
Abstract
Background/Objectives
This study aims to elucidate the genetic causes of congenital hypogonadotropic hypogonadism (CHH), a rare genetic disorder resulting in GnRH deficiency, in six families from Pakistan.
Methods
Eighteen DNA samples from six families underwent genome sequencing followed by standard evaluation for pathogenic single nucleotide variants (SNVs) and small indels. All families were subsequently analyzed for pathogenic copy number variants (CNVs) using CoverageMaster.
Results
Novel pathogenic homozygous SNVs in known CHH genes were identified in four families: two families with variants in GNRHR, and two others harboring KISS1R variants. Subsequent investigation of CNVs in the remaining two families identified novel unique large deletions in ANOS1.
Conclusion
A combined, systematic analysis of single nucleotide and CNVs helps to improve the diagnostic yield for variants in patients with CHH.
Similar content being viewed by others


Introduction
Congenital hypogonadotropic hypogonadism (CHH) is a rare genetic endocrine disorder resulting in partial or absent puberty and infertility due to defects in gonadotropin-releasing hormone (GnRH) secretion and/or action. The frequency of CHH is estimated to be between 1:86,000 [1] and 1:10,000 [2], and a reported male predominance from 5:1 [3] to 2:1 [4]. Clinically, CHH is difficult to distinguish from constitutional delay of growth and puberty (CDGP) during early adolescence, but the presence of micropenis and/or cryptorchidism in male neonates may be early clues for long-term GnRH deficiency. The clinical diagnosis of CHH is primarily a diagnosis of exclusion after other common causes of hypogonadotropic hypogonadism have been ruled out [5]. Approximately 50% of CHH patients present with anosmia – Kallmann Syndrome – due to defects affecting the olfactory and GnRH neuron systems during fetal development [6].
Pathogenic variants in more than 65 genes [5, 7, 8] have been associated with both non-syndromic and syndromic CHH. While these variants are present in up to 50% of cases [9], these variants only fully explain the CHH phenotype in closer to 25% of patients [10]. Previous studies focused primarily on patients from European descent [1, 2], however one recent study found a similar frequency of genetic variants in a non-European population [11]. Dominant, recessive, X-linked inheritance, and uniparental disomy (UPD) have been observed in CHH families, and variable expressivity and incomplete penetrance are prevalent in this disorder [12, 13]. Furthermore, the rapid discovery of new CHH genes coupled with advances in high-throughput sequencing (HTS), namely whole exome sequencing (WES) and genome sequencing (WGS), have increased our genetic understanding of CHH and uncovered a notable number of CHH patients with oligogenic inheritance [14].
Several of the known CHH genes were discovered through the detection of copy number variants (CNVs—large insertion/deletion variants), most notably ANOS1 [15] and FGFR1, [16] among others. Despite this, routine screening evaluation of CHH genes for CNVs is lacking, primarily due to the high cost and time for traditional assays such as karyotyping, fluorescence in situ hybridization (FISH), multiplex ligation-dependent probe amplification (MLPA) or array comparative genomic hybridization (CGH). Although HTS has been available for over a decade, recent advances in bioinformatic analysis of HTS data demonstrated its value to detect CNVs [17].
The current study evaluates six families segregating CHH and originating from remote areas of Punjab, Pakistan—an underrepresented population in the genetic studies of this disorder. Using a combination of traditional single nucleotide variants (SNVs) and advances in CNV detection, we successfully determined the underlying genetic causes of CHH in all families.
Results
Clinical findings
We present six families (A-F) with 24 affected individuals (20 males and 4 females) of which 12 CHH men have been studied. All participants were evaluated at multiple stages of their development, and the final diagnosis was given after a detailed medical consultation and hormonal blood tests at 18 years old or later to confirm the absence of pubertal development. Eight participants were normosmic CHH (nCHH) and four were hyposmic (KS). All presented with prepubertal testicular volume (< 3 mL), and one patient had a history of cryptorchidism (Family C, V-8). No hypothalamic or pituitary anomalies were detected on brain MRI. No other CHH-associated phenotypes (e.g. renal agenesis, synkinesia, etc.) were observed in our study population. The clinical evaluations of the participants are summarized in Table 1 and Fig. 1a. All unaffected family members had normal hormonal evaluations, and no defects in smell and/or fertility.

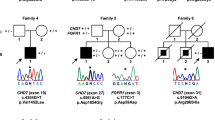
Consanguineous families’ pedigrees and CNVs detected in ANOS1. a Pedigrees of the 6 consanguineous families. b Output of CoverageMaster on WGS for the region around ANOS1 in Subject III:6. The top panel shows the exon–intron map of ANOS1 in the genomic space. The bottom panels show the coverage for subject III:6 compared to the sequencing batch and controls in the genomic space. c Output of CoverageMaster on WGS for the region around ANOS1 in Subject IV:9. The top panel shows the exon–intron map of ANOS1 in the genomic space. The bottom panels show the coverage for subject IV:9 compared to the sequencing batch and controls in the genomic space
SNV analysis
Homozygous pathogenic variants in GNRHR were identified in affected individuals of Families A and B (Fig. 1a, Table 1). All affected members were normosmic, which is consistent with a defect in GnRH action at the pituitary level. Family A is a large consanguineous pedigree including 8 affected individuals with nCHH (5 males and 3 females). DNA was available on two affected males (21.IV & 26.IV), their unaffected brother (18.IV) and parents (10.III & 11.III). A novel homozygous p.[Pro320Gln];[Pro320Gln] variant was identified in both affected patients in Family A (Fig. 1a). This variant is absent from gnomAD, has a CADD of 27.5, and predicted as damaging by SIFT. Additionally, an alternative variant at the same position results in binding defects (with GnRH), as shown in transiently transfected cells resulting in a total loss of function [18]. Consequently, p.Pro320Gln meets the PP3, PM5, PM2, PP2 and is classified as likely pathogenic according to ACMG standards. Family B also resulted from consanguineous marriages. Three brothers are affected with nCHH. DNA analyses were performed on two affected brothers (6.IV & 8.IV), one unaffected brother (11.IV) and the unaffected mother (10.III) Similarly, all affected patients in Family B harbored homozygous p.[Phe309del];[Phe309del] GNRHR variants (MAF = 6.72 × 10–5 in gnomAD with no homozygotes observed). This variant is predicted to be damaging with SIFT, has a CADD of 21.8, meaning it is within the top 0.66% of the most pathogenic variants in humans, and co-segregates perfectly with the CHH phenotype. This variant meets the PM4, PM2 and PP1 strong ACMG criteria leading to a likely pathogenic classification.
Homozygous truncating variants in KISS1R were identified in the affected individuals evaluated in both Families C and D, consistent with the autosomal recessive inheritance mode. Family C is segregating a p.[Trp108Ter];[Trp108Ter] variant, while Family D is segregating a p.[Ser262Ter];[Ser262Ter] variant. Neither of these variants is found in gnomAD nor in Clinvar [19], and both are considered pathogenic by ACMG classification due to their likely undergoing nonsense mediated decay (NMD) resulting in complete loss-of-function.
The SNVs observed in Families A-D were confirmed by Sanger sequencing in all available samples, and segregation analysis was consistent with their involvement. The homozygous nature of the pathogenic variants in all four families (Families A-D) is consistent with the consanguineous matings present. No additional SNVs in CHH genes were found in the affected individuals from Families A-D. Furthermore, no putative causative SNVs in CHH genes were identified in Families E and F.
CNV analysis
As expected given the SNV results, no relevant CNVs were detected in CHH genes in Families A-D. However, hemizygous CNVs were present in ANOS1 on chromosome X in Family E and F (Fig. 1a,b and Table 1). In Family E, a 140 kb deletion beginning 33 kb upstream of ANOS1 and extending through the first exon and intron of the gene was detected. A 100 kb deletion was also observed in Family F and encompassed the last 11 of the 14 exons of ANOS1. Similar CNVs were absent in both DGV and gnomAD, and are considered pathogenic according to ACMG classification [20] given their truncating nature. The segregation of these pathogenic ANOS1 variants in both families is consistent with the known X-linked mode of inheritance for this gene. All male affected family members tested for olfactory defects have CHH with anosmia/hyposmia, also known as Kallmann syndrome (Fig. 1, Table 2).
Discussion
Previous studies have demonstrated that pathogenic SNVs and small insertion/deletion variants can be found in up to 50% of CHH patients [7, 9, 21] However, the diagnostic yield is much lower [10]. The current study uncovered novel pathogenic or likely pathogenic variants in known CHH genes that explain the patients’ phenotype in all 6 families. One key factor of this diagnostic success is the use of Whole Genome Sequencing (WGS) to subsequently evaluate patient DNA for Copy Number Variations (CNVs) when standard Single Nucleotide Variant (SNV) analysis is negative. It’s important to note that CNVs are better detected in WGS compared to Whole Exome Sequencing (WES), making WGS a more effective tool for comprehensive genetic investigation [22, 23]. This is particularly useful when further investigation is needed to find genetic causes for CHH. CNVs were evaluated in two prior CHH studies, the first one utilizing the relatively expensive and unscalable MLPA method [24], and the second [25] using WES data with 7.4% and 2% diagnostic yield, respectively. It’s worth noting that increased diagnostic yield has been observed in other diseases as well when using similar genomic analysis techniques. This underscores the broad utility and effectiveness of these methods in enhancing our understanding of various diseases [17, 26, 27]. As shown in our study and many others, the advent of WGS and its rapidly decreasing cost now allows for more efficient and productive evaluation of CNVs in the increasing number of CHH-associated genes in both exonic and intronic regions.
Intriguingly, two Pakistani families harbored novel homozygous pathogenic variants in KISS1R—a notable finding since the frequency of KISS1R variants in CHH patients is quite rare (< 1.0%) [10]. To date, only nine loss-of-function variants have been described in this gene [28] (Fig. S1) since its discovery two decades ago [29]. KISS1R-deficient individuals are normosmic and exhibit severe GnRH deficiency. This is in line with the crucial role of the galanin-like G protein-coupled receptor encoded by KISS1R on the regulation of GnRH secretion. The patients’ symptoms are consistent with those previously reported for KISS1R variants [7]. Two families harbored homozygous missense variants in GNRHR, the first gene identified to cause nCHH in 1997 [30] and underlying 4% of CHH cases [31]. One of them, p.Pro320Gln, is not found in any control databases. GNRHR encodes for the type 1 GnRH receptor, GnRHR, a G protein-coupled receptor expressed in the gonadotrophs. Natural GnRHR mutants are frequently recognized by the cellular quality control system as misfolded and are retained in the endoplasmic reticulum [32, 33]. Of interest, pharmacochaperones rescue misfolded Gnrhr in murine models, disable the ability of Gnrhr mutants to reach the plasma membrane, and restore their ability to respond to endogenous Gnrh ligand, thus being a promising strategy to treat CHH patients with a genetic profile similar to the affected individuals in Family A [34].
It is important to remark that the genetic causes of CHH have been widely studied in patients mainly from European and North American populations. Indeed, only one publication has specifically evaluated CHH genes in a single Pakistani family [35]. The current manuscript not only demonstrates once more the improved diagnostic potential of WGS over the classical hybridisation-based sequencing methods, but also highlights the diagnostic utility of medical genetics in under-represented populations.
Materials and methods
Patients
This study included 24 individuals from six Pakistani families with at least two members affected by CHH. Five of the families reported consanguinity. Patients were diagnosed with CHH in accordance with the guidelines presented in the European Consensus Statement on CHH [5]. In short, CHH diagnosis included: (1) absent or partial puberty by age 17 years, (2) low or normal gonadotropin levels in the context of low serum levels of sex steroids (testosterone or estradiol), (3) a normal hypothalamus and pituitary on imaging, and (4) otherwise normal anterior pituitary function [5]. Smell tests were performed using UPSIT [36].
DNA extraction and sequencing
DNA from the 24 participating family members was extracted in Pakistan. Eighteen of the samples had DNA of sufficient quality for WGS sequencing. DNA from the remaining seven individuals was retained for subsequent SNV or CNV confirmation (see SNV and CNV analysis, below) once a pathogenic variant was found to be segregating in the family. Paired-end WGS was performed using the DNBSEQ technology through the Denmark facility of BGI (Beijing Genomics Institute) Global. The DNBs (DNA nanoballs) were loaded into a patterned nanoarray, and paired-end reads of 100–150 bases were generated by probe-anchor synthesis (cPAS). Each sample was sequenced to a minimal depth of 30X.
The resulting raw sequences (BGI fastq files) were processed by an in-house bioinformatics analysis workflow which relies on Sentieon DNASeq (v202112.05), a GATK compliant toolbox [37, 38] that maps the reads to the human reference sequence (GRCh37) and detects SNVs and short insertions/deletions (Indels), smaller than 50 bp. Identified variants were then annotated with minor allele frequencies (MAFs) from gnomAD (v2.1.1) [39] and with multiple pathogenicity prediction techniques including CADD (v1.6) [40] and SpliceAI (v1.3) [41] using ANNOVAR (v2020-06–07) [42].
SNV and CNV analysis
Selected variants satisfied at least one of the following criteria: nonsense (stop gain, frameshift, and acceptor–donor splice sites ± 2 bp from an exon), missense, inframe indels, and variants with a probability higher than 0.8 of causing a splicing defect as determined by the SpliceAI algorithm [41].
All the variants present in one of 65 CHH genes (see Supplementary Table S1) passed GATK filters, including a minimum quality score (QS) of 50. In line with inheritance patterns observed in rare diseases, variants with a minor allele frequency (MAF) of less than 1% were deemed potentially pathogenic if homozygous, and those with a MAF of less than 0.01% if heterozygous. Variants passing these filters were further annotated using Varsome [43] for classification of pathogenicity according to the American College of Medical Genetics (ACMG) standards. Sanger sequencing was used to confirm and evaluate segregation in all families, including members not sent for WGS.
CoverageMaster (v1.0) [44] was used to detect CNVs in CHH genes. In brief, this program uses depth of coverage from WGS or WES, and compresses these data into a multiscale wavelet space. The output is then analyzed through an iterative Hidden Markov Model to detect insertions or deletions > 50 bp at nucleotide-scale resolution. In addition, 30 unrelated samples sequenced with the same technology were used as controls for the CoverageMaster analysis. This helps to identify errors of sequencing/assembly and frequent CNVs. The Database of Genomic Variants [45] and gnomAD for structural variants [46] were also used as controls. MLPA was used to confirm CNVs in all available DNA samples. For ANOS1, the SALSA MLPA probemix P132-A4 kallmann-1 kit (MRC Holland) was used according to the manufacturer’s protocol to validate the variants.
Availability of data and materials
The genetic data pertinent to this study, specifically the BAM files, have been deposited in the European Nucleotide Archive (ENA). The project can be accessed using the accession number PRJEB76310. Individual samples within this project are identifiable with unique accession numbers, which range from ERR13194595 to ERR13194658. The nomenclature for the sample names aligns with the identities presented in this article, adhering to the format: family_generation_individual. For further details or queries, please reach out to the corresponding author, Nelly Pitteloud.
References
Filippi G. Klinefelter’s syndrome in Sardinia. Clinical report of 265 hypogonadic males detected at the time of military check-up. Clin Genet. 1986;30(4):276–84.
Fromantin M, Gineste J, Didier A, Rouvier J. Impuberism and hypogonadism at induction into military service. Statistical study. Probl Actuels Endocrinol Nutr. 1973;16:179–99.
Waldstreicher J, Seminara SB, Jameson JL, Geyer A, Nachtigall LB, Boepple PA, Holmes LB, Crowley WF Jr. The genetic and clinical heterogeneity of gonadotropin-releasing hormone deficiency in the human. J Clin Endocrinol Metab. 1996;81(12):4388–95.
Bonomi M, Vezzoli V, Krausz C, Guizzardi F, Vezzani S, Simoni M, Bassi I, Duminuco P, Di Iorgi N, Giavoli C, et al. Characteristics of a nationwide cohort of patients presenting with isolated hypogonadotropic hypogonadism (IHH). Eur J Endocrinol. 2018;178(1):23–32.
Boehm U, Bouloux PM, Dattani MT, de Roux N, Dode C, Dunkel L, Dwyer AA, Giacobini P, Hardelin JP, Juul A, et al. Expert consensus document: European consensus statement on congenital hypogonadotropic hypogonadism-pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2015;11(9):547–64.
Schwanzel-Fukuda M, Pfaff DW. Origin of luteinizing hormone-releasing hormone neurons. Nature. 1989;338(6211):161–4.
Oleari R, Massa V, Cariboni A, Lettieri A. The Differential Roles for Neurodevelopmental and Neuroendocrine Genes in Shaping GnRH Neuron Physiology and Deficiency. Int J Mol Sci. 2021;22(17):9425.
Maione L, Dwyer AA, Francou B, Guiochon-Mantel A, Binart N, Bouligand J, Young J. Genetics in endocrinology: genetic counseling for congenital hypogonadotropic hypogonadism and Kallmann syndrome: new challenges in the era of oligogenism and next-generation sequencing. Eur J Endocrinol. 2018;178(3):R55–80.
Young J, Xu C, Papadakis GE, Acierno JS, Maione L, Hietamaki J, Raivio T, Pitteloud N. Clinical management of congenital hypogonadotropic hypogonadism. Endocr Rev. 2019;40(2):669–710.
Cassatella D, Howard SR, Acierno JS, Xu C, Papadakis GE, Santoni FA, Dwyer AA, Santini S, Sykiotis GP, Chambion C, et al. Congenital hypogonadotropic hypogonadism and constitutional delay of growth and puberty have distinct genetic architectures. Eur J Endocrinol. 2018;178(4):377–88.
Patil VA, Lila AR, Shah N, Arya S, Sarathi V, Shah R, Jadhav SS, Memon SS, Karlekar M, Bandgar T. Genetic spectrum of Kallmann syndrome: single-center experience and systematic review. Clin Endocrinol. 2022;97(6):804–13.
Sykiotis GP, Plummer L, Hughes VA, Au M, Durrani S, Nayak-Young S, Dwyer AA, Quinton R, Hall JE, Gusella JF, et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci USA. 2010;107(34):15140–4.
Cioppi F, Riera-Escamilla A, Manilall A, Guarducci E, Todisco T, Corona G, Colombo F, Bonomi M, Flanagan CA, Krausz C. Genetics of ncHH: from a peculiar inheritance of a novel GNRHR mutation to a comprehensive review of the literature. Andrology. 2019;7(1):88–101.
Pitteloud N, Quinton R, Pearce S, Raivio T, Acierno J, Dwyer A, Plummer L, Hughes V, Seminara S, Cheng YZ, et al. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J Clin Investig. 2007;117(2):457–63.
Franco B, Guioli S, Pragliola A, Incerti B, Bardoni B, Tonlorenzi R, Carrozzo R, Maestrini E, Pieretti M, Taillon-Miller P, et al. A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature. 1991;353(6344):529–36.
Dode C, Levilliers J, Dupont JM, De Paepe A, Le Du N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33(4):463–5.
Yuan B, Wang L, Liu P, Shaw C, Dai H, Cooper L, Zhu W, Anderson SA, Meng L, Wang X, et al. CNVs cause autosomal recessive genetic diseases with or without involvement of SNV/indels. Genet Med. 2020;22(10):1633–41.
Meysing AU, Kanasaki H, Bedecarrats GY, Acierno JS Jr, Conn PM, Martin KA, Seminara SB, Hall JE, Crowley WF Jr, Kaiser UB. GNRHR mutations in a woman with idiopathic hypogonadotropic hypogonadism highlight the differential sensitivity of luteinizing hormone and follicle-stimulating hormone to gonadotropin-releasing hormone. J Clin Endocrinol Metab. 2004;89(7):3189–98.
Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–7.
Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, Raca G, Ritter DI, South ST, Thorland EC, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2020;22(2):245–57.
Vezzoli V, Hrvat F, Goggi G, Federici S, Cangiano B, Quinton R, Persani L, Bonomi M. Genetic architecture of self-limited delayed puberty and congenital hypogonadotropic hypogonadism. Front Endocrinol. 2022;13:1069741.
Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, Willemsen MH, Kwint M, Janssen IM, Hoischen A, Schenck A, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511(7509):344–7.
Meienberg J, Zerjavic K, Keller I, Okoniewski M, Patrignani A, Ludin K, Xu Z, Steinmann B, Carrel T, Rothlisberger B, et al. New insights into the performance of human whole-exome capture platforms. Nucleic Acids Res. 2015;43(11):e76.
Pedersen-White JR, Chorich LP, Bick DP, Sherins RJ, Layman LC. The prevalence of intragenic deletions in patients with idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Mol Hum Reprod. 2008;14(6):367–70.
Stamou MI, Brand H, Wang M, Wong I, Lippincott MF, Plummer L, Crowley WF, Talkowski M, Seminara S, Balasubramanian R. Prevalence and phenotypic effects of copy number variants in isolated hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2022;107(8):2228–42.
Pfundt R, Del Rosario M, Vissers L, Kwint MP, Janssen IM, de Leeuw N, Yntema HG, Nelen MR, Lugtenberg D, Kamsteeg EJ, et al. Detection of clinically relevant copy-number variants by exome sequencing in a large cohort of genetic disorders. Genet Med. 2017;19(6):667–75.
Sun D, Liu Y, Cai W, Ma J, Ni K, Chen M, Wang C, Liu Y, Zhu Y, Liu Z, et al. Detection of disease-causing SNVs/Indels and CNVs in single test based on whole exome sequencing: a retrospective case study in epileptic encephalopathies. Front Pediatr. 2021;9:635703.
Alzahrani AJ, Ahmad A, Alhazmi T, Ahmad L. An isolated hypogonadotropic hypogonadism due to a L102P inactivating mutation of KISS1R/GPR54 in a large family. Case Rep Pediatr. 2019;2019:3814525.
de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA. 2003;100(19):10972–6.
de Roux N, Young J, Misrahi M, Genet R, Chanson P, Schaison G, Milgrom E. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N Engl J Med. 1997;337(22):1597–602.
Gianetti E, Hall JE, Au MG, Kaiser UB, Quinton R, Stewart JA, Metzger DL, Pitteloud N, Mericq V, Merino PM, et al. When genetic load does not correlate with phenotypic spectrum: lessons from the GnRH receptor (GNRHR). J Clin Endocrinol Metab. 2012;97(9):E1798-1807.
Brothers SP, Cornea A, Janovick JA, Conn PM. Human loss-of-function gonadotropin-releasing hormone receptor mutants retain wild-type receptors in the endoplasmic reticulum: molecular basis of the dominant-negative effect. Mol Endocrinol. 2004;18(7):1787–97.
Millar RP, Lu ZL, Pawson AJ, Flanagan CA, Morgan K, Maudsley SR. Gonadotropin-releasing hormone receptors. Endocr Rev. 2004;25(2):235–75.
Janovick JA, Stewart MD, Jacob D, Martin LD, Deng JM, Stewart CA, Wang Y, Cornea A, Chavali L, Lopez S, et al. Restoration of testis function in hypogonadotropic hypogonadal mice harboring a misfolded GnRHR mutant by pharmacoperone drug therapy. Proc Natl Acad Sci USA. 2013;110(52):21030–5.
Hussain HMJ, Murtaza G, Jiang X, Khan R, Khan M, Kakakhel MBS, Khan T, Wahab F, Zhang H, Zhang Y, et al. Whole exome sequencing revealed a novel nonsense variant in the gnrhr gene causing normosmic hypogonadotropic hypogonadism in a Pakistani family. Horm Res Paediatr. 2019;91(1):9–16.
Doty RL, Shaman P, Dann M. Development of the University of Pennsylvania Smell Identification Test: a standardized microencapsulated test of olfactory function. Physiol Behav. 1984;32(3):489–502.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8.
Kendig KI, Baheti S, Bockol MA, Drucker TM, Hart SN, Heldenbrand JR, Hernaez M, Hudson ME, Kalmbach MT, Klee EW, et al. Sentieon DNASeq variant calling workflow demonstrates strong computational performance and accuracy. Front Genet. 2019;10:736.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43.
Rentzsch P, Schubach M, Shendure J, Kircher M. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021;13(1):31.
Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, Kosmicki JA, Arbelaez J, Cui W, Schwartz GB, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176(3):535-548 e524.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, Massouras A. VarSome: the human genomic variant search engine. Bioinformatics. 2019;35(11):1978–80.
Rapti M, Zouaghi Y, Meylan J, Ranza E, Antonarakis SE, Santoni FA. CoverageMaster: comprehensive CNV detection and visualization from NGS short reads for genetic medicine applications. Brief Bioinform. 2022;23(2).
Church DM, Lappalainen I, Sneddon TP, Hinton J, Maguire M, Lopez J, Garner J, Paschall J, DiCuccio M, Yaschenko E, et al. Public data archives for genomic structural variation. Nat Genet. 2010;42(10):813–4.
Collins RL, Brand H, Karczewski KJ, Zhao X, Alfoldi J, Francioli LC, Khera AV, Lowther C, Gauthier LD, Wang H, et al. A structural variation reference for medical and population genetics. Nature. 2020;581(7809):444–51.
Acknowledgements
We thank the patients and family members who participated in this research study.
Funding
Open access funding provided by University of Lausanne This work was supported by the Swiss National Science Foundation Project SNF IZSTZ0_202612 (to N.P.) and by the Punjab University Research grant 2021–2022.
Author information
Authors and Affiliations
Contributions
SI, AF, MS, AM, NP, and NN conceived and designed the research. AMC was responsible for patient recruitment. The clinical assessment of patients was carried out by KR, NiN, MA, and FDAC. Genetic data preparation and processing were handled by YZ and AB. Family analysis was conducted by YZ, JA, and IH. YZ prepared the figures. The manuscript was edited and revised by AMC, SI, AF, MS, FS, and AM. Manuscript writing was done by YZ, JA, NP, and AF. The final version of the manuscript was reviewed and approved by all authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the ethics review committees of both the Lausanne University Hospital and Punjab University, and all participants provided written informed consent in accordance with the Declaration of Helsinki. Anonymized data was shared between the University of the Punjab (Lahore, Pakistan) and the Lausanne University Hospital (Lausanne, Switzerland) in the context of a registered study (clinicaltrials.gov, NCT01601171).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zouaghi, Y., Choudhary, A.M., Irshad, S. et al. Genome sequencing reveals novel causative structural and single nucleotide variants in Pakistani families with congenital hypogonadotropic hypogonadism. BMC Genomics 25, 787 (2024). https://doi.org/10.1186/s12864-024-10598-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10598-3