
Abstract
Background
Swamp-type buffaloes with varying degrees of white spotting are found exclusively in Tana Toraja, South Sulawesi, Indonesia, where spotted buffalo bulls are highly valued in accordance with the Torajan customs. The white spotting depigmentation is caused by the absence of melanocytes. However, the genetic variants that cause this phenotype have not been fully characterized. The objective of this study was to identify the genomic regions and variants responsible for this unique coat-color pattern.
Results
Genome-wide association study (GWAS) and selection signature analysis identified MITF as a key gene based on the whole-genome sequencing data of 28 solid and 39 spotted buffaloes, while KIT was also found to be involved in the development of this phenotype by a candidate gene approach. Alternative candidate mutations included, in addition to the previously reported nonsense mutation c.649 C > T (p.Arg217*) and splice donor mutation c.1179 + 2T > A in MITF, a nonsense mutation c.2028T > A (p.Tyr676*) in KIT. All these three mutations were located in the genomic regions that were highly conserved exclusively in Indonesian swamp buffaloes and they accounted largely (95%) for the manifestation of white spotting. Last but not the least, ADAMTS20 and TWIST2 may also contribute to the diversification of this coat-color pattern.
Conclusions
The alternative mutations identified in this study affect, at least partially and independently, the development of melanocytes. The presence and persistence of such mutations may be explained by significant financial and social value of spotted buffaloes used in historical Rambu Solo ceremony in Tana Toraja, Indonesia. Several de novo spontaneous mutations have therefore been favored by traditional breeding for the spotted buffaloes.
Similar content being viewed by others

Background
Coat color is one of the most visible phenotypic traits in animals and is under continuous selection in most domesticated animal species. It is largely determined by melanocytes, which produce melanins [1, 2]. After originating in the neural crest, melanoblasts migrate to various destinations, including the iris, dermis, and epidermis, where they differentiate into melanocytes determining the color of skin [2, 3]. The development of melanoblasts relies on the regulation of numerous transcription factors and signaling pathways, including the transcription factors PAX3, SOX10, LEF1 and MITF, G protein-coupled endothelin receptor B (EDNRB) and its ligand endothelin 3 (EDN3), and receptor tyrosine kinase KIT and KIT-ligand (KITLG) [3, 4]. However, disruption in the survival, migration, proliferation, and differentiation of melanoblasts may result in the absence of mature melanocytes, leading to white spotting [3, 5]. Due to the interaction of multiple genes that regulate the development of functional melanocytes, this phenotype was considered as a complex trait with patterns ranging from partially to completely white [6, 7]. A number of genes including MITF, KIT, PAX3, EDN3, EDNRB, SNAI2, TRPM1, ADAMTS20 and TWIST2 have been identified as candidate genes associated with white spotting in cattle [8,9,10], horses [11, 12], sheep [13, 14], and mice [15,16,17,18].
The swamp buffalo (Bubalus bubalis carabanensis) in Tana Toraja, South Sulawesi, Indonesia exhibit three coat color phenotypes, including solid black (wild type), spotted, and pure white coats (Figure S1) [19]. In the Toraja classification system, there are four types of spotted patterns, Saleko, Lotong Boko, Bonga, and Toddi [19]. Spotted buffalo bulls are sacrificed in funeral ceremonies and are valued ten times the price of solid black buffaloes [19,20,21]. The Toraja peoples’ perception and classification of buffaloes underlie the Rambu Solo ceremony, which is a traditional ritual for parents to become ancestors and reside in Puya, a life after death. For the Toraja people, this sacred ceremony is also related to prestige, dignity, and social status [22,23,24,25,26], which depend on the number and type of buffaloes that are sacrificed [22, 24,25,26,27]. The Toraja people search for buffaloes with a unique coat color pattern, large body size, and long and widespread horns [27]. After the slaughter, the distribution of meat while showing the buffalo heads highlight the social status of the owners [22, 24,25,26,27]. Therefore, the color pattern is under strong culture-driven selection in Indonesian swamp buffaloes [19].
A previous candidate gene study on Indonesian swamp buffaloes has identified two independent loss-of-function mutations in the MITF gene associated with white spotting [19]. However, these mutations cannot explain all the white-spotted phenotypes [19], implicating other mutations affecting the white spotting in swamp buffaloes.
In this study, we performed whole-genome sequencing (WGS) and applied a genome-wide association study (GWAS), combined with a candidate gene approach, to identify novel genetic variants associated with the white spotting in swamp buffaloes.
Results
MITF as a potential and functional candidate gene for the white spotting
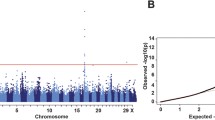
Totally, 13,467,488 single nucleotide polymorphisms (SNPs) were retained for GWAS after quality control to identify the genes responsible for white spotting in swamp buffaloes (Figure S2A). We detected 208 genome-wide significant SNPs and 25 annotated genes associated with the white spotting (Table S1). Furthermore, a linkage disequilibrium-based cross-population extended haplotype homozygosity (XP-EHH) detection of selection signatures identified 853 annotated genes with the top 5% value as threshold (Figure S3). Enrichment analyses of the significant genes identified by XP-EHH indicated 232 gene ontology (GO) functional terms, including positive regulation of Wnt signaling pathway and pigmentation (Table S2). Notably, nine genes including MITF, TBC1D4, LOC102412347, LOC102400532, LOC102399551, AURKC, ZNF805, TMEM132C, and OSBPL10 were identified both by GWAS and XP-EHH analysis. Among these genes, MITF was the only functional gene regulating the development of melanocytes and the transcription of melanogenic enzyme genes (Figure S2B, C) [3, 28].
Nonsense and splice donor mutations in MITF associated with the white spotting
The buffalo MITF gene spanned approximately 230 kb and comprised 10 coding exons (Fig. 1A, Figure S4A). A total of 1,399 SNPs were detected in the MITF genomic region, including 1,390 intron variants, three synonymous variants, four 3′ untranslated region (UTR) variants, one splice donor variant, and one nonsense variant (Fig. 1B).

Identification of the nonsense and splice donor mutations in the MITF of the spotted buffaloes. AMITF gene and transcript structure. B Types of variants annotated in MITF. C The Sanger sequencing of gDNA in solid and spotted buffaloes reveals a nonsense mutation in exon 4 and a splice donor mutation in intron 9 of MITF
The nonsense mutation (BBU21:31637770, c.649 C > T, Fig. 1C) led to a premature termination codon (PTC) (p.Arg217*) in the MITF exon 4. This variant is significantly association with the white-spotted phenotype (P = 3.45 × 10− 8, Table 1) and is also in linkage disequilibrium (LD) with the two adjacent variants (Figure S4B). The haplotype containing the nonsense mutation occurred exclusively in Indonesian swamp buffaloes and is one of the major haplotypes in spotted buffaloes (Figure S4C, D).
In addition, a splice donor variant (BBU21:31613452, c.1179 + 2T > A, Fig. 1C) was identified in intron 9 of the MITF gene. This variant was predicted to impact splicing with a SpliceAI donor loss delta score of 0.99 (Table 1). Again, this mutation is only present in Indonesian spotted buffaloes. Although this mutation with a non-reference allele frequency of 0.03 is not significantly associated with the white spotting (P = 0.11, Table 1), it was only found in one spotted buffalo without the MITF or KIT mutations and in three complete white buffaloes that were heterozygous for the MITF nonsense mutation. This may suggest that it is an alternative causative mutation for the white spotting and, together with a heterozygous nonsense genotype, for the complete white coat color. This observation supports a previous study in which the splice donor variant of MITF was found to be significantly associated with spotted color in swamp buffalo [19].
Identification of nonsense mutation in KIT associated with the white spotting
Given the benefit of WGS data in exploring the contribution of rare variants to phenotypic variation [29, 30], we utilized the candidate gene approach to identify other functional mutations that may affect the white spotting.
Based on previous studies, we identified 11 plausible genes (see Methods). The interaction among these genes, as well as MITF, suggested their association with the white spotting (Table S3, Figure S5). Overall, 5,638 SNPs were found in the genomic regions of these 11 genes (Table S4). Functional annotations revealed 37 potential mutations affecting gene coding regions, including one nonsense mutation, 27 missense mutations, and nine splice region mutations. Based on the functional annotation and association analysis, these variants were further filtered as shown in Table 1.
In addition to the nonsense mutation in KIT, which was very likely to have a high impact, only two mutations in ADAMTS20 and one in TRPM1 were predicted to be deleterious. In addition, splice region mutations in EDN3 and PAX3 were predicted to have a low impact with a new splice donor site in EDN3. We further analyzed the association of these mutations with the white spotting and found only KIT and PAX3 to be significantly associated.
KIT encoded a tyrosine kinase receptor indispensable for the migration and survival of melanoblasts [31]. The buffalo KIT gene extended across 85.8 kb, encompassing 21 coding exons. Within the KIT genomic region, 628 SNPs were discovered, including 608 intron variants, seven synonymous variants, one 5′ UTR variant, nine 3′ UTR variants, two missense variants, and one nonsense variant (Fig. 2A). The nonsense mutation (BBU7:47207784, c.2028T > A, Fig. 2B) was located in its exon 14, altering the codon for tyrosine into a stop codon (p.Tyr676*), which truncated the tyrosine kinase domain (Fig. 2C). This mutation occurred exclusively in Indonesian spotted buffaloes and the genomic region encompassing the nonsense mutation was highly conserved. It was significantly associated with the white spotting (P = 0.01, Table 1).

Identification of the nonsense mutation in the KIT of the spotted buffaloes. A Types of variants annotated in KIT. B The Sanger sequencing of gDNA in solid and spotted buffaloes reveals a nonsense mutation in exon 14 of KIT. C The domain structure of KIT
The splice region mutation (BBU2:163527488, c.452–3 C > T) in PAX3 was significantly associated with the white spotting (P = 0.013, Table 1). Although it was predicted to have a small effect that did not change protein structure (Table 1), it may be associated with a regulatory mutation to affect gene expression.
MITF and KIT jointly explained most spotted phenotypes
Since both MITF and KIT were significantly associated with the white spotting, the genotype combinations of three candidate mutations were examined for their distribution in solid black and spotted buffaloes. Due to the lack of coverage at specific loci and the unavailability of DNA from one sample, we finally used 66 buffaloes for this analysis. Among the 39 spotted individuals, 37 carried one or more candidate mutations, while the remaining two were homozygous for the wild-type alleles (Table 2). Remarkably, the mutations in MITF and KIT jointly explained 95% of the white spotting in swamp buffaloes.
To further identify potential mutations affecting the remaining two spotted buffaloes, we filtered all mutations in the candidate genes, except MITF and KIT, and found 39 mutations in SOX10, ADAMTS20, EDNRB, and TWIST2 to potentially contribute to the white spotting (Table S5). Of these mutations, four were significantly associated with the white spotting, but these were located in introns of ADAMTS20 and TWIST2 (P < 0.05, Table S6). One of these two spotted individuals was homozygous for mutations in ADAMTS20 (BBU4:83215693, c.4294-1882T > A, Table 2) and TWIST2 (BBU6:117577951, c.*35 + 19893G > A, Table 2), while another was homozygous for two mutations in TWIST2 (BBU6:117578304, c.*35 + 20246G > A; BBU6:117580907, c.*35 + 22849G > A; Table 2). The ADAMTS20 mutation also occurred in two solid black buffaloes (Table 2) and was therefore not causative for the coat color. It is noteworthy that the frequent heterozygotes of MITF 21:31637770 was only observed in white-spotted buffaloes if accompanied by a KIT mutation which also occurred in white-spotted animals together with the MITF homozygous wild-type genotype or one of the three TWIST2 mutations (Table 2), supporting a potential causative role of these TWIST2 mutations. Although our data indicated the involvement of other genes than MITF and KIT in the white-spotted trait, further investigation is warranted to identify different combinations of genotypes responsible for the white-spotted coat color in extended samples.
However, the analysis of WGS data often excludes intron variants or classifies them as variants of uncertain significance due to difficulties in predicting or determining their impacts [32]. Given the additional challenges in interpreting deep intronic mutations, the integration of multi-omics datasets will be critical to confirm their functions.
Discussion
The white spotting, as a unique coat color pattern, has been mainly investigated in horses [11], cattle [8, 9], sheep [13, 14], and mice [16]. In this study, we integrated GWAS, selection signature analysis, and candidate gene approach to explore the molecular mechanism of the white spotting in swamp buffaloes. Previous study demonstrated that the two MITF mutations cannot fully account for the white spotting [19]. We successfully validated these two previously identified mutations in MITF [19] and detected the KIT gene as another candidate responsible for the manifestation of the white spotting in swamp buffaloes.
To date, a number of variants in MITF and KIT have been shown to cause the white spotting in domestic animals such as cattle [8, 9, 33, 34] and horses [7, 11, 35,36,37,38,39]. MITF is a transcription factor that has a bHLH-Zip domain, which not only influences melanocyte development, but also regulates the expression of color genes, including TYR and TYRP1 [40]. Interestingly, a previous study of swamp buffalo MITF has demonstrated that the nonsense mutation resulted in mRNA degradation by the nonsense-mediated mRNA decay (NMD) pathway, while the splice donor mutation affected the binding efficiency of the mutant protein to the MITF binding site [19]. KIT, a type III receptor protein tyrosine kinase, plays a crucial role in the migration, survival, and proliferation of melanocytes and in the cell survival signaling of melanoblasts [31, 41,42,43]. Insufficient KIT expression in melanoblasts and melanocytes triggers apoptosis and results in the white spotting [38, 44]. Therefore, the nonsense mutation identified in the buffalo KIT gene in this study truncates the tyrosine kinase domain of KIT via the NMD pathway, which impacts the MAPK signaling pathway and thereby regulates melanocyte development. Most spotted buffaloes in this study can be classified mainly into three categories: those with MITF or KIT mutation or with both MITF and KIT mutations. These mutations might lead to reduction in MITF or KIT protein levels, which impair melanocyte development and cause the white spotted coat color.
The white phenotype in mammals can also be caused by the disruption of melanin synthesis [38, 45, 46]. The causal mutation for dominant white coat color in swamp buffaloes from China is a LINE-1 insertion in the ASIP gene [47]. To determine whether the four completely white Indonesian buffaloes in our study were caused by the ASIP mutation, we investigated their whole genome sequencing reads, but did not find such LINE-1 insertion (Figure S6). ASIP and MITF play different roles in the formation of white coat color. Specifically, over-expression of ASIP prevents melanocytes from undergoing terminal differentiation [47, 48], while MITF participates in melanocyte development by modulating the expression of target genes [49,50,51,52]. In mice, early deletion of functional MITF protein leads to the termination of melanocyte development [53], with individuals harboring a homozygous mutation presenting with a white coat and small eyes [54, 55]. In our study, buffaloes homozygous for the nonsense mutation in MITF are viable and typically have a more pronounced depigmentation phenotype than heterozygous buffaloes. One of the four white individuals was homozygous for the MITF nonsense mutation, while the remaining three were carriers of the nonsense mutation and splice donor mutation in MITF, which most likely resulted in an abnormal MITF protein through NMD and abnormal splicing. Therefore, we propose that the white buffaloes in our study lacked normal MITF protein and thus their melanocyte development was disrupted.
A previous study reported a significant association between the splice donor mutation of MITF and white-spotted coat color in Indonesian swamp buffalo [19]. This study, however, detected only a limited number of individuals carrying this mutation and reported a non-significant association with white spotting phenotype. In addition, the candidate mutations in ADAMTS20 and TWIST2 were found at a low frequency. Therefore, these candidate functional variants require the validation on the basis of a larger panel of spotted and white swamp buffaloes.
Conclusions
Our results first emphasized previous findings that suggested the involvement of the MITF gene in the white spotted phenotype in water buffaloes and then revealed a new mutation in the KIT gene. These mutations jointly explained 95% of the white spotting in swamp buffaloes and may regulate melanocyte development by impacting gene function, ultimately resulting in the white spotting. We also found evidence for other candidate genes relevant to the white spotting. The remarkable diversity of spotted-associated genotypes may reflect a strong selection of de novo mutations that confer the highly prized spotted phenotype. However, genomic analysis of more white-spotted buffaloes is necessary to confirm and/or identify other causal variants that could fully explain the four different spotting patterns according to the Toraja classification system.
Methods
Samples included in this study
A total of 67 swamp buffaloes were sampled from local village farms on Sulawesi and Sumbawa islands (Figure S7A) in Indonesia, consisting of 28 solid black (wild type), 35 spotted, and four pure white individuals (Table S7). Hair follicle samples were collected from the tail. Pedigree information was available for 16 of these individuals (Table S8). White spotting was scored as the presence or absence of white spots on the coat. The complete whiteness can be regarded as an extreme form of white spotting [56].
Sequencing and variant calling
Genomic DNA was extracted from hair follicles using the phenol/chloroform method. The quality and quantity of the genomic DNA were assessed and verified using agarose gel electrophoresis and a Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Paired-end libraries were prepared with an average insert size of 350 bp and sequenced on the Illumina HiSeq X Ten Platform (Illumina, San Diego, CA, USA). Raw reads were filtered using the QualityTrim software (https://bitbucket.org/arobinson/qualitytrim). The reads were then aligned to the water buffalo reference genome (UOA_WB_1 at https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_003121395.1/) [57] using the BWA-MEM algorithm [58]. The UOA_WB_1 is a chromosome level reference genome with N50 and L50 scores similar to those of a recently published water buffalo reference genome (https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_019923935.1/). After sorting the mapping reads using Samtools [59], potential PCR duplicates were marked with the “MarkDuplicates” of Picard tools v2.9.0 (http://broadinstitute.github.io/picard) and local realignment around indels was performed by the Genome Analysis Toolkit (GATK, v3.7) [60]. We obtained sequence data with an average depth of 9.73× and a coverage of 98.27% (Table S7). SNPs were identified using the “UnifiedGenotyper” of GATK. Finally, hard filters were applied to the raw SNPs according to the following parameters: QUAL < 30; QualByDepth (QD) < 2.0; RMSMappingQuality (MQ) < 40.0; MappingQualityRankSumTest (MQRankSum) < − 12.5; ReadPosRankSumTest (ReadPosRankSum) < − 8.0; and HaplotypeScore > 13.0.
Quality control
The variant call format (VCF) file generated in GATK was converted to the Plink format using the PLINK software v1.90 [61]. All the variants were filtered by PLINK with the following options “--mind 0.1, --maf 0.05, --geno 0.1, --hwe 0.0000000001 in the cases and --hwe 0.000001 in the controls”. Using these parameters, we excluded samples with missing call rates exceeding 0.1 and removed SNPs with minor allele frequencies (MAF) below 0.05, SNP missing call rates exceeding 0.1, and Hardy-Weinberg equilibrium (HWE) exact test p-values below 10− 10 in cases and below 10− 6 in controls. Autosomal SNPs were pruned using the PLINK software v1.90 [61], with a window size of 50 SNPs, a step of 5 SNPs, andr2 threshold of 0.1, resulting in 388,771 independent SNPs for calculating the GWAS threshold.
Genome-wide association study
A principal component analysis (PCA) revealed no clear distinct differentiation between the spotted and solid black buffaloes (Figure S7B), ensuring that the association test was not influenced by potential population stratification. The association test was performed with a univariate linear mixed model using the GEMMA software v0.98.3 [62] with statistical model y = Wα + xβ + u + ε, where y is a vector of phenotype values for 67 individuals, coded as 0 for solid and 1 for spotted; W is an n × c matrix of covariates (PC1, PC2); α is a c × 1 vector of the corresponding coefficients, including the intercept; x is an n × 1 vector of marker genotypes; β is the effect size of the marker; u is an n × 1 vector of random polygenic effects with a covariance structure as u ~ N (0, KVg), where K is an n × n marker-based additive genetic relatedness matrix and Vg is the polygenic additive variance; and ε is an n × 1 vector of residual errors with ε ~ N (0, IVe), where I is an n × n identity matrix and Ve is the residual variance.
The threshold P-value for suggestive association was 2.57 × 10–6 (1/388,771). The Manhattan and quantile–quantile (Q-Q) plots were created using the ggplot2 [63] and qqman [64] packages in R, respectively.
Detection of selection signature
To identify potential selection signatures across the genome, XP-EHH was employed to identify differences between populations using Selscan v1.2.0a [65]. Haplotype phasing was first implemented in BEAGLE [66]. SNPs with minor allele count (MAC) lower than 10 and samples with a fraction of missing genotypes (F_MISSING) higher than 0.1 were removed using the bcftools v1.17 [59]. We identified regions under selection in the spotted buffaloes in contrast to the solid black buffaloes based on the extended haplotype statistics via the XP-EHH approach, using 50 kb sliding windows with a step size of 20 kb. For the XP-EHH selection scan, our test statistic was the average XP-EHH score in each 50 kb region. Positive or negative values of XP-EHH indicated selection in spotted or solid black buffaloes, respectively. The top 5% windows with average XP-EHH values were considered as the candidate selection regions and were annotated by the ANNOVAR [67]. The clusterProfiler R package [68] was used for the GO analysis. We selected GO terms with P < 0.05.
Selection of candidate genes responsible for white-spotted coat color
Candidate genes for white spotting selected from published studies included KIT [33], KITLG [69], EDN3 [15], EDNRB [16], SNAI2 [17], TRPM1 [70], ADAMTS20 [18], TWIST2 [10], and the MITF transcription factors PAX3 [71], SOX10 [72], and LEF1 [28] (Table S9). A protein-protein interaction (PPI) network was predicted by the candidate gene products using the STRING database [73] and GeneMANIA web site [74]. The interactions were categorized into four groups in the STRING database on the basis of the combined score of each interaction: low, medium, high, and the highest confidences at 0.15, 0.4, 0.7, and 0.9, respectively.
Transcript assembly
The RNA-seq data of skin tissues for three solid black and three white buffaloes [47] were collected to analyze the transcripts of candidate genes. Genome-guided transcript assembly was performed using the StringTie [75]. The RNA-seq reads were visualized in the Integrative Genomic Viewer (IGV) [76].
Functional annotation and filtering of mutations
The SnpEff [77] was used to annotate and predict the variants’ effects in candidate genes with default parameters. Nonsense, missense, and splice site mutations were selected by the SnpSift [78] with the parameter “ANN[*].EFFECT”. The pathogenicity of missense mutations was analyzed using the SIFT 4G [79] and PolyPhen-2 [80]. The effects of variants on splicing were predicted using the SpliceAI and the resulting scores were evaluated as 0.2 for high recall, 0.5 for recommended, and 0.8 for high precision [81]. For the spotted buffaloes that did not carry the MITF and KIT candidate mutations, the vcfR R package [82] was used to analyze their genotype frequencies along with the solid black buffaloes at all candidate mutations, based on dominant and recessive inheritance patterns. The Fisher’s exact test (http://vassarstats.net/) was performed to verify the association between the allele frequency of SNPs and the coat color patterns (spotted and solid).
Linkage disequilibrium and haplotype analyses
The Haploview software v4.2 [83] was used to analyze the LD and haplotypes of candidate genes. LD blocks were defined according to the definition of Gabriel et al. [84].
Verification, frequency, and conservation of candidate mutations
PCR primers (Table S10) were designed by Primer3 v0.4.0 [85, 86]. PCR products amplified in a 25-µl reaction containing 50 ng genomic DNA, 21 µl Golden Mix (Tsingke, Beijing, China), and 10 pmol forward and reverse primers were used for the Sanger sequencing.
Genomic data from 45 buffalo populations (348 individuals, Table S11) were used to calculate the frequency of candidate SNPs by the PLINK v1.90 [61]. Multiple protein alignments were visualized and analyzed using the Unipro UGENE v38.1 [87].
Data availability
The newly generated genome sequences of Indonesian buffalo are available from the Sequence Read Archive (SRA) with the Bioproject accession number PRJNA1053598 and PRJNA1135737.
Abbreviations
- GWAS:
-
Genome-Wide Association Study
- WGS:
-
Whole-Genome Sequencing
- SNPs:
-
Single Nucleotide Polymorphisms
- PCA:
-
Principal Component Analysis
- VCF:
-
Variant Call Format
- GATK:
-
Genome Analysis Toolkit
- Q-Q:
-
Quantile–Quantile
- MAC:
-
Minor Allele Count
- F_MISSING:
-
Fraction of Missing Genotypes
- XP-EHH:
-
Cross-population extended haplotype homozygosity
- GO:
-
Gene Ontology
- PPI:
-
Protein-Protein Interaction
- LD:
-
Linkage Disequilibrium
- IGV:
-
Integrative Genomic Viewer
- UTR:
-
Untranslated Region
- PTC:
-
Premature Termination Codon
- NMD:
-
Nonsense-mediated mRNA decay
References
Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445(7130):851–7.
Lin JY, Fisher DE. Melanocyte biology and skin pigmentation. Nature. 2007;445(7130):843–50.
Hou L, Pavan WJ. Transcriptional and signaling regulation in neural crest stem cell-derived melanocyte development: do all roads lead to Mitf? Cell Res. 2008;18(12):1163–76.
Wang XP, Liu YL, Mei LY, He CF, Niu ZJ, Sun J, Zhao YL, Feng Y, Zhang H. Wnt signaling pathway involvement in genotypic and phenotypic variations in Waardenburg syndrome type 2 with MITF mutations. J Hum Genet. 2018;63(5):639–46.
Baxter LL, Hou L, Loftus SK, Pavan WJ. Spotlight on spotted mice: a review of white spotting mouse mutants and associated human pigmentation disorders. Pigment Cell Res. 2004;17(3):215–24.
Hauswirth R, Haase B, Blatter M, Brooks SA, Burger D, Drogemuller C, Gerber V, Henke D, Janda J, Jude R, et al. Mutations in MITF and PAX3 cause splashed white and other white spotting phenotypes in horses. PLoS Genet. 2012;8(4):404–12.
Magdesian KG, Tanaka J, Bellone RR. A de novo MITF deletion explains a novel splashed white phenotype in an American paint horse. J Hered. 2020;111(3):287–93.
Hofstetter S, Seefried F, Häfliger IM, Jagannathan V, Leeb T, Drögemüller C. A non-coding regulatory variant in the 5′-region of the mitf gene is associated with white-spotted coat in Brown Swiss cattle. Anim Genet. 2019;50(1):27–32.
Jivanji S, Worth G, Lopdell TJ, Yeates A, Couldrey C, Reynolds E, Tiplady K, McNaughton L, Johnson TJJ, Davis SR et al. Genome-wide association analysis reveals QTL and candidate mutations involved in white spotting in cattle. Genet Selection Evol. 2019;51(1).
Awasthi Mishra N, Drögemüller C, Jagannathan V, Keller I, Wüthrich D, Bruggmann R, Beck J, Schütz E, Brenig B, Demmel S. A structural variant in the 5’-flanking region of the TWIST2 gene affects melanocyte development in belted cattle. PLoS ONE. 2017;12(6):e0180170.
Brooks SA, Bailey E. Exon skipping in the KIT gene causes a Sabino spotting pattern in horses. Mamm Genome. 2005;16(11):893–902.
Bellone RR, Forsyth G, Leeb T, Archer S, Sigurdsson S, Imsland F, Mauceli E, Engensteiner M, Bailey E, Sandmeyer L, et al. Fine-mapping and mutation analysis of TRPM1: a candidate gene for leopard complex (LP) spotting and congenital stationary night blindness in horses. Brief Funct Genomics. 2010;9(3):193–207.
Lühken G, Fleck K, Pauciullo A, Huisinga M, Erhardt G. Familiar hypopigmentation syndrome in sheep associated with homozygous deletion of the entire endothelin type-B receptor gene. PLoS ONE. 2012;7(12).
Pauciullo A, Fleck K, Lühken G, Di Berardino D, Erhardt G. Dual-color high-resolution fiber-FISH analysis on lethal white syndrome carriers in sheep. Cytogenet Genome Res. 2013;140(1):46–54.
Baynash AG, Hosoda K, Giaid A, Richardson JA, Emoto N, Hammer RE, Yanagisawa M. Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell. 1994;79(7):1277–85.
Hosoda K, Hammer RE, Richardson JA, Baynash AG, Cheung JC, Giaid A, Yanagisawa M. Targeted and natural (piebald-lethal) mutations of endothelin-B receptor gene produce megacolon associated with spotted coat color in mice. Cell. 1994;79(7):1267–76.
Perez-Losada J, Sanchez-Martin M, Rodriguez-Garcia A, Sanchez ML, Orfao A, Flores T, Sanchez-Garcia I. Zinc-finger transcription factor slug contributes to the function of the stem cell factor c-kit signaling pathway. Blood. 2002;100(4):1274–86.
Silver DL, Hou L, Somerville R, Young ME, Apte SS, Pavan WJ. The secreted metalloprotease ADAMTS20 is required for melanoblast survival. PLoS Genet. 2008;4(2).
Yusnizar Y, Wilbe M, Herlino AO, Sumantri C, Noor RR, Boediono A, Andersson L, Andersson G. Microphthalmia-associated transcription factor mutations are associated with white-spotted coat color in swamp buffalo. Anim Genet. 2015;46(6):676–82.
Cockrill WR. The husbandry and health of the domestic buffalo. Rome: Food and Agriculture Organization of the United Nations; 1974.
Nooy-Palm H. The Sa’dan-Toraja: A Study of Their Social Life and Religion. Volume I. Organization, Symbol and Beliefs: Brill;; 1979.
Kambuno D. Adat Istiadat, Seni Budaya, Kekayaan Alam. Tana Toraja: Yayasan Lepongan Bulan; 2005.
Anggraeni AS, Putri GA. Makna Upacara adat pemakaman rambu solo’di Tana Toraja. Visual Heritage: Jurnal Kreasi Seni Dan Budaya. 2020;3(1):72–81.
Handayani¹ R, Ahimsa-Putra HS, Budiman C. Out of crisis: maintaining hegemony through rambu solo ritual in toraja. 2020.
Aulia GR, UPACARA ADAT RAMBU SOLO. Jurnal Ushuluddin: Media Dialog Pemikiran Islam. 2022;24(2).
Embon D, Suputra I. Sistem simbol dalam upacara adat Toraja Rambu Solo: Kajian Semiotik. Jurnal Bahasa Dan Sastra. 2018;3(7):1–10.
Tulung AM. Terong: Jenis Dan Makan Simbolik Kerbau Di Toraja Utara = Tedong: types and symbolic meaning of Buffalo in North Toraja. Universitas Hasanuddin; 2022.
Goding CR. Mitf from neural crest to melanoma: signal transduction and transcription in the melanocyte lineage. Gene Dev. 2000;14(14):1712–28.
Bansal V, Libiger O, Torkamani A, Schork NJ. Statistical analysis strategies for association studies involving rare variants. Nat Rev Genet. 2010;11(11):773–85.
David S. A current guide to candidate gene association studies. Trends Genet. 2021;37(12):1056–9.
Grichnik JM. Kit and melanocyte migration. J Invest Dermatol. 2006;126(5):945–7.
Ellingford JM, Ahn JW, Bagnall RD, Baralle D, Barton S, Campbell C, Downes K, Ellard S, Duff-Farrier C, FitzPatrick DR et al. Recommendations for clinical interpretation of variants found in non-coding regions of the genome. Genome Med. 2022;14(1).
Hafliger IM, Hirter N, Paris JM, Hofstetter SW, Seefried FR, Drogemuller C. A de novo germline mutation of KIT in a white-spotted Brown Swiss cow. Anim Genet. 2020;51(3):449–52.
Kttel L, Letko A, Hfliger IM, Signer-Hasler H, Joller S, Hirsbrunner G, Mszros G, Slkner J, Flury C, Leeb T, et al. A complex structural variant at the KIT locus in cattle with the Pinzgauer spotting pattern. Anim Genet. 2019;50(5):423–9.
Henkel J, Lafayette C, Brooks SA, Martin K, Patterson-Rosa L, Cook D, Jagannathan V, Leeb T. Whole-genome sequencing reveals a large deletion in the MITF gene in horses with white spotted coat colour and increased risk of deafness. Anim Genet. 2019;50(2):172–4.
Druml T, Grilz-Seger G, Neuditschko M, Horna M, Ricard A, Pausch H, Brem G. Novel insights into Sabino1 and splashed white coat color patterns in horses. Anim Genet. 2018;49(3):249–53.
Durig N, Jude R, Holl H, Brooks SA, Lafayette C, Jagannathan V, Leeb T. Whole genome sequencing reveals a novel deletion variant in the KIT gene in horses with white spotted coat colour phenotypes. Anim Genet. 2017;48(4):483–5.
Haase B, Brooks SA, Schlumbaum A, Azor PJ, Bailey E, Alaeddine F, Mevissen M, Burger D, Poncet PA, Rieder S, et al. Allelic heterogeneity at the equine KIT locus in dominant white (W) horses. PLoS Genet. 2007;3(11):2101–8.
Haase B, Brooks SA, Tozaki T, Burger D, Poncet PA, Rieder S, Hasegawa T, Penedo C, Leeb T. Seven novel KIT mutations in horses with white coat colour phenotypes. Anim Genet. 2009;40(5):623–9.
Gelmi MC, Houtzagers LE, Strub T, Krossa I, Jager MJ. Mitf in normal melanocytes, cutaneous and uveal melanoma: a delicate balance. Int J Mol Sci. 2022;23(11).
Chabot B, Stephenson DA, Chapman VM, Besmer P, Bernstein A. The proto-oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature. 1988;335(6185):88–9.
Geissler EN, Ryan MA, Housman DE. The dominant-white spotting (W) locus of the mouse encodes the c-kit proto-oncogene. Cell. 1988;55(1):185–92.
Lev S, Blechman JM, Givol D, Yarden Y. Steel factor and c-kit protooncogene: genetic lessons in signal transduction. Crit Rev Oncog. 1994;5(2–3):141–68.
Henkel J, Saif R, Jagannathan V, Schmocker C, Zeindler F, Bangerter E, Herren U, Posantzis D, Bulut Z, Ammann P et al. Selection signatures in goats reveal copy number variants underlying breed-defining coat color phenotypes. PLoS Genet. 2019;15(12).
Cieslak M, Reissmann M, Hofreiter M, Ludwig A. Colours of domestication. Biol Rev. 2011;86(4):885–99.
Damé MCF, Xavier GM, Oliveira JP, Borges AS, Oliveira HN, Riet-Correa F, Schild AL. A nonsense mutation in the tyrosinase gene causes albinism in water buffalo. Bmc Genet. 2012;13.
Liang D, Zhao P, Si J, Fang L, Pairo-Castineira E, Hu X, Xu Q, Hou Y, Gong Y, Liang Z, et al. Genomic analysis revealed a convergent evolution of LINE-1 in coat color: a case study in water buffaloes (Bubalus bubalis). Mol Biol Evol. 2021;38(3):1122–36.
Manceau M, Domingues VS, Mallarino R, Hoekstra HE. The developmental role of Agouti in color pattern evolution. Science. 2011;331(6020):1062–5.
Haq R, Yokoyama S, Hawryluk EB, Jonssond GB, Frederick DT, McHenry K, Porter D, Tran TN, Love KT, Langer R, et al. BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition. P Natl Acad Sci USA. 2013;110(11):4321–6.
Levy C, Khaled M, Robinson KC, Veguilla RA, Chen PH, Yokoyama S, Makino E, Lu J, Larue L, Beermann F, et al. Lineage-specific transcriptional regulation of DICER by MITF in melanocytes. Cell. 2010;141(6):994–1005.
McGill GG, Horstmann M, Widlund HR, Du JY, Motyckova G, Nishimura EK, Lin YL, Ramaswamy S, Avery W, Ding HF, et al. Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell. 2002;109(6):707–18.
McGill GG, Haq R, Nishimura EK, Fisher DE. c-Met expression is regulated by Mitf in the melanocyte lineage. J Biol Chem. 2006;281(15):10365–73.
Opdecamp K, Nakayama A, Nguyen MT, Hodgkinson CA, Pavan WJ, Arnheiter H. Melanocyte development in vivo and in neural crest cell cultures: crucial dependence on the Mitf basic-helix-loop-helix-zipper transcription factor. Development. 1997;124(12):2377–86.
Gruneberg H. The relations of microphthalmia and white in the mouse. J Genet. 1953;51(2):359–62.
Hodgkinson CA, Moore KJ, Nakayama A, Steingrimsson E, Copeland NG, Jenkins NA, Arnheiter H. Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein. Cell. 1993;74(2):395–404.
McFadden A, Vierra M, Martin K, Brooks SA, Everts RE, Lafayette C. Spotting the pattern: a review on White Coat Color in the domestic horse. Animals-Basel. 2024;14(3).
Low WY, Tearle R, Bickhart DM, Rosen BD, Kingan SB, Swale T, Thibaud-Nissen F, Murphy TD, Young R, Lefevre L, et al. Chromosome-level assembly of the water buffalo genome surpasses human and goat genomes in sequence contiguity. Nat Commun. 2019;10(1):260.
Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv Preprint arXiv:13033997. 2013.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10(2).
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The genome analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75.
Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44(7):821–U136.
Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer-; 2016.
Turner S. Qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. J Open Source Softw. 2018.
Szpiech ZA, Hernandez RD, Selscan. An efficient multithreaded program to perform EHH-based scans for positive selection. Mol Biol Evol. 2014;31(10):2824–7.
Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81(5):1084–97.
Wang K, Li MY, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16).
Yu GC, Wang LG, Han YY, He QY. ClusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–7.
Wehrle-Haller B. The role of kit-ligand in melanocyte development and epidermal homeostasis. Pigm Cell Res. 2003;16(3):287–96.
Su MY, Miao F, Jiang S, Shi Y, Luo LF, He XL, Wan J, Xu SZ, Lei TC. Role of the p53-TRPM1/miR-211-MMP9 axis in UVB-induced human melanocyte migration and its potential in repigmentation. Int J Mol Med. 2020;45(4):1017–26.
Kubic JD, Young KP, Plummer RS, Ludvik AE, Lang D. Pigmentation PAX-ways: the role of Pax3 in melanogenesis, melanocyte stem cell maintenance, and disease. Pigm Cell Melanoma R. 2008;21(6):627–45.
Seberg HE, Van Otterloo E, Cornell RA, Beyond MITF. Multiple transcription factors directly regulate the cellular phenotype in melanocytes and melanoma. Pigm Cell Melanoma R. 2017;30(5):454–66.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–13.
Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38:W214–20.
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–.
Robinson JT, Thorvaldsdottir H, Wenger AM, Zehir A, Mesirov JP. Variant review with the Integrative Genomics Viewer. Cancer Res. 2017;77(21):E31–4.
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu XY, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w(1118); iso-2; iso-3. Fly. 2012;6(2):80–92.
Cingolani P, Patel VM, Coon M, Nguyen T, Land SJ, Ruden DM, Lu X. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front Genet. 2012;3:35.
Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. 2016;11(1):1–9.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Jaganathan K, Panagiotopoulou SK, McRae JF, Darbandi SF, Knowles D, Li YI, Kosmicki JA, Arbelaez J, Cui WW, Schwartz GB, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176(3):535–.
Knaus BJ, Grünwald NJ. VCFR: a package to manipulate and visualize variant call format data in R. Mol Ecol Resour. 2017;17(1):44–53.
Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–5.
Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, et al. The structure of haplotype blocks in the human genome. Science. 2002;296(5576):2225–9.
Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23(10):1289–91.
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012;40(15).
Okonechnikov K, Golosova O, Fursov M, Team U. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics. 2012;28(8):1166–7.
Acknowledgements
We thank Dong Liang for assistance in DNA extraction and J. Stuart F. Barker for assistance in editing the English used in this article.
Funding
This study was supported by National Key Research and Development Program of China (2021YFD1200904), National Natural Science Foundation of China (31561143010) and the earmarked fund for CARS36.
Author information
Authors and Affiliations
Contributions
YZ, EMS and JH conceived and designed the experiments, EMS, HA, JH and MIAD performed formal investigation and sample collection, DD prepared the DNA samples for sequencing, DD, JS, EMS, HA and MIAD analyzed the data, DD, HA, AP, JAL, JH and YZ wrote and revised the manuscript. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The sample collection was performed in strict accordance with the protocol approved by the Animal Welfare Committee of China Agricultural University (AW42303202-2-1). We obtained informed consent from each owner to use the animals in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dai, D., Sari, E.M., Si, J. et al. Genomic analysis reveals the association of KIT and MITF variants with the white spotting in swamp buffaloes. BMC Genomics 25, 713 (2024). https://doi.org/10.1186/s12864-024-10634-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10634-2