
Abstract
Background
Effective population size (Ne) is a pivotal parameter in population genetics as it can provide information on the rate of inbreeding and the contemporary status of genetic diversity in breeding populations. The population with smaller Ne can lead to faster inbreeding, with little potential for genetic gain making selections ineffective. The importance of Ne has become increasingly recognized in plant breeding, which can help breeders monitor and enhance the genetic variability or redesign their selection protocols. Here, we present the first Ne estimates based on linkage disequilibrium (LD) in the pea genome.
Results
We calculated and compared Ne using SNP markers from North Dakota State University (NDSU) modern breeding lines and United States Department of Agriculture (USDA) diversity panel. The extent of LD was highly variable not only between populations but also among different regions and chromosomes of the genome. Overall, NDSU had a higher and longer-range LD than the USDA that could extend up to 500 Kb, with a genome-wide average r2 of 0.57 (vs 0.34), likely due to its lower recombination rates and the selection background. The estimated Ne for the USDA was nearly three-fold higher (Ne = 174) than NDSU (Ne = 64), which can be confounded by a high degree of population structure due to the selfing nature of pea.
Conclusions
Our results provided insights into the genetic diversity of the germplasm studied, which can guide plant breeders to actively monitor Ne in successive cycles of breeding to sustain viability of the breeding efforts in the long term.
Similar content being viewed by others

Introduction
Dry pea (Pisum sativum L.) is a diploid, cool-season legume and a member of the Leguminosae family [1]. Pea is one of the most important pulse crops grown in more than 100 countries, where 7,043,605 hectares of dry pea were planted around the world with a total production of 12,403,522 tonnes [2]. In the USA alone, the pea production reached one million tonnes in 2019 [3]. In recent years, pea protein has become more popular in the market for plant-based diets e.g., Beyond® Meat Burger [4]. Pea seeds have earned a reputation as a dietary goldmine with around 15 – 32% protein content, vitamins, folate, fibers, potassium and minerals, which is good for human health and helps prevent cardiovascular and specific cancer diseases [4, 5]. The increasing popularity of plant-based proteins in the market has further propelled the demand for peas. Therefore, the study of genetic diversity should expand to accelerate the genetic gain of pea varieties to meet future demands, maintaining the diversity in peas is the top priority for plant breeders [4, 6].
Estimation of effective population size (Ne) determines the rate of inbreeding [7, 8] and genetic changes due to genetic drift [9]. Ne is an important parameter in population genetics and breeding introduced by Sewall Wright in 1931, which helps breeders to maintain and monitor the level of genetic diversity in their species [10]. The estimated Ne is expected to be smaller than the census size (N), as it influences the rate at which genetic diversity decreases within a population [11, 12]. Relatively smaller Ne indicates limited population diversity, which, in turn, can restrict genetic advancement within a breeding program [13]. Moreover, Ne parameter retrieves the population dynamics of the genes [14].
The effective size of a population refers to the hypothetical number of individuals in an idealized population that would exhibit a comparable genetic response to stochastic processes, similar to that observed in a real-world population which is based on the Wright-Fisher model [15,16,17]. This model shows genetic drift as the main operating factor, and that changes in allelic and genotypic frequencies over generations are solely influenced by the population size (N) [15]. In real-world breeding populations, factors such as mutation, migration, natural selection, and non-random mating come into play [15]. These factors affect the actual rates of inbreeding and changes in gene frequency variance observed in a population [18]. This will indeed impact Ne and therefore, reduce the genetic variation and diversity. The most commonly used extensions for effective population size theory are variance effective size and inbreeding effective size [15]. The variance effective size reflects the rate of change in gene frequency variance, while inbreeding effective size corresponds to the rate of inbreeding observed in a population [19]. These measures allow us to quantify the consequences of genetic drift in a real population, based on the characteristics and dynamics of the idealized Wright-Fisher population [15].
While Ne of a population can be estimated either from demographic data or genetic markers, the latter is preferred [20,21,22]. Demographic data involves using census size and variance of reproductive success whereas genetic markers reveal changes in allele frequencies over time and are based on linkage disequilibrium (LD). When the pedigree or demographic data is not available, Ne can be estimated using genetic markers [23]. The most popular and widely-employed genetic approach has been the temporal method, which relies on temporal fluctuations in allele frequencies observed on multiple samples collected from the same population [14]. Ne, however, can also be directly estimated using LD between loci at various distances along the genome [13, 24]. Recent advancements in high-throughput sequencing and the availability of high-density markers such as single nucleotide polymorphisms (SNPs) have increased over the past decade, contributing to the LD-based approach now being acknowledged as more reliable, robust [25], cost and time effective than the temporal approach [9].
Linkage disequilibrium (represented as r2) is a phenomenon characterized by the non-random association of alleles at various loci [26] which became popular in recent years for predicting Ne [27]. Correlations between alleles are generated by genetic drift when it is inversely proportional to Ne [9], which changes the allele frequencies in a population over time. The biggest advantage of LD over the temporal method [28], is the strength of associations between markers that can be used to calculate Ne at any time (generations) from a single population accurately without relying on longitudinal data. This makes LD a valuable tool for studying populations where temporal information may be limited or unavailable. Recombination and mutation rates are fundamental processes that shape the genetic landscape [29] and by analyzing LD, we can better understand their history and apply it to plant breeding and population genetics [30].
In this study, we estimated the extent of LD decay in the dry pea genome and utilized the relationship between LD and recombination frequency, as initially described by Sved J [24], to estimate Ne which is convenient as it only requires one sampling time [31, 32]. We used two sets of populations: 1) NDSU modern breeding lines, hereafter referred to as NDSU set, and 2) USDA diversity panel, hereafter referred to as USDA set. Our objectives were two-fold: (i) to estimate Ne for these two germplasms set in dry pea and (ii) to compare the genetic variation between these germplasms. To achieve these goals, we developed a comprehensive R package that implements the Sved J [24] formula for Ne prediction. This package not only caters to the specific needs of dry pea research but can also be adapted for use in other crop species. Since there has been no information on Ne for peas, our findings serve as a valuable reference for researchers seeking to determine the minimum number of lines required for designing experiments. Furthermore, comparing the genetic variation between NDSU modern breeding lines and USDA multi-environmental lines provides valuable information about the diversity and potential of these germplasm collections. This knowledge can guide breeding programs and conservation efforts, ensuring the maintenance and enhancement of genetic resources in dry pea cultivation.
Methods
Plant materials
In this study, we used plant materials from two distinct germplasms pool. The first population comes from the NDSU Pulse Breeding Program (NDSU set) where 300 advanced elite lines were generated from multiple bi-parental populations. The NDSU breeding lines represented a set of pre-selected, non-structured, elite advanced lines at the preliminary yield testing stage, which were carefully chosen and contained both contemporary and past elite germplasm [33, 34]. The breeding lines were built using modern and historical elite cultivars and germplasm in the breeding program, which are representative of a decade of continuous genetic improvement. Further, these selected lines were created specifically with a focus on phenotypes including high yield, grain quality, resistance to disease and some other desirable agronomic traits [33, 34].
The second population is from a USDA diversity panel (USDA set), and contained 482 accessions, of which 292 samples were from the Pea Single Plant Plus Collection (Pea PSP) [4, 35, 36]. The USDA set was composed of accessions that represent most of available diversity within the USDA pea germplasm collection based on the knowledge of geography, taxonomy, morphology and genotyping-by-sequencing data generated previously [35].
DNA extraction, sequencing and variant calling
Leaf tissues from the greenhouse were collected at different stages for all NDSU elite lines and USDA accessions. The DNA from the lyophilized tissues were extracted using the DNeasy Plant Mini Kit (Qiagen). Detailed information regarding the tissue collections and extractions are provided in Bari M [4, 33]. Both NDSU set and USDA set were sequenced using genotyping-by-sequencing (GBS). Using the restriction enzyme ApeKI, dual-indexed GBS libraries for both populations were prepared [37]. Samples were sequenced using NovaSeq S1 × 100 Illumina sequencing technologies. The NDSU set sequenced libraries were retrieved with a quality score ≥ 30. For USDA set, FASTQC [38] was utilized to perform quality check and removed reads with lengths < 50 bases. All reads that passed the quality check were aligned with the reference genome [39] (https://www.pulsedb.org). Finally, the aligned reads were analyzed using SAMtools (v1.10) and generated the variant files (VCF) using FreeBayes (V1.3.2).
The amount of single nucleotide polymorphisms (SNPs) identified for the NDSU set was 28,832, while 380,527 SNP markers were identified in the USDA set [4, 34]. For these marker datasets, we filtered minor allele frequency (MAF), since alleles with < 5% could produce bias to the LD and Ne calculations [40, 41]. We also removed markers with more than 20% missing values using Plink v1.9 [42] and heterozygosity > 20% using Tassel v5.0 [43]. The resulting marker sets consisted of 7,157 (NDSU set) and 19,826 (USDA set) SNP markers that were used for downstream analysis.
Calculation of linkage disequilibrium (r.2)
LD was calculated using Plink v1.9 [42] with a maximum distance of 750 kb. Using “ggplot2” R package, the genome-wide and chromosome-wide LD-decay (r2) were visualized against the physical distance (kb) to show the recombination history (see Figs. 1 & 2).

Genome-wide linkage disequilibrium—decay of NDSU set and USDA set

Chromosome-wide linkage disequilibrium—decay of NDSU set and USDA set
LD scores were also estimated using Genome-wide Complex Trait Analysis (GCTA) software for window size of 1000 kb and r2 cutoff of 0 [44]. This approach was employed to visualize the distribution of mean LD throughout the genome.
Calculation of effective population size
Effective population size (Ne) for both the NDSU set and the USDA set were estimated based on LD using the Sved J [24] equation. The recombination rate (cM) was calculated using cM/Mb conversion ratio from a recent pea genetic linkage map [45] and then transformed to Morgan’s (c).
where, \({N}_{e}\) = effective population size.
\(c\) = genetic distance in Morgan’s
The expected r2 was predicted by linear regression model using least square estimation (LSE),
Prediction of \({r}^{2}\):
The mean r2 from the Y parameter was calculated by LD (r2) for the genetic distance ‘c’ using ‘group by’ mean function in R Environment [46]. Now with the availability of all required parameters, we finally estimated Ne from Eq. (1) using LSE.
According to the formula (Eq. 1), we assigned the variables as predictor (X) and response (Y) and calculated the coefficient \({{\varvec{\upbeta}}}_{1}\) without the intercept term \({{\varvec{\upbeta}}}_{0}\), following Juma R [47].
Again, we used Eq. (3) to calculate the coefficient \({{\varvec{\upbeta}}}_{1}\) which represents Ne.
Results
Linkage disequilibrium decay rate and scores
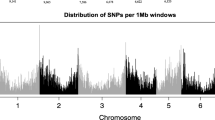
The decay of linkage disequilibrium (r2) was examined in both NDSU set and USDA set by utilizing 7,157 and 19,826 SNP markers, respectively. This analysis allowed for the identification of the physical distance at which the decay rate occurred. Supplementary Fig. 1 depicts the distribution of SNPs within and across chromosomes for both populations, providing an illustration of the marker density. The NDSU set’s genome-wide LD-decay plot (Fig. 1) demonstrates that the r2 reached its peak value of 0.57 within the initial kilobases and subsequently exhibited a gradual decline. The r2 showed a decrease from 0.3 to 0.25 when the genomic distance increased from 150 to 250 kb. Following that, the LD within each chromosome was observed visually in Fig. 2 in order to improve comprehension of the decay pattern. Chromosomes 1 and 6 exhibited a rapid decay at approximately 175 kb, while chromosomes 2 and 5 demonstrated a comparatively slower decay rate of around 350 kb. Furthermore, it is worth noting that chromosome 5 had the higher r2 value of 0.61 compared to other chromosomes. Whereas, the genome-wide LD of USDA set showed that r2 started at a lower value of 0.34 and dropped rapidly and reached 0.2 and 0.1 at 100 kb and 200 kb (Fig. 1). From the chromosome-wide LD-decay (Fig. 2), we observed that chromosome 3 dropped faster around ~ 150 kb, but the r2 decreased below 0.1 for chromosomes 4 and 7. Also, chromosomes 1, 5 and 6 decayed slowly (~ 250 kb) and reached r2 0.1. We also observed that chromosome 1 exhibited a higher r2 of 0.37. LD-decay figures show the trend of the r2 decaying from LD to linkage equilibrium (LE).
Additionally, we performed calculations of LD scores as an alternative metric for inferring LD. The analysis of local LD in the NDSU set indicates a notable rise in the average r2 of 0.6 across all chromosomes. The average r2 of chromosomes 5 and 6 was the highest with 0.8. The genomic interval encompassing the centromeric region of chromosome 2 was missing. In contrast, the USDA set exhibited low average r2, with chromosome 2 hardly reaching 0.4, and chromosomes 1, 4, and 7 having few sets that reached 0.3. It is worth noting that the LD density of the NDSU set is comparatively lower than the USDA set (Fig. 3).

The Mean LD scores estimated in 1000 kb windows. There is a significant increase in LD of NDSU set compared to USDA set
With respect to recombination rate (centimorgans—cM), the genome-wide r2 on average decayed from 0.54 to 0.27 at 0.7 cM for the NDSU set, indicating a moderate level of correlation within this specific genetic distance across the genome. In contrast, the USDA set had lower average r2 (0.28) which dropped within a shorter genetic distance (0.5 cM). This implies that as the distance between the markers increases to 0.5 cM, they tend to be less correlated with each other (Supplementary Fig. 2).
The level of LD exhibited significant variation across distinct genomic regions and populations of dry peas. The impracticality of conducting whole-genome scanning can be attributed to the excessive number of markers required for such studies, particularly in cases where there is a low level of linkage disequilibrium [48]. The USDA set reported a low LD value, indicating a higher occurrence of recombination events. In contrast, the NDSU set showed a higher LD score, suggesting a greater frequency of linked markers presumably due to limited recent recombination to date [49].
Effective population size (N e )
Based on LD, the estimated effective population size (Ne) for both the populations are shown in Fig. 4. The smaller Ne and high LD in NDSU set indicates that it has undergone selective pressures leading to reduced diversity and increased correlation between the markers. Given NDSU set’s population history and marker density, it is acceptable to state that despite lower Ne, it holds a reasonable level of diversity that may help maintain its genetic variability which is essential for long-term viability and adaptability. The USDA set resulted in lower LD and higher Ne, meaning it has more diversity and has encountered relatively fewer instances of selective pressures or genetic bottlenecks. It is important to note that the low LD can also be observed in a population with high Ne. Thus, it was expected to see NDSU set with lower Ne compared to USDA set. These estimates explain how genetic drift and selections have shaped these populations over time.

Estimated effective population size (Ne) for NDSU set is 64 and USDA set is 174
Discussion
The importance of Ne has become increasingly recognized in plant breeding as it describes the rate of inbreeding and can reflect the contemporary status of genetic diversity in breeding populations [50]. When Ne is low, the population can become quickly inbred with little potential for genetic gain making long-term selection ineffective. Therefore, plant breeders should be cognizant of the effective population size of their breeding program [10]. Actively monitoring Ne in successive cycles of breeding can enhance the viability of the breeding efforts and help sustain long-term genetic gain. In this study, we presented the first estimation of Ne in dry pea using two distinct germplasm sets: 1) the NDSU set consisting of elite breeding lines within the NDSU breeding program, and 2) the USDA set comprised of landraces and plant introductions collected all over the world [35, 36]. The former represents breeding lines and germplasm in an active breeding program that releases new modern cultivars, while the latter represents germplasm accessions in a repository. As expected, the estimated Ne for the USDA set (Ne = 174) was higher than the NDSU set (Ne = 64). The selection and derivation of closely related breeding lines from multiple breeding populations likely resulted to a lower Ne estimation in the NDSU set, presumably due to increased inbreeding. The genetic diversity for the USDA set is higher than the NDSU set as it represents most of the available diversity in the USDA pea germplasm collection [35, 36].
The Ne estimate for the NDSU set was within the same range as those reported in other self-pollinating crops such as rice (Oryza sativa) and soybean (Glycine max), with calculated Ne ranging from 20 to 60. Juma R [47] estimated the Ne in rice to be 22 using an elite core panel comprised of 72 lines, but Ne may have been underestimated due to limited marker information used in the analysis. Similar studies in rice also had the same range of Ne, with calculated values ranging from 23–57 and 40–60; these were estimated based on breeding populations from recurrent selection programs [51] and pedigree data [52]. The estimated Ne of USDA set was within the range of Ne values reported in studies conducted on other crops. In soybean, Xavier A [53] estimated Ne for the USDA soybean germplasm collection comprised of 19,652 accessions from Bandillo N [54] and reported it to be 106 individuals. Recent studies have shown that soybean possess several genetic bottlenecks [55] and its genetic diversity has been reduced [56, 57]. The Ne estimate of USDA set is relatively higher than soybean, implying greater diversity. Zhao Y [58] estimated Ne in wild rice using 11 Chinese Oryza rufipogon populations including 32 landraces and reported it between 96–158, which is in a similar range to the USDA set. Thus, the Ne of USDA set offers greater potential for adaptation, maintaining rare alleles, population stability, and reduced risk for inbreeding.
The results of our study also suggest that the use of GBS holds good potential for making inferences of Ne regardless of the germplasm type. Using GBS-based markers, we approximated the LD pattern within and across chromosomes of both germplasms and then used the LD information for estimation of Ne. Genome-wide LD (r2) of the USDA set decayed from lower LD at 200 kb, while the NDSU set had the highest LD declined at a longer distance of around 250 kb. These results provided consistency of higher genetic variations of the former over the latter. Similar LD findings have been observed in previous studies conducted on peas, wherein both wild and spring peas exhibited a decay distance of approximately 200 kb, whereas wild/landrace peas were around 100 kb [49] which is a bit lower than the USDA set. Comparing the LD of USDA set and the NDSU set to other selfing crops such as rice, soybeans, and barley, the physical distances found were more or less similar depending on the populations. For instance, Huang X [59] estimated LD using O. indica and O. japonica landraces of rice at 123 and 167 kb, respectively, with r2 declining to 0.25 and 0.28. Additionally, soybean landraces extended from 90 to 500 kb [60] while improved cultivars hit 133 kb [61] which is similar to the USDA set. Alternatively, a recent LD analysis from soybean USDA germplasm revealed that the r2 dropped intragenically within a few kilobases [61] and the one in barley’s landraces hit 90 kb [62], both shorter than the USDA set. The LD-decay of the NDSU set was also found to be in a similar range with elite varieties of barley which extended to at least 212 kb [62] and O. japonica elite lines at ~ 318 kb [63], but had a higher distance compared to O. indica elite lines (~ 124 kb) [63]. The LD-decay rate of a crop does depend on the genetic background of the populations being studied, and it can be affected due to mutations, genetic drift, non-random mating, and a small Ne [64].
Effective population size helps breeders preserve and remodel their selection strategies to enhance the stability and variability in their breeding populations [10]. Breeders can also implement marker-based mating experiments known as optimum contribution selection (OCS) [47] in order to maintain diversity in selection candidates for long-term gain. As pulse crop breeders navigate through challenges in their breeding programs, the information from this study provides valuable insights by demonstrating the strength of contemporary populations and possibly contributing to the long-term goal of increasing genetic gain while maintaining diversity in breeding programs.
Conclusions
We provided insights of effective population size (Ne) in field pea which can guide plant breeders to actively monitor Ne in successive cycles of breeding to sustain viability of the breeding efforts in the long term. Our estimations revealed that the Ne of USDA set (174) was larger than the NDSU set (64), providing insights into the extent of inbreeding and available genetic diversity in both germplasm pool. For future estimation of Ne, researchers could incorporate additional biological information (e.g., gene expression, metabolomics, etc.) along with DNA markers and demographic history, that will likely increase the understanding of plant breeders regarding the population dynamics and potential for adaptation to different ever-changing environments.
Availability of data and materials
The SNP data used in this study were uploaded in a public repository and is available at this link: https://www.ncbi.nlm.nih.gov/sra/PRJNA730349 (Submission ID: SUB9608236). All the codes and the R package developed and used in this study are publicly available in the “EffectivePopSize” GitHub repository, https://github.com/PrincyJohnson/EffectivePopSize.
References
Abbo S, Gopher A, Lev-Yadun S. The domestication of crop plants. In: Thomas B, Murray BG, Murphy DJ, editors. Encyclopedia of applied plant sciences. 2nd ed. Oxford: Academic Press; 2017. p. 50–4.
FAOSTAT. Food and Agricultural Organization of the United Nations. 2021. https://www.fao.org/faostat/ . Accessed 6 Jul 2023 .
USDA. United States Acreage. National Agricultural Statistics Service. 2020. https://www.nass.usda.gov/Publications/Todays_Reports/reports/acrg0620.pdf. Accessed 15 Aug 2023.
Bari MAA, Zheng P, Viera I, Worral H, Szwiec S, Ma Y, et al. Harnessing genetic diversity in the USDA pea germplasm collection through genomic prediction. Front Genet. 2021;12:707754.
Tayeh N, Klein A, Le Paslier M-C, Jacquin F, Houtin H, Rond C, et al. Genomic prediction in pea: effect of marker density and training population size and composition on prediction accuracy. Front Plant Sci. 2015;6:941.
Gali KK, Sackville A, Tafesse EG, Lachagari VBR, McPhee K, Hybl M, et al. Genome-wide association mapping for agronomic and seed quality traits of field pea (Pisum sativum L.). Front Plant Sci. 2019;10:1538.
Rahimmadar S, Ghaffari M, Mokhber M, Williams JL. Linkage disequilibrium and effective population size of buffalo populations of Iran, Turkey, Pakistan, and Egypt using a medium density SNP array. Front Genet. 2021;12:608186.
Tenesa A, Navarro P, Hayes BJ, Duffy DL, Clarke GM, Goddard ME, et al. Recent human effective population size estimated from linkage disequilibrium. Genome Res. 2007;17:520–6.
Gargiulo R, Decroocq V, González-Martínez SC, Paz-Vinas I, Aury JM, Kupin IL, et al. Estimation of contemporary effective population size in plant populations: limitations of genomic datasets. Evol Appl. 2024;17:e13691.
Cobb JN, Juma RU, Biswas PS, Arbelaez JD, Rutkoski J, Atlin G, et al. Enhancing the rate of genetic gain in public-sector plant breeding programs: lessons from the breeder’s equation. Theor Appl Genet. 2019;132:627–45.
Lonsinger RC, Adams JR, Waits LP. Evaluating effective population size and genetic diversity of a declining kit fox population using contemporary and historical specimens. Ecol Evol. 2018;8:12011–21.
Hare MP, Nunney L, Schwartz MK, Ruzzante DE, Burford M, Waples RS, et al. Understanding and estimating effective population size for practical application in marine species management. Conserv Biol. 2011;25:438–49.
Hayes BJ, Visscher PM, McPartlan HC, Goddard ME. el multilocus measure of linkage disequilibrium to estimate past effective population size. Genome Res. 2003;13:635–43.
Nei M, Tajima F. Genetic drift and estimation of effective population size. Genetics. 1981;98:625–40.
Wang J, Santiago E, Caballero A. Prediction and estimation of effective population size. Heredity. 2016;117:193–206.
Wright S. Evolution in Mendelian populations. Genetics. 1931;16:97–159.
Fisher RA. The genetical theory of natural selection. Oxford: Oxford University Press; 1930.
Charlesworth B. Fundamental concepts in genetics: effective population size and patterns of molecular evolution and variation. Nat Rev Genet. 2009;10:195–205.
Crow JF, Kimura M. An introduction to population genetics theory. New York: Harper & Row; 1970.
Gilbert KJ, Whitlock MC. Evaluating methods for estimating local effective population size with and without migration. Evolution. 2015;69:2154–66.
Luikart G, Ryman N, Tallmon DA, Schwartz MK, Allendorf FW. Estimation of census and effective population sizes: the increasing usefulness of DNA-based approaches. Conserv Genet. 2010;11:355–73.
Fernández J, Villanueva B, Pong-Wong R, Toro MA. Efficiency of the use of pedigree and molecular marker information in conservation programs. Genetics. 2005;170:1313–21.
Wang J. Estimation of effective population sizes from data on genetic markers. Philos Trans R Soc Lond B Biol Sci. 2005;360:1395–409.
Sved JA. Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor Popul Biol. 1971;2:125–41.
Novo I, Santiago E, Caballero A. The estimates of effective population size based on linkage disequilibrium are virtually unaffected by natural selection. PLoS Genet. 2022;18:e1009764.
Hill WG, Robertson A. Linkage disequilibrium in finite populations. Theor Appl Genet. 1968;38:226–31.
Antao T, Pérez-Figueroa A, Luikart G. Early detection of population declines: high power of genetic monitoring using effective population size estimators. Evol Appl. 2011;4:144–54.
Pollak E. A new method for estimating the effective population size from allele frequency changes. Genetics. 1983;104:531–48.
Ardlie KG, Kruglyak L, Seielstad M. Patterns of linkage disequilibrium in the human genome. Nat Rev Genet. 2002;3:299–309.
Sved JA, Hill WG. One hundred years of linkage disequilibrium. Genetics. 2018;209:629–36.
García-Cortés LA, Austerlitz F, de Cara MAR. An evaluation of the methods to estimate effective population size from measures of linkage disequilibrium. J Evol Biol. 2019;32:267–77.
Hill WG. Estimation of effective population size from data on linkage disequilibrium1. Genet Res. 1981;38:209–16.
Bari MAA, Fonseka D, Stenger J, Zitnick-Anderson K, Atanda SA, Morales M, et al. A greenhouse-based high-throughput phenotyping platform for identification and genetic dissection of resistance to Aphanomyces root rot in field pea. Plant Phenome Journal. 2023;6:e20063.
Atanda SA, Steffes J, Lan Y, Al Bari MA, Kim J-H, Morales M, et al. Multi-trait genomic prediction improves selection accuracy for enhancing seed mineral concentrations in pea. Plant Genome. 2022;15:e20260.
Holdsworth WL, Gazave E, Cheng P, Myers JR, Gore MA, Coyne CJ, et al. A community resource for exploring and utilizing genetic diversity in the USDA pea single plant plus collection. Hortic Res. 2017;4:17017.
Cheng P, Holdsworth W, Ma Y, Coyne CJ, Mazourek M, Grusak MA, et al. Association mapping of agronomic and quality traits in USDA pea single-plant collection. Mol Breed. 2015;35:75.
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6:e19379.
Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010. https://www.bioinformatics.babraham.ac.uk/Projects/Fastqc/ .
Kreplak J, Madoui M-A, Cápal P, Novák P, Labadie K, Aubert G, et al. A reference genome for pea provides insight into legume genome evolution. Nat Genet. 2019;51:1411–22.
Toosi A, Fernando RL, Dekkers JCM. Genomic selection in admixed and crossbred populations. J Anim Sci. 2010;88:32–46.
Lee Y-S, Woo Lee J, Kim H. Estimating effective population size of thoroughbred horses using linkage disequilibrium and theta (4Nμ) value. Livest Sci. 2014;168:32–7.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559.
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–5.
Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82.
Sawada C, Moreau C, Robinson GHJ, Steuernagel B, Wingen LU, Cheema J, et al. An integrated linkage map of three recombinant inbred populations of pea (Pisum sativum L.). Genes. 2022;13:196.
R Core Team. A language and environment for statistical computing. 2023. https://www.r-project.org/ .
Juma RU, Bartholomé J, Thathapalli Prakash P, Hussain W, Platten JD, Lopena V, et al. Identification of an elite core panel as a key breeding resource to accelerate the rate of genetic improvement for irrigated rice. Rice. 2021;14:92.
Kruglyak L. Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet. 1999;22:139–44.
Siol M, Jacquin F, Chabert-Martinello M, Smýkal P, Le Paslier M-C, Aubert G, et al. Patterns of genetic structure and linkage disequilibrium in a large collection of pea germplasm. G3. 2017;7:2461–71.
Onda Y, Mochida K. Exploring genetic diversity in plants using high-throughput sequencing techniques. Curr Genomics. 2016;17:358–67.
Grenier C, Cao T-V, Ospina Y, Quintero C, Châtel MH, Tohme J, et al. Accuracy of genomic selection in a rice synthetic population developed for recurrent selection breeding. PLoS One. 2015;10:e0136594.
Morais Júnior OP, Breseghello F, Duarte JB, Morais OP, Rangel PHN, Coelho ASG. Effectiveness of recurrent selection in irrigated rice breeding. Crop Sci. 2017;57:3043–58.
Xavier A, Thapa R, Muir WM, Rainey KM. Population and quantitative genomic properties of the USDA soybean germplasm collection. Plant Genet Resour. 2018;16:513–23.
Bandillo N, Jarquin D, Song Q, Nelson R, Cregan P, Specht J, et al. A population structure and genome-wide association analysis on the USDA soybean germplasm collection. Plant Genome. 2015;8:eplantgenome2015.04.0024.
Guo J, Wang Y, Song C, Zhou J, Qiu L, Huang H, et al. A single origin and moderate bottleneck during domestication of soybean (Glycine max): implications from microsatellites and nucleotide sequences. Ann Bot. 2010;106:505–14.
Li Y-H, Zhao S-C, Ma J-X, Li D, Yan L, Li J, et al. Molecular footprints of domestication and improvement in soybean revealed by whole genome re-sequencing. BMC Genomics. 2013;14: 579.
Min W, Run-zhi L, Wan-ming Y, Wei-jun D. Assessing the genetic diversity of cultivars and wild soybeans using SSR markers. Afr J Biotech. 2010;9:4857–66.
Zhao Y, Vrieling K, Liao H, Xiao M, Zhu Y, Rong J, et al. Are habitat fragmentation, local adaptation and isolation-by-distance driving population divergence in wild rice Oryza rufipogon? Mol Ecol. 2013;22:5531–47.
Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y, et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet. 2010;42:961–7.
Hyten DL, Choi I-Y, Song Q, Shoemaker RC, Nelson RL, Costa JM, et al. Highly variable patterns of linkage disequilibrium in multiple soybean populations. Genetics. 2007;175:1937–44.
Zhou Z, Jiang Y, Wang Z, Gou Z, Lyu J, Li W, et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat Biotechnol. 2015;33:408–14.
Caldwell KS, Russell J, Langridge P, Powell W. Extreme population-dependent linkage disequilibrium detected in an inbreeding plant species, Hordeum vulgare. Genetics. 2006;172:557–67.
Li X, Chen Z, Zhang G, Lu H, Qin P, Qi M, et al. Analysis of genetic architecture and favorable allele usage of agronomic traits in a large collection of Chinese rice accessions. Sci China Life Sci. 2020;63:1688–702.
Flint-Garcia SA, Thornsberry JM, Buckler ES 4th. Structure of linkage disequilibrium in plants. Annu Rev Plant Biol. 2003;54:357–74.
Acknowledgements
We would also like to acknowledge the contributions of Jérôme Bartholomé who provided technical guidance on the implementation of Sved’s (1971) equation.
Funding
The authors would like to acknowledge the funding provided by USDA-NIFA (Hatch Project #: ND01513). The genotyping of the NDSU materials was funded by the North Dakota Department of Agriculture through the Specialty Crop Block Grant Program (19–429) and Northern Pulse Growers Associations. The genotyping of the USDA germplasm was partially supported through funding from USDA Plant Genetic Resource Evaluation, USA Dry Pea and Lentil Council Research Committee, USDA ARS Pulse Crop Health Initiative and USDA ARS Project: 5348–21000-017-00D (CJC), and 5348–21000-024-00D (RJM). This investigation used resources of the Center for Computationally Assisted Science and Technology (CCAST) at North Dakota State University, Fargo, ND, USA which were made possible in part by NSF MRI Award No. 2019077.
Author information
Authors and Affiliations
Contributions
JPJ: Conceptualization; Data curation; Pipeline development; Formal analysis; Investigation; Methodology; Writing – original draft, review and editing, LP: Methodology; Review and editing, HW: Methodology; Review and editing, SAA: Writing—review and editing, CJC: Funding acquisition; Resources; Review and editing, KM: Funding acquisition; Resources; Review and editing, RJM: Funding acquisition; Resources; Review and editing, and NB: Conceptualization; Supervision; Funding acquisition; Resources; Validation; Writing—review and editing.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Johnson, J.P., Piche, L., Worral, H. et al. Effective population size in field pea. BMC Genomics 25, 695 (2024). https://doi.org/10.1186/s12864-024-10587-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10587-6