -80 °C for further analysis. Each sample contained husk tissues from three independent maize, and three samples at each time were conducted. Total RNA was extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. Approximately 1 µg of RNA per sample representing each library was used for the transcriptome preparation. The libraries were sequenced on an Illumina Hiseq PE150 platform (Illumina, SanDiego, CA, USA), and 150-bp paired-end reads were generated. Raw data were filtered to remove the adaptors and low-quality reads (≤ 20% of the bases with a quality score ≤ 10). The high-quality clean reads were then aligned to the maize B73 reference genome (ZmB73_RefGen_V5) using TopHatv 2.0.9 software with default parameters [30]. Only the unique mapping reads (reads only mapped to unambiguous unique location of B73 genome) were retained for calculating the gene. Furthermore, alignments from each library were processed with Cufflinks v0.9.3 to assemble transcript isoforms and quantify expression values, and then normalized as fragments per kilobase of exon model per million mapped reads (FPKM) [31]. To detect genes involved in GER resistance, DEGs were screened using the DESeq2 R package with a False Discovery Rate (FDR) threshold of 0.05 and a Log 2 fold change [32]. To further assign and annotate the DEGs, Gene Ontology (GO) terms and the significantly enriched pathways in the Kyoto Encyclopedia of Genes and Genomes (KEGG) were performed using the genes within each cluster.
Defense-related phytohormones measurement
Extraction and analysis of defense-related phytohormones, including JA, MEJA, H2JA, JA-ILE, OPDA, JA-Phe, JA-Val, OPC-4, OPC-6, SA and SAG, were carried out in Metware Biotechnology Co. Ltd. (Wuhan, China). 50 mg of the inoculated husk tissues at 0, 12, 24, 48, 72 and 96 hpi were grounded to powder, then dissolved in 70% methanol solution, vortexed and placed at 4 °C for overnight. 10 µL internal standard mixed solution was added into the extract as internal standards for the quantication. The standard for the defense-related phytohormones were purchased from Olchemim Ltd. (Olomouc, Czech Republic) and isoReag (Shanghai, China). After centrifugation, the supernatant of the mixtures were filtered through a 0.22 mm pore size membrane. Subsequently, the contents of the phytohormones were analysed by an UPLC-MS/MS platform (UPLC, ExionLC™ AD; MS, Applied Biosystems 6500 Triple Quadrupole). The mobile phase consisted of 0.1% formic acid water (A) and acetonitrile (B) with elution gradient at a flow rate of 0.35 ml/min as follows: started at 5% B (0–1 min), increased to 95% B (1–8 min), 95% B (8–9 min), finally ramped back to 5% B (9–12 min). The analysis parameters were optimized for the production of characteristic precursor-to-product ion transitions in negative or positive ionization modes. Data acquisitions were performed using Analyst 1.6.3 software (Sciex) to determine analyte responses based on peak area integrations relative to the internal standard. Samples were then quantified based on standard curves to determine the concentrations. To identify specific defense-related phytohormones associated with GER, the differentially expressed phytohormones were screened using log2 fold change (log2FC) ≥ 1 and further analyzed using orthogonal partial least squares discriminant analysis (OPLS-DA) method in R package. Variable importance in the projection (VIP) ranks was used in the OPLS-DA model. The phytohormonal alterations were selected on the basis of the combination of a statistically significant threshold of VIP > 1 and p-value < 0.05 from the OPLS-DA model [33].
Weighted gene co-expression network analysis
Weighted gene co-expression network analysis (WGCNA) was used for identifying genes with similar expression patterns that may participate in specific biological functions [34]. The obtained DEGs from transcriptomic analysis were imported into the R package WGCNA (version 1.47) to construct co-expression modules with following parameters: variance data expression > 0; no missing data expression < 0.1; soft threshold = 8 (estimate value); max block size = 200; deep split = 2; min module size = 3; merge cut height = 0.1. The hierarchical clustering of genes in the dissimilarity matrix was performed by the hclust function, and the resulting clustering tree was cut by the dynamic tree cut to define the modules [35]. Then, the calculated module eigengenes and Pearson’s correlation coefficient values were used to determine the association of modules among the samples. In each module, the genes with eigengenebased connectivity value (|KME|) > 0.9 and topological overlap measure (TOM) value > 0.2 were regarded as hub genes [36, 37]. The visualization of co-expression networks and the identification of hub genes in each module were implemented by Cytoscape software [38]. Genes annotation were processed on MaizeGDB website (http://www.maizegdb.org).
Gene expression validation
Quantitative real-time PCR (qRT-PCR) was conducted using a SYBR Premix Ex Taq™ kit (TaKaRa, Dalian, China) according to the manufacturer’s instructions. The actin gene was used as the internal control. Specific primers used in sets for PCR were designed for target genes using Primer Premier 5.0. Thermal cycle conditions were as follows: 2 min at 95 °C followed by 40 cycles of 15 s at 95 °C, 30 s at 56 °C, and 30 s at 72 °C. Each PCR reaction was repeated at least three times. The relative expression levels were calculated according to the Eq. 2−ΔΔCT.
Statistical analysis
All values are expressed as the mean ± standard deviation. The data were processed by one-way analysis of variance (ANOVA) using SPSS version 21.0 software (SPSS Inc., Chicago, IL, USA) at test significant difference (p < 0.05) to evaluate significant difference between groups. The transcriptome and phytohormone visualizations (Venn diagrams, heat maps, volcano maps, etc.) were made using an online platform (www.majorbio.com).
Results
Symptom severity of GER between Nov-82 and H10
The two maize inbred lines, Nov-82 and H10, were further assessed for GER severity via field inoculation. At maturity, symptom of the both lines were visually scored for GER scale. Results showed that a great different severity was observed between the two lines in response to the F. graminearum inoculation, in which Nov-82 was more resistant with less disease symptom compared to H10 (Fig. 1a). The line Nov-82 (GER score = 1.45) showed lower disease severity than that of H10 (GER score = 6.81), exhibiting a distinct resistant level of GER severity.

Phenotypic variation of GER severity in inbred lines Nov-82 and H10 after F. graminearum inoculation. (a), Representative ears of resistant phenotypes in inbred lines Nov-82 and H10. (b), Representative leaves show the disease phenotype of inbred lines Nov-82 and H10 after F. graminearum inoculation at 72 h post inoculation (hpi). Left, 3,3′-Diaminobenzidine (DAB) staining for ROS detection. Right, trypan blue straining to visualize cell death. The inoculated positions are indicated in white circles. Scale bar = 5 cm
To further characterize the resistant performance between the two lines, the DAB (3,3′-Diaminobenzidine) staining was conducted to assess their ROS accumulation after F. graminearum inoculation. It appeared that Nov-82 produced more ROS accumulation compared to H10 in response to the pathogen infection. Similarly, Nov-82 leaves displayed a more intensive hypersensitive response (HR)-like cell death in the infected areas following trypan blue staining, further strengthening the stronger resistant reaction in Nov-82 than that in H10 (Fig. 1b). These observations implied that the line Nov-82 exhibited higher resistant performance compared with H10 during F. graminearum infection.
Transcriptomic analysis under F. graminearum infection
To get the transciptional response in GER resistance, a high throughput RNA-seq was conducted between Nov-82 and H10 at 0, 12, 24, 48, 72 and 96 hpi during F. graminearum infection. With three biological replicates per time point, the 36 samples (18 samples from the Nov-82 and 18 samples from the H10) yielded more than 1,669 million high-quality raw data with an average Q30 value of 94.38% (Supplementary Materials 1: Table S1). A range from 43.43 to 47.93 million clean reads filtered from the raw reads of each library were obtained and then aligned against the B73 reference genome (B73 RefGen_v5). In total, an average of 82.91% of clean data were mapped, indicating that the RNA-seq quality was sufficient and efficient for further analyses.
Subsequently, a comparative analysis of the transcriptional difference between Nov-82 and H10 at different time point was conducted to reveal the dynamic changes in response to F. graminearum infection. After the statistical test (false discovery rate (FDR) < 0.01 and fold change (FC) ≥ 2), a total of 4,417 (2765 up-regulated and 1652 down-regulated) differentially expressed genes (DEGs) in Nov-82, and 4,313 (2734 up-regulated and 1579 down-regulated) DEGs in H10, were found, respectively (Fig. 2a, b). As a result, the number of up-regulated DEGs is higher than down-regulated ones in both lines. Notably, there were 647 common DEGs between the two lines by comparing the transcriptome profile (Fig. 2c). These common DEGs were believed to be specific responsive genes involved in regulating GER resistance.

Global gene expression profiling in the two inbred lines Nov-82 and H10 after F. graminearum inoculation. (a), Numbers of expressed genes in inbred lines Nov-82. (b), Numbers of expressed genes in inbred lines H10. (c), The Upset diagram of comparison differentially expressed genes (DEGs) between the two inbred lines at different time points
Functional enrichment analysis of DEGs
By functional enrichment analysis of the obtained DEGs in Nov-82 and H10, GO terms revealed that these DEGs were mainly distributed in plant disease resistant pathways, such as abiotic stimulus, response to oxidative stress and defense response (Supplementary Materials 2: Fig. S1). At 12 hpi, DEGs were involved primarily in response to stress and chemical. Besides, DEGs at 24 hpi was remarkably enriched in regulation of protein serine/threonine phosphatase activity, oxidative stress and oxidoreductase activity. Furthremore, extracellular region, membrane part and defense response were significantly enriched for DEGs from 48, 72 and 96 hpi, respectively. KEGG pathway analysis showed that phenylpropanoid biosynthesis, secondary metabolites biosynthesis, metabolic pathways, diterpenoid biosynthesis and photosythesis were most significantly enriched in different time points, suggesting that these pathways were deserved as vital pathways in GER resistance (Supplementary Materials 2: Fig. S1). The results of GO and KEGG demonstrated that the specific DEGs and pathways might be helpful to confer GER defense response, thus enabling plant elevating protective ability against pathogens invasion.
Differential defense-related phytohormones profile in the contrasting lines
In order to deeply elucidate the defense mechanisms of GER resistance, we performed the UPLC-MS/MS analysis to detect the dynamic changes of defense-related phytohormones between Nov-82 and H10 at the six time points during F. graminearum infection. Nine jasmonates (JAs) including JA, MEJA, H2JA, JA-ILE, OPDA, JA-Phe, JA-Val, OPC-4, OPC-6, and two salicylates (SAs) containing SA and SAG, were detected across each time point. On the whole, the defense-related phytohormones were differentially induced in both lines (Fig. 3). Altogether, the contents of JAs were more higher accumulation in H10 than those in Nov-82, except for few groups (MEJA, OPDA) or individual time points. On the contrary, the SA displayed higher levels in Nov-82 compared with H10, but the SAG was seemed irregular expression between the two contrasting lines. To better distinguish the different resistance among the two lines, the OPLS-DA approach was applied to analyze significant alterations of the defense-related phytohormones (Supplementary Materials 3: Fig. S2). As a result, 28 and 43 differentially phytohormones were obtained in Nov-82 and H10, respectively (Table 1). The OPLS-DA results suggested that various genotypes could induce different levels of defense-related phytohormones during F. graminearum infection.

Differentially induced levels of defense-related phytohormones, including jasmonates (JAs) and salicylates (SAs), at six time points after F. graminearum inoculation between the two contrasting lines Nov-82 and H10. Each data point represents mean (3 replicates) ± standard deviation. R, inbred line Nov-82; S, inbred line H10
Identification of key modules by weighted gene co-expression network
To obtain key genes highly associated with GER resistance, the specific DEGs from Nov-82 and H10 were conducted to build a gene co-expression network through WGCNA strategy. After filtering out the low-expression genes, a total of 3,654 DEGs were effectively obtained (FPKM ≥ 1) and then inputted to construct the global co-expression network accroding to genes expression patterns (Fig. 4a). Based on pairwise correlations analysis of gene expression, these DEGs were assigned to 18 distinct co-expression modules with different colors (Fig. 4b). According to the eigengene connectivity values, the grey and turquoise modules were the largest items, containing 791 and 420 DEGs, respectively (Supplementary Materials 4: Fig. S3). The lightgreen module had the fewest genes.

Weighted gene co-expression network analysis (WGCNA) of effectively expressed genes in the two inbred lines Nov-82 and H10 after F. graminearum inoculation. (a), Scale-free topology model and mean connectivity. (b), Hierarchical cluster tree showing co-expression modules identified by WGCNA. Each leaf in the tree represents one gene. The major tree branches constitute 18 modules labeled with different colors
Furthermore, the correlation between the 18 particular modules and 11 defense-related phytohormones was further analyzed to characterize module-trait relationships (Fig. 5a). As a result, the red module was positively correlated with JA-Val (r = 0.85, p < 0.05), OPC-4 (r = 0.93, p < 0.05) and OPC-6 (r = 0.95, p < 0.05). Meanwhile, the turquoise module was positively associated with H2JA, JA-Val and OPC-4 (r = 0.79, 0.75, 0.78, p < 0.05, respectively), but negatively correlated with SA (r = -0.77, p < 0.05). The green module was also positively associated with H2JA and JA (r = 0.84, 0.79, p < 0.05, respectively). The salmon and brown modules had strongly positive correlations with H2JA (r = 0.9, p < 0.05) and SA (r = 0.89, p < 0.05), respectively. Overall, a total of five key co-expression modules (red, turquoise, green, salmon and brown) were highly correlated with the appointed defense-related phytohormonal changes (Fig. 5b). Therefore, the alterations of the phytohormones and transcript patterns of the DEGs were highly consistent in the key modules, suggesting that the co-expression modules could be full reliable.

Identification of key modules associated with maize GER resistance by WGCNA. (a), Heat map of correlations between gene co-expression network modules and defense-related phytohormonal alterations. The horizontal axis represents different defense-related phytohormones, and the vertical axis represents the module eigengenes in each module. The values in each cell represent the correlation coefficients (r), and the p-values (in parentheses) of the module-trait association. The deeper color in a cell, the higher positive or negative correlation between the module eigengene and a trait feature. (b), Heat maps show the expression profiles of all the co-expressed genes in the five key co-expression modules. Bar graphs show the expression pattern of module eigengenes in each module
Integrative analyses to reveal hub genes associated with GER resistance
The integrated analysis of transcriptome and defense-related phytohormones revealed five key co-expression modules. Based on GO enrichment analysis, the DEGs in the key modules were remarkably enriched in the oxidation-reduction process (GO:0055114), response to stimulus (GO:0009607) and defense response (GO:0006952), suggesting these specific DEGs may play vital roles in protection of maize against F. graminearum infection (Fig. 6). These results reflected that the maize might be activate simultaneously defense processes at transcriptional and metabolic levels, thus building comprehensive defensive barriers for improving plant resistance in GER disease.

GO enrichment analysis of genes in key co-expression modules. (a), The top ten GO terms in the red module. (b), The top ten GO terms in the salmon module. (c), The top ten GO terms in the rrown module. (d), The top ten GO terms in the green module. (E), The top ten GO terms in the turquoise module
To identify candidate genes tightly associated with GER resistance, the gene co-expression networks of the key modules were visualized to search hub genes (Supplementary Materials 5: Fig. S4). Those genes with more connections were thought to be of great importance in each module. The top 12 genes with the highest connectivity in each module were captured as hub genes and may play central roles in GER resistance (Table 2; Supplementary Materials 5: Fig. S4). In detail, the hub genes encoding fatty acid desaturase, subtilisin-like protease, ethylene-responsive transcription factor, 1-aminocyclopropane-1-carboxylate oxidase, sugar transport protein and unknown genes, may play crucial roles in regulating GER resistance. Next, we employed qRT-PCR to validate the expression patterns of selected eight hub genes in the two lines under F. graminearum infection (Fig. 7). The expression patterns had a good correlation with RNA-seq results (Supplementary Materials 6: Fig. S5). High agreement of expression patterns between the RNA-seq and the qRT-PCR strongly supported the obtianed hub genes tightly associated with GER resistance in maize.

Confirmation of the eight hub genes by qRT-PCR. The qRT-PCR results of the selected eight hub genes at six time points after F. graminearum inoculation in the two inbred lines Nov-82 and H10
Discussion
Maize GER resistance is a complex quantitative trait caused by multi-genes and influenced by environmental conditions, host genotypes and pathogenic races [2, 6]. An in-depth exploration of specific mechanisms conferring GER resistance is of great importance for implementing a genetic-based breeding strategy in maize. Here, the two contrasting lines Nov-82 and H10 were further evaluated in the field test with a distinct resistant level of GER severity in which a lower GER score in Nov-82 compared to H10 (Fig. 1a). The ROS accumulation and dead cells were also stronger in Nov-82 than those in H10 during the F. graminearum infection (Fig. 1b). These results indicated that the stronger ability to produce ROS might be lead to the better resistant phenotype of Nov-82. The line with excellent resistant performance should be served as elite germplasm for breeding GER-resistant cultivars.
Resistance to GER is complex because it is characterized by a quantitative inheritance, requiring comprehensive consideration and multi-perspective analysis [39,40,41,42]. In our study, the transcriptional profiles and defense-related phytohormonal alterations among the two contrasting lines in response to F. graminearum infection were investigated through multi-omics aproaches. Many DEGs related to phenylpropanoid biosynthesis, secondary metabolites biosynthesis, metabolic process and defense-related pathways were obtained in this study (Supplementary Materials 1: Table S1), indicating that these genes may play a critical role in protecting maize against the pathogen invasion. In addition, the accumulation patterns of defense-related phytohormones was dramatically induced during F. graminearum infection, suggesting that up-regulated JAs and SAs could be a common regulatory process of diverse miaze genotypes to biotic stress (Fig. 3). Meanwhile, it has been found that the JAs were relatively higher accumulation in H10 than those in Nov-82, and an opposite pattern for SA, indicating that various genotypes may induce different levels of defense-related phytohormones during defense processes. Thus, it was proposed that the higher expression of SA and lower level JAs probably led to maintain an excellent resistant performance for Nov-82, and also inferred that SAs might play particular roles in maize against F. graminearum. Overall, the alterations of defense-related phytohormones in both lines might trigger different level defensive reaction due to their various resistant mechanisms of GER resistance.
Large dynamic profiling of transcriptomics and metabolomics have expanded our view of plant complex resistance mechanism [9]. Combination the two-perspective data could gain a better understanding of the molecular and physiological responses in plant resistance [39]. Based on the integration analysis of the transcriptomic profiles and defensive phytohormones alterations, five key co-expression modules (red, turquoise, green, salmon and brown) were identified based on the integrated analysis (Fig. 5a, b). Among the five key modules, the DEGs were closely correlated with the defense-related phytohormones, suggesting that these particular genes may coordinate with the hormones to participate in GER defense (Fig. 6). The results were consistent with several previous studies that the expression of resistant genes and related phytohormones was usually induced together in plant defensive response [16, 21, 42]. Overall, our results showed that the integrated analyses of transcriptomic and phytohormonal expression profiles could provide informative clues for the elucidation of comprehensive defense responses of maize against F. graminearum infection, as well as provide a basis for the identification of GER resistance genes.
Owing to hub genes as central location within network, these kind genes are considered to play the most critical regulatory roles in specific physiological processes [25, 43]. Herein, 12 hub genes were captured from the identified five key modules, which may play central roles in response to F. graminearum–induced GER resistance (Table 2; Supplementary Materials 5: Fig. S4). Several studies have highlighted the roles of these hub genes in plant resistance. For instance, fatty acid desaturase was reported with important signaling roles in regulating plant resistance. In a previous study, the surppression of fatty acid desaturase related gene OsSSI2 could enhance resistance to the blast and leaf blight diseases and involve in the negative regulation of defense responses in rice [44]. Here, two hub genes Zm00001eb409700 and Zm00001eb409690 encoding fatty acid desaturase were significantly up-regulated in the both lines, suggesting that the hub genes may play particular roles in maize GER resistance. Another hub gene annotated as subtilisin-like protease (Zm00001eb235950), was widely decribed to be involved in plant immunity via activating downstream immune signaling processes in regulation of plant defense [45]. The hub gene Zm00001eb397130 encoding LOB domain-containing protein was decribed as a key transcription factor, which is required for plant growth and development, hormone response, metabolic regulation and disease resistance [46]. Encoding 1-aminocyclopropane-1-carboxylate oxidase gene Zm00001eb357070 is an enzyme involved in phytohormonal signal pathway in both plant development and resistance [47]. In additon, the hub gene Zm00001eb052520 encoding protein P21 was considered as pathogenesis-related protein 5 family particularly involved in defensive activities [48]. The other hub genes, Zm00001eb006160 encoding ethylene-responsive transcription factor, Zm00001eb008810 encoding sugar transport protein, Zm00001eb218990 encoding B3 domain-containing protein, Zm00001eb015500 encoding WUSCHEL-related homeobox and two uncharacterized genes (Zm00001eb086370 and Zm00001eb333200), may be have their specific roles in plant resistance. In general, these obtained hub genes should be further focused on their essential roles in response to F. graminearum infection. A possible next step on the hub genes with potential function and specific resistant mechanisms for GER resistance is required.

Proposed model of maize defense activation in response to Fusarium graminearum infection. The model is based on the differently expression genes of the transcriptomic analysis and alterations of defense-related phytohormones in this study. SA, salicylic acid; JA, jasmonic acid; PR, pathogenesis-related; R, resistant; ROS, reactive oxygen species
Conclusions
In the present study, the transcriptomic profiles and defensive phytohormonal alterations revealed that hundreds of DEGs and different alteration of phytohormones participated in GER defense. Co-expression network analysis of the DEGs and phytohormones, we proposed a possible working model for the cellular response to F. graminearum in maize (Fig. 8). Additionally, the DEGs involved in defense-related pathways and the differentially indued phytohormones, as welll as the hub genes associated with disease resistance, were suggested as essential factors in GER resistance. The results provided insights into the host-specific mechanisms of maize GER resistance and potential resources of defense genes that would benefit the following resistance breeding in GER disease.
Data availability
The raw data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA014990) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa.
Abbreviations
- GER:
-
Gibberella ear rot.
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes.
- DEGs:
-
Differentially expressed genes.
- GO:
-
Gene Ontology
- DAB 3:
-
3′-Diaminobenzidine.
- WGCNA:
-
Weighted gene co-expression network analysis.
References
Reid LM, Nicol RW, Ouellet T, Savard M, Miller JD, Young JC, et al. Interaction of Fusarium Graminearum and F. moniliforme in maize ears: Disease progress, fungal biomass, and mycotoxin accumulation. Phytopathology. 1999;89:1028–37.
Yang Q, Balint-Kurtil P, Xu ML. Quantitative disease resistance: dissection and adoption in maize. Mol Plant. 2017;10:402–13.
Martin M, Dhillon BS, Miedaner T, Melchinger AE. Inheritance of resistance to Gibberella ear rot and deoxynivalenol contamination in five flint maize crosses. Plant Breed. 2012;131:28–32.
Vigier B, Reid LM, Dwyer LM, Stewart DW, Sinha RC, Arnason JT, et al. Maize resistance to Gibberella ear rot: symptoms, deoxynivalenol, and yield. Can J Plant Pathol. 2001;23:99–105.
Munkvold GP. Epidemiology of Fusarium diseases and their mycotoxins in maize ears. Eur J Plant Pathol. 2003;109:705–13.
Mesterházy Á, Lemmens M, Reid LM. Breeding for resistance to ear rots caused by Fusarium spp in maize-a review. Plant Breed. 2012;131:1–19.
Duan C, Qin Z, Yang Z, Li W, Sun S, Zhu Z, et al. Identification of pathogenic Fusarium spp. causing maize ear rot and potential mycotoxin production in China. Toxins. 2016;8:186.
Geng ZY, Zhu W, Su H, Zhao Y, Zhang KQ, Yang JK. Recent advances in genes involved in secondary metabolite synthesis, hyphal development, energy metabolism and pathogenicity in Fusarium graminearum (teleomorph Gibberella Zeae). Biotechnol Adv. 2014;32:390–402.
Lanubile A, Maschietto V, Borrelli VM, Stagnati L, Logrieco AF, Marocco A. Molecular basis of resistance to Fusarium ear rot in maize. Front Plant Sci. 2017;8:1774.
Bari R, Jones J. Role of plant hormones in plant defence responses. Plant Mol Biol. 2009;69:473–88.
Han X, Kahmann R. Manipulation of phytohormone pathways by effectors of filamentous plant pathogens. Front Plant Sci. 2019;10:822.
Xu Z, Zhou Z, Cheng Z, Zhou Y, Wang F, Li M, et al. A transcription factor ZmGLK36 confers broad resistance to maize rough dwarf disease in cereal crops. Nat Plants. 2023;9:1720–33.
Santiago R, Cao A, Malvar RA, Butrón A. Genomics of maize resistance to Fusarium ear rot and fumonisin contamination. Toxins. 2020;12:431.
Berens ML, Berry HM, Mine A, Argueso CT, Tsuda K. Evolution of hormone signaling networks in plant defense. Annu Rev Phytopathol. 2017;55:401–25.
Wang Y, Zhou Z, Gao J, Wu Y, Xia Z, Zhang H, et al. The mechanisms of maize resistance to Fusarium verticillioides by comprehensive analysis of RNA-seq data. Front Plant Sci. 2016;7:1654.
Christensen SA, Nemchenko A, Park YS, Borrego E, Huang PC, Schmelz EA, et al. The novel monocot-specific 9-lipoxygenase zmlox12 is required to mount an effective jasmonate-mediated defense against Fusarium verticillioides in maize. Mol Plant-Microbe in. 2014;27:1263–76.
Maschietto V, Colombi C, Pirona R, Pea G, Strozzi F, Marocco A, et al. QTL mapping and candidate genes for resistance to Fusarium ear rot and fumonisin contamination in maize. BMC Plant Biol. 2017;17:20.
Campos-Bermudez VA, Fauguel CM, Tronconi MA, Casati P, Presello DA, Andreo CS. Transcriptional and metabolic changes associated to the infection by Fusarium verticillioides in maize inbreds with contrasting ear rot resistance. PLoS ONE. 2013;8:e61580.
Wilson RA, Gardner HW, Keller NP. Cultivar-dependent expression of a maize lipoxygenase responsive to seed infecting fungi. Mol Plant-Microbe in. 2001;14:980–7.
Christensen SA, Kolomiets MV. The lipid language of plant–fungal interactions. Fungal Genet Biol. 2011;48:4–14.
Yuan GS, He XJ, Li H, Xiang K, Liu L, Zou CY, et al. Transcriptomic responses in resistant and susceptible maize infected with Fusarium Graminearum. Crop J. 2020;8:153–63.
Schaefer RJ, Michno JM, Jeffers J, Hoekenga O, Dilkes B, Baxter I, et al. Integrating co-expression networks with GWAS to prioritize causal genes in maize. Plant Cell. 2018;30:2922–42.
Wan Q, Tang J, Han Y, Wang D. Co-expression modules construction by WGCNA and identify potential prognostic markers of uveal melanoma. Exp Eye Res. 2018;166:13–20.
Wu Z, Wang M, Yang S, Chen S, Chen X, Liu C, et al. A global coexpression network of soybean genes gives insights into the evolution of nodulation in nonlegumes and legumes. New Phytol. 2019;223:2104–19.
Ma L, Zhang M, Chen J, Qing C, He S, Zou C, et al. GWAS and WGCNA uncover hub genes controlling salt tolerance in maize (Zea mays L.) seedlings. Theor Appl Genet. 2021;134:3305–18.
Azam M, Zhang S, Li J, Ahsan MA, Boateng KG, Qi J, et al. Identification of hub genes regulating isoflavone accumulation in soybean seeds via GWAS and WGCNA approaches. Front Plant Sci. 2023;14:1120498.
Saito K. Phytochemical genomics-a new trend. Curr Opin Plant Biol. 2013;16:373–80.
Yuan GS, He DD, Shi JX, Li YL, Yang Y, Du J, et al. Genome-wide association study discovers novel germplasm resources and genetic loci with resistance to Gibberella ear rot caused by Fusarium Graminearum. Phytopathology. 2023;113:1317–24.
Daudi A, O’Brien JA. Detection of hydrogen peroxide by DAB staining in Arabidopsis leaves. Bio-Protocol. 2012;2:e263.
Trapnell C, Pachter L, Salzberg SL. TopHat, discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van-Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106.
Zhang R, Watson DG, Wang L, Westrop GD, Coombs GH, Zhang T. Evaluation of mobile phase characteristics on three zwitterionic columns in hydrophilic interaction liquid chromatography mode for liquid chromatography-high resolution mass spectrometry based untargeted metabolite profiling of leishmania parasites. J Chromatogr A. 2014;1362:168–79.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559.
Zhan J, Thakare D, Ma C, Lloyd A, Nixon NM, Arakaki AM, et al. RNA sequencing of lasercapture microdissected compartments of the maize kernel identifies regulatory modules associated with endosperm cell differentiation. Plant Cell. 2015;27:513–31.
Buckberry S, Bianco-Miotto T, Bent SJ, Clifton V, Shoubridge C, Shankar K. Placental transcriptome co-expression analysis reveals conserved regulatory programs across gestation. BMC Genomics. 2017;18:1–13.
Zhang B, Horvath S. A general framework for weighted gene coexpression network analysis. Stat Appl Genet Mol Biol. 2005;4:1–45.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504.
Gaikpa D, Miedaner T. Genomicsassisted breeding for ear rot resistances and reduced mycotoxin contamination in maize: methods, advances and prospects. Theor Appl Genet. 2019;132:2721–39.
Reinprecht Y, Wu X, Yan S, Labey L, Dasliva E, Martin J, et al. A microarray-based approach for identifying genes for resistance to Fusarium graminearum in maize (Zea mays L). Cereal Res Commun. Suppl 2008;6:253–9.
Kebede AZ, Woldemariam T, Reid LM, Harris LJ. Quantitative trait loci mapping for Gibberella ear rot resistance and associated agronomic traits using genotypingbysequencing in maize. Theor Appl Genet. 2016;129:17–29.
Liu Y, Guo Y, Ma C, Zhang D, Wang C, Yang Q, et al. Transcriptome analysis of maize resistance to Fusarium Graminearum. BMC Genomics. 2016;17:477.
Luo Y, Pang D, Jin M, Chen J, Kong X, Li W, et al. Identification of plant hormones and candidate hub genes regulating flag leaf senescence in wheat response to water deficit stress at the grain-filling stage. Plant Direct. 2019;3:e00152.
Jiang CJ, Shimono M, Maeda S, Inoue H, Mori M, Hasegawa M, et al. Suppression of the rice fatty-acid desaturase gene OsSSI2 enhances resistance to blast and leaf blight diseases in rice. Mol Plant-Microbe in. 2009;22:820–9.
Figueiredo J, Sousa SM, Figueiredo A. Subtilisin-like proteases in plant defence: the past, the present and beyond: subtilases in plant defence. Mol Plant Pathol. 2018;19:1017–28.
Zhang Y, Li Z, Ma B, Hou Q, Wan X. Phylogeny and functions of LOB domain proteins in plants. Int J Mol Sci. 2020;21:2278.
Kim YS, Choi D, Lee MM, Hi LS, Taek KW. Biotic and abiotic stress-related expression of 1-aminocyclopropane-l-carboxylate oxidase gene family in Nicotiana glutinosa L. Plant Cell Physiol. 1998;39:565–73.
Pechanova O, Pechan T. Maize-pathogen interactions: an ongoing combat from a proteomics perspective. Int J Mol Sci. 2015;16:28429–48.
Acknowledgements
We are grateful to Dr Peng Liu for technical assistance in the laboratory.
Funding
This study was supported by the National Natural Science Foundation of China (32272177) and the Natural Science Foundation of Sichuan (24NSFSC0323).
Author information
Authors and Affiliations
Contributions
GY, GP and YS conceived and designed the research; JS, CZ (C Zeng) and HS performed the experiment; YY, CZ (C Zeng), TM, MW and ZJ analyzed the data; JD, CZ (C Zou) and LM contributed to manuscript preparation and data curation. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The authors declare that all the experimental research studies on plants, including the collection of plant material, were carried out in accordance with relevant institutional, national, and international guidelines and legislation.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it.The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

{kind=link}
Cite this article
Yuan, G., Shi, J., Zeng, C. et al. Integrated analysis of transcriptomics and defense-related phytohormones to discover hub genes conferring maize Gibberella ear rot caused by Fusarium Graminearum. BMC Genomics 25, 733 (2024). https://doi.org/10.1186/s12864-024-10656-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10656-w