
Abstract
Analyzing the genetic diversity and selection characteristics of sheep (Ovis aries) holds significant value in understanding their environmental adaptability, enhancing breeding efficiency, and achieving effective conservation and rational utilization of genetic resources. In this study, we utilized Illumina Ovine SNP 50 K BeadChip data from four indigenous sheep breeds from the southern margin of the Taklamakan Desert (Duolang sheep: n = 36, Hetian sheep: n = 74, Kunlun sheep: n = 27, Qira black sheep: n = 178) and three foreign meat sheep breeds (Poll Dorset sheep: n = 105, Suffolk sheep: n = 153, Texel sheep: n = 150) to investigate the population structure, genetic diversity, and genomic signals of positive selection within the indigenous sheep. According to the Principal component analysis (PCA), the Neighbor-Joining tree (NJ tree), and Admixture, we revealed distinct clustering patterns of these seven sheep breeds based on their geographical distribution. Then used Cross Population Extended Haplotype Homozygosity (XP-EHH), Fixation Index (FST), and Integrated Haplotype Score (iHS), we identified a collective set of 32 overlapping genes under positive selection across four indigenous sheep breeds. These genes are associated with wool follicle development and wool traits, desert environmental adaptability, disease resistance, reproduction, and high-altitude adaptability. This study reveals the population structure and genomic selection characteristics in the extreme desert environments of native sheep breeds from the southern edge of the Taklimakan Desert, providing new insights into the conservation and sustainable use of indigenous sheep genetic resources in extreme environments. Additionally, these findings offer valuable genetic resources for sheep and other mammals to adapt to global climate change.
Similar content being viewed by others
Introduction
Sheep (Ovis aries) play a crucial role in providing various products for humans, including wool, skin, meat, and milk, making them one of the most economically significant agricultural species [1]. Domesticated approximately 11,000 years ago, Sheep have spread to different continents and adapted to diverse agricultural ecological conditions accompanied human migration [2], including extreme weather such as highlands and deserts. The Taklamakan Desert, located in the Tarim Basin within the Eurasian hinterland [3], features gravel deserts surrounded by several mountains, forming a ‘C’ type mountain-basin pattern [4]. The average annual precipitation in this basin is less than 50 millimeters, while evaporation averages around 2600 millimeters [5]. The region experiences frequent windy and dusty weather [6], large diurnal temperature variations, and abundant sunlight, typical of a temperate continental desert climate. The extreme environmental conditions pose significant challenges to livestock production [7]. Studying how domesticated animals adapt to extreme environments in a short period of time can help formulate appropriate breeding plans to mitigate the impact of the future environmental changes.
Natural selection plays a crucial role in shaping individuals better adapted to new environments. In addition to natural selection, artificial selection has been extensively implemented in livestock breeding to cultivate more desirable and advantageous individuals [8]. Notably, compared to traditional artificial selection methods, genomic selection expedites the process of identifying superior genotypes and accelerating the breeding cycle [9]. The employment of the Illumina Ovine SNP 50 K BeadChip in conjunction with selective trait detection not only uncovers the underlying mechanisms of modern breeding and artificial selection but also provide possibilities to identify the candidate genes associated with adaptively and economically important traits [10, 11]. In past studies of ruminants by others, a number of genes that directly cause trait variation when subjected to positive selection have been identified. Lei et al. [12]. analyzed 27 different populations of sheep with varying wool types based on the Illumina Ovine SNP 50 K Genotyping BeadChip. They identified four potential genes (PRX, SOX18, TGM3, and TCF3) directly or indirectly influencing wool follicle development and wool shedding. Cheng et al. [13]. analyzed the whole-genome sequencing data from 1167 sheep (1098 domestic sheep and 69 wild sheep) among the 161 breeds (154 breeds and 7 Ovis species). They highlighted the haplotypes closely associated with spiral horn characteristics (RXFP2), ear morphology (MSRB3), and facial features (VPS13B). Cao et al. [14]. used whole-genome single nucleotide polymorphism (SNP) data from 111 populations of domestic sheep (n = 3447) and 7 populations of wild sheep (n = 403) to detect clear signals of introgression from the wild relatives into the domestic populations. Ultimately, they identified signals of introgression associated with olfaction (ADCY3, TRPV1) and innate immunity (PADI2). Zhang et al. [15] examined the selection signatures of 5 sheep breeds with three different agricultural geographic characteristics based on the Illumina Ovine SNP 50 K Genotyping BeadChip. They found genetic information associated with immunity (DNTT, FEN1, POLL, PRKDC, XRCC4), high reproductive rate (IGFBP7, STC1, TFAP2), and adaptability (PNPLA6, MITF) in indigenous sheep breeds adapted to the Taklamakan desert environment using four complementary genomic selection signals (FST, XP-EHH, Rsb, and iHS). Kim et al. [16]. compared the genetic variation patterns of five indigenous African cattle populations from regions south of the Sahara with those of 53 commercial taurine breeds using the Illumina Bovine SNP 50 K Genotyping BeadChip. They revealed the genetic variations in African indigenous cattle related to gene selection characteristics associated with heat tolerance (BTA22, HSPA4, SOD1, PRLH), disease resistance (HCRTR1, BOLA-DRB3), coat color (MC1R, KIT, MITF, PDGFRA), and horn development (MAP3K5, PPP2R2C, FGF18, FRS3, ACVRL1, CASR, TLX3, ACVR1B, RUNX3).
Although the genetic basis underlying the important economic traits such as reproduction and year-round estrus in the indigenous sheep from the southern edge of the Taklamakan Desert has been reported [17], our understanding of their interpopulation genetic structure and the genetic mechanisms relating to their adaptation to the local environment is currently limited. This study aims to employ high-density 50 K SNP genetic data to uncover the population structure of the indigenous sheep from the southern edge of the Taklamakan Desert and assess the genomic signatures indicative of selective pressures in the extreme environmental conditions. Of greater significance, this investigation promises to offer new insights into the conservation and sustainable utilization of local sheep breeds’ genetic resources in the extreme desert environment. Additionally, it has the potential to deliver valuable resources for other mammals to cope with global climate change.
Materials and methods
Ethics statement
This work was conducted in accordance with the standards set by the Ethics Committee of the College of Animal Science and Technology of Tarim University (SYXK2020-009). Written informed consent was obtained from the owners for the participation of their animals in this study.
Animal samples
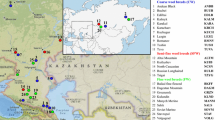
In this study, a total of 723 samples from seven different breeds were included (Fig. 1; Table 1). To be specific, we used four native sheep breeds inhabiting the southern border of the Taklamakan Desert, including 36 Duo-lang sheep (DUO) from the Duo-lang Sheep Breeding Farm in Maigaiti County, Xinjiang; 178 Qira black sheep (QIR) from the Qira Black Sheep Breeding Farm in Cele County, Xinjiang; 74 Hetian sheep (HET) from Hetian District in Xinjiang; and 27 Kunlun sheep (KUN) from the Kunlun Sheep Breeding Farm in Qiemo County, Xinjiang. Samples were selected on a random sampling basis from different breeding groups, different pens, and different batches to ensure their unrelatedness. All samples underwent assessments based on factors such as dental wear, size development, and developmental stage to initially determine their age. Subsequently, the age data collected were then cross-referenced in detail with on-farm management records to verify the accuracy of the animals’ age. Female specimens were deliberately chosen to minimize any potential confounding effects stemming from sexual disparities. Furthermore, we obtained data of Poll Dorset sheep (POL, 105), Suffolk sheep (SUF, 153), and Texel sheep (TEX, 150) from the International Sheep Genomics Consortium (ISGC) Footnote 1.

Map showing the different geographic distribution of the four indigenous sheep breeds from the southern edge of the taklamakan desert, china, analyzed in this study
Genotyping and quality control
Blood samples were collected from the jugular vein, and DNA was extracted using a DNA extraction kit (Tiangen Biotech Co Ltd., Beijing, China). DNA samples were genotyped using the Illumina Ovine SNP50K BeadChip (Compass Biotechnology Co Ltd., Beijing, China). The quality of the single nucleotide polymorphism genotyping data was evaluated using Plink V.1.90 software [18], and unqualified SNPs were excluded. The quality control criteria for this study were as follows: (1) individual detection rate > 0.95; (2) SNPs detection rate > 0.95; (3) Minor allele frequency (MAF) > 0.05; (4) Hardy-Weinberg equilibrium (HWE) p-value ≥ 1 × 10− 6.
Genetic diversity
The inbreeding coefficient (F), observed heterozygosity (Ho), expected heterozygosity (He), and minor allele frequency (MAF) were calculated for the seven sheep breeds using Plink V. 1.90 software.
Population structure analyses
After completing quality control, the genotypic data was subjected PCA analysis using Plink V. 1.90 software. The P distance matrix was calculated using VCF2Dis V. 1.09 software Footnote 2, which was then used to construct an NJ-tree via the ATGC: Montpellier Bioinformatics platform Footnote 3. Lastly, the NJ-tree was visualized using iTOL Footnote 4 online tool [19]. Genetic admixture calculations were executed using ADMIXTURE 1.3.0 [20], and the population genetic structure was analyzed by using K values ranging from 2 to 10. Additionally, respective cross-validation (CV) errors were calculated.
Genome-wide selective signatures
To accurately reveal the genetic imprints left by indigenous sheep during their evolution at the southern edge of the Taklamakan Desert, we utilized several selection signal detection methods [21, 22]. Initially, we identified candidate gene regions in indigenous sheep that may have undergone strong natural selection utilizing FST and XP-EHH methods. Subsequently, we conducted an in-population genome scan on each of the four local sheep breeds using iHS. Finally, by cross-comparing the results obtained from FST and XP-EHH with the iHS scans, we identified positively selected alleles in each local sheep breed.
XP-EHH statistic is designed to detect ongoing or nearly fixed selection features by comparing haplotypes between two populations [23]. The formula is as follows:
XP-EHH values were calculated using Selscan v1.3.0 [24]. Positive values indicate selection signals in population I, while negative values indicate selection signals in population Ii.
Fst is a measure that quantifies the degree of genetic differentiation between populations, making it suitable for comparing diversity among different subpopulations of the same species. The formula is as follows:
MSG represents the mean squared error within populations, MSP represents the mean squared error between populations, and nc is the average sample size corrected for the entire population. Vcftools was used to calculate the Fst values for each window (window size of 50 Kb, sliding step size of 25 Kb). The thresholds of 1% of these two selective signatures were considered as potential candidate regions under selection.
IHS is a statistic based on haplotype frequency to detect selection signals within the genome [25]. The formula is as follows:
IHS denotes the integrated haplotype value, iHHA denotes the EHH integration score of the ancestral allele, and iHHD denotes the EHH integration score of the derived allele. The obtained iHS values were corrected using Voight’s method with the following formula:
represents the expected iHS value, and
represents the standard deviation. A large iHS value suggests that the haplotype likely carries ancestral alleles, whereas a small iHS value indicates the likely possess of evolved genes. Candidate regions under selection were determined based on the 5% threshold of iHS.
Enrichment analysis of candidate genes
Gene annotation was performed utilizing the sheep genome Oar_v4.0. Gene functional annotation was conducted referencing the NCBI Footnote 5 and OMIM Footnote 6 databases. Gene clustering analysis was carried out using DAVID Footnote 7. Pathway enrichment analysis was performed utilizing Gene Ontology Footnote 8(GO) and Kyoto Encyclopedia of Genes and Genomes Footnote 9(KEGG). Performed gene networking analysis on the overlapping candidate genes obtained through paired comparisons.
Results
SNP quality control
In the experiment, a total of 46,900 SNPs from 723 sheep underwent genotypic quality control. After removing SNPs that did not meet the quality control criteria, 41,831 SNPs remained for further analysis.
Genetic diversity
The genetic diversity results for the seven sheep breeds are as follows (Table 1):
KUN has the lowest F (-0.0746), while HET has the highest (0.0464).
KUN has the lowest MAF (0.2665), while HET has the highest (0.2977).
KUN has the lowest Ho (0.5898), while QIR has the highest (0.6450).
HET has the lowest He (0.6034), while QIR has the highest (0.6371).
Population genetic structure
We utilized PCA, NJ tree, and ADMIXTURE methods to analyze the genetic relationships and structure among four native sheep breeds from the southern edge of the Taklamakan Desert and three sheep breeds from other countries.
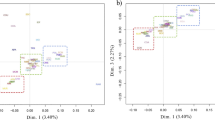
In the PCA results (Fig. 2) revealed a distinct separation between the sheep breeds from other countries (POL, SUF, TEX) and the native sheep breeds from the southern edge of the Taklamakan Desert (DUO, QIR, KUN, HET). Furthermore, the native sheep breeds from the southern edge of the Taklamakan Desert exhibited further differentiation based on their geographic distribution, with HET appearing to be divided into three subgroups. The NJ tree accurately differentiated the seven sheep breeds based on their geographic origins (Fig. 3). The native sheep breeds from the southern edge of the Taklamakan Desert coalesced into one branch, whereas the sheep breeds from other countries constituted the other branch. Notably, the NJ tree revealed that HET was subdivided into three clusters, with one cluster aligning with DUO, QIR, and KUN, while the other two clusters formed separate groups. The results closely align with those obtained from the PCA analysis. The ADMIXTURE results showed (Fig. 4) that the sheep breeds from other countries and the native sheep from the southern edge of the Taklamakan Desert possess different ancestral components. At K = 4, QIR predominantly belonged to the genomic cluster specific to the native sheep breeds from the southern edge of the Taklamakan Desert. HET showed the highest level of admixture, with no dominant genomic cluster among the four clusters. At K = 5, a new genomic cluster emerged within certain portions of HET. Then, at K = 6, KUN was delineated as a new genomic cluster. As K increased, DUO, HET, KUN, and QIR no longer exhibited obvious separation, while noticeable differentiation emerged among the three breeds POL, SUF, and TEX. This suggests that POL, SUF, and TEX may represent three hybrid varieties resulting from artificial selection processes.

The PCA analysis chart of sheep breeds from two different geographical regions, the X-axis represents PC1, and the Y-axis is PC2. The picture on the left shows the genetic distances of seven sheep breeds, and that on the right shows the genetic distances of four sheep breeds from the southern edge of the taklimakan desert

Evolutionary tree of seven sheep breeds. Gray represents QIR, Red represents KUN, Blue represents DUO, Green represents HET, Yellow represents POL, Purple represents SUF, and Brown represents TEX

Admixture analysis of seven sheep breeds. Results for inferred numbers of clusters k = 2–10 were shown. Different colors represent different ancestral components. From left to right are DUO, QIR, HET, KUN, POL, SUF and TEX
Detection of selection signatures
The intersection and union relations of all selective scanning results obtained through the XP-EHH, FST, and iHS methods are presented in the Appendix Table (Table S1). In the XP-EHH analysis, a total of 203 candidate genes were identified across 15 autosomes at a threshold of 1% (Fig. 5a, Table S2). Subsequent GO enrichment analysis on these candidate genes revealed the top three significant GO terms: keratin filament (GO:0045095; P-Value = 9.22024E-06), anterior/posterior pattern specification (GO:0009952; P-Value = 2.86195E-05), and chromatin binding (GO:0003682; P-Value = 0.001262505) (Table S3). In the FST analysis, a total of 738 candidate genes were identified across 26 autosomes at a threshold of 1% (Fig. 5b, Table S4). Subsequent GO enrichment analysis on these candidate genes revealed the top three significant GO terms: transcription factor activity, sequence-specific DNA binding (GO: 0003700; P-Value = 0.000151821), regulation of cardiac muscle cell action potential (GO:0098901; P-Value = 0.000176044), and regulation of heart rate (GO:0002027; P-Value = 0.000330621) (Table S5). At the threshold of 1%, both XP-EHH and FST analyses detected 57 overlapping candidate genes (Fig. 5c, Table S1).

Manhattan plots of selection signatures determined by comparing the four Chinese local sheep breeds with three foreign sheep breeds using FST (a) and XP-EHH (b) methods. Wenn diagram(c) of XP-EHH intersection with Fst
In the iHS test, we separately detected strongly selected regions in each local sheep breed, using a threshold of 5% (Table S6). Within the iHS-screened regions, we identified 1302, 1545, 1159 and 1241 candidate genes undergoing positive selection in the four indigenous sheep breeds (DUO, HET, KUN and QIR), respectively. By intersecting these results with those obtained from XP-EHH and FST, we obtained 14, 24, 21 and 19 intersection genes, respectively (Fig. 6, Table S1).

iHS Manhattan chart of DUO, HET, KUN and QIR. The intersection with FST and XP-EHH results obtained 14 (a), 24 (b), 21 (c) and 19 (d) genes, respectively
Ultimately, using the three methods of detecting selection signals, XP-EHH, FST and iHS, we identified a total of 32 overlapping candidate genes across four indigenous sheep breeds inhabiting the southern edge of the Taklamakan Desert (Fig. 7a). These candidate genes are associated with wool follicle development and wool traits, adaptation to desert environments, reproductive traits, adaptation to high-altitude environment, and immune response in sheep (Table 2). Pathway enrichment analysis was performed on the candidate genes obtained by the intersection. (Table S7) Genes with similar functions within the overlapping candidate genes cluster aggregate and interact with each other (Fig. 7b).

(a) 32 Overlapping genes subject to positive selection identified in four indigenous sheep breeds utilizing XP-EHH, FST, and iHS Analyses (b) Gene networking analysis of 32 overlapping genes
Discussion
Population genetic diversity
Genetic variability serves as a crucial response mechanism of organisms adapting to environmental changes and stands as a pivotal aspect in biodiversity research [26]. Assessing genetic variation within species or population provides a theoretical bedrock for the preservation and enhancement of sheep germplasm resources [27].
MAF results revealed that the genetic variability in four indigenous sheep breeds from the southern edge of the Taklamakan Desert was comparable to that in other foreign sheep populations. Within our study cohorts, KUN exhibited the lowest F value (-0.0746), predominantly attributed to contemporary breeding strategies and the introduction of new individuals with diverse genetic backgrounds, effectively mitigating inbreeding. However, genetic diversity within the KUN remained relatively limited, primarily owing to their isolation in high-altitude mountainous regions and the small size of their core population. The implementation of rigorous scientific management practices is essential for conserving KUN ‘s genetic resources. Regular exchange of breeding rams among KUN could simultaneously enhance effective population size and enrich intraspecific genetic diversity [10].
It is notable that Ho closely approximated He across the four indigenous sheep breeds examined. This indicates that these populations are currently in a state of genetic equilibrium, neither undergoing significant selection pressure nor experiencing substantial inbreeding. Moreover, this finding indicates their substantial potential for selective breeding. In compared to DUO and KUN, HET and QIR exhibit slightly higher Ho than He. Considering their predominant distribution in the Hotan region of Xinjiang, a key junction along the Silk Road [28], we speculate that gene flow may significantly contribute to intraspecific genetic variation. This underscores the urgency of implementing genetic conservation programs aimed at preserving the unique genetic diversity of these breeds.
KUN demonstrated the lowest genetic variation, necessitating the implementation of a rational mating system, establishment of a gene bank, and promotion of gene flow among diverse populations as key strategies for conserving its genetic diversity. Conversely, HET and QIR showed high genetic variation, facilitating their adaptation to diverse environments and potential challenges. However, this diversity also complicates inter-population gene exchange. Future efforts for HET and QIR should focus on breeding individuals with superior traits through selective breeding or crossbreeding, while minimizing excessive artificial intervention.
The current study elucidated the genetic variation in indigenous sheep populations at the southern edge of the Taklamakan Desert through the analysis of genetic variation parameters within the populations. However, the small sample sizes of KUN and DUO may not sufficiently capture the genetic diversity within their populations. Additionally, selecting only adult ewes as samples may not fully reveal all the genetic characteristics within the populations. To enhance the representation of genetic diversity within the populations, future studies should expand the sample size of each sheep type and include male samples for analysis.
Population genetic structure
Population genetic structure indicates a clear separation of indigenous sheep in Xinjiang from other sheep populations. The four indigenous sheep breeds in southern Xinjiang form two distinct groups with three sheep breeds from other countries, and the genetic resources of the four indigenous sheep breeds are closely related to each other. This clustering pattern is significantly associated with their geographical environment and genetic distance. Additionally, PCA and NJ tree analysis confirmed the presence of three distinct clusters within the HET, consistent with previous findings of Abied et al. [10]. on indigenous sheep breeds in China. Admixture analysis revealed that QIR displayed the highest proportion of Xinjiang sheep ancestry, while the genetic diversity in HET correlated with its geographic distribution [27]. HET living at intermediate and lower altitudes exhibited considerable genetic diversity, likely influenced by their geographic locations. Hetian, situated along the Silk Road, serves as a vital trade junction, possibly facilitating gene admixture in HET at intermediate and lower altitudes [29, 30]. The study also indicates that high-altitude HET have lower genetic diversity, primarily because they inhabit mountainous regions at higher altitudes. Geographical isolation is the main factor contributing to this lower genetic diversity in high-altitude HET populations [31, 32], implying a closer genetic relationship with their ancestral HET population.
Selection signatures of candidate genes
To enhance the robustness of our results, we initially identified candidate genes with strong selection traits in local sheep by intersecting selection signals identified by the XP-EHH and FST methods. These candidate genes were than integrated with those identified through iHS analysis to detect alleles undergoing positive selection in each local sheep breed [21, 22]. Ultimately, the identified genes were associated with economic traits and adaptation to harsh environments.
Candidate genes associated with wool follicle development and wool traits
Wool, a key economic trait in wool-producing sheep, depends on the growth and development of hair follicles attached to the skin [33].
In this study, we identified five overlapping genes involved in hair follicle growth and development: BMP7 [34, 35], SOX2 [36, 37], SPTLC3 [38], LEF-1, and TRPS1. LEF-1, a transcription factor, promotes the maturation of secondary hair follicles by regulating hair follicle development [39]. It has been reported that SOX2 regulate hair growth by controlling BMP signaling [40]. TRPS1 and SPTLC3 are associated with cashmere wool characteristics [38].
Additionally, we also identified seven overlapping genes associated with wool traits. KRT71, KRT72, KRT73, and KRT74 are specifically expressed in the inner root sheath of the hair follicles and play a vital role in the formation of wool fibers [41, 42]. Studies have shown a strong correlation between KRT71 and curly hair phenotypes in sheep [43], dogs [44, 45], and cats [46]. KRT72 is a major component of wool fibers [47], and KRT74 is positively correlated with wool growth [48]. EPHA5 and SCFD2 are significantly associated with the curling characteristics and coat color of wool [49, 50].
Adaptive mechanism of the desert environment
The indigenous sheep breeds from the southern edge of the Taklimakan Desert are exposed to various biological and physical stressors throughout the year, such as high temperatures, drought, intense ultraviolet radiation, and parasites. Therefore, our candidate regions encompass several genes associated with adaptation to the desert environment and specific immune responses.
In arid or hot environments, many breeds typically exhibit smaller body sizes as an adaptation to cope with scarce water sources and temperature regulation [51]. This can explain the occurrence of genes such as RAPSN, CNBD2, KCNJ16, KCNMB2, PLCG1, and BMP7 in the candidate regions. For instance, RAPSN is significant correlation with body mass in sheep [52], while CNBD2 is associated with short stature, a trait also observed in human [53]. Additionally, KCNJ16 [54], KCNMB2 [55], and PLCG1 [56] have been linked to vascular growth and dilation. Skin vascular dilation increases blood flow, facilitating heat dissipation, which explains the cooling mechanism in hot environments. BMP7 is crucial for kidney function and development [51], as the kidney serves as a core adaptation for water retention and reabsorption in desert environments. Genes related to pigment deposition and eye lens development have been identified in the candidate regions. For instance, CELF1 play vital roles in lens development [57], aiding in light refraction and filtering out ultraviolet rays. TECRL has a positive effect on pigment deposition [58]. These findings likely stem from natural selection resulting from prolonged exposure to intense sunlight and ultraviolet radiation.
We have identified CSMD3 genes associated with cytokine signaling and immune infiltration [59], along with candidate genes involved in host defense, disease resistance, and inflammatory responses. For instance, PCDH7 is a potential biomarker for host resistance to parasite infection [60], while CDH26 regulates leukocyte activation and adhesion during allergic reactions [61].
Candidate genes associated to reproduction traits
Reproduction is an important trait in sheep breeding, influenced by various factors like environment, nutrition, and genetic factors. We have identified several candidate genes associated with reproductive traits, including those in gonadal development, gametogenesis, litter size, and embryonic development.
IGFBP7 and BMP7 are pivotal in reproductive processes, with IGFBP7 [62]is highly expressed in ovaries, regulating follicular development and ovulation, while BMP7 [63], a core BMP subfamily member, influences hormone production, granulosa cell development, and follicular development. BMP7 is also a potential candidate gene for ovulation rate and litter size traits [64]. EPG5 influences gonadal formation, gametogenesis and early embryonic differentiation [65, 66]. Targeted disruption of CELF1 impairs spermatogenesis, causing male infertility [67]. GRID2 is associated with litter size in Dazu Black goats [68]. RBM38, a target of the p53 family, affects embryonic development by modulating p53 accumulation via the Rbm38-p53 axis, particularly in a p21 (a conventional target of the p53 pathway) dependent manner at the morula/blastocyst stage [69,70,71].
EPB41L1/4.1 N, located within the hypothalamic-pituitary-gonadal (HPG) axis, regulates reproductive neuroendocrine function by facilitating the release of hormones (gonadotropin-releasing hormone/GnRH, growth hormone-releasing hormone/GHRH) in the hypothalamic-pituitary pathway [72]. GnRH acts on GnRH receptors (G-protein-coupled receptors) in the anterior pituitary, stimulating gonadotropin secretion (follicle-stimulating hormone/FSH, luteinizing hormone/LH, prolactin/PRL), thereby modulating reproductive system development.
Adaptive mechanism of the high-altitude environment
Animals inhabiting high-altitude areas face significant environmental challenges such as low oxygen, rugged terrain, and limited food availability. This study identified several genes associated with adaptability to high-altitude environments within the candidate regions.
TMTC2 is a positively selected gene in high-altitude environments [73]. SOX2 has been associated with extreme hypoxia and cold adaptation in Himalayan marmots [74].
Good motor coordination is crucial for sheep to navigate rugged high-altitude environments. TBL1XR1, associated with various developmental disorders affecting neural system functions [75], play a role in neural progenitor cell proliferation and differentiation. Loss or mutations in TBL1XR1 can lead to abnormal motor coordination. GABRA1 and GABRA6, exclusive to the nervous system, regulate brain development mechanisms [76, 77], and reduced levels may compromise motor function. GRID2, expressed in layer V cortical neurons involved in motor function formation, underpins motor control and limb coordination. Moreover, GRID2 has been identified as a potential candidate gene for adipogenesis [78, 79].
In high-altitude conditions with low oxygen, sympathetic nervous system activation increases heart rate and cardiac output to enhance tissue oxygenation. We identified two candidate genes associated with cardiac function: CELF1 deletion affects cardiac function and morphology in newborns, reducing cardiac vitality [80, 81]. DNAJC19 (DnaJ heat shock protein family member C19), exhibits high expression in the heart’s mitochondria [82, 83], providing a significant amount of ATP for cardiac pumping function.
Adipose tissue serves as a crucial energy reserve for animals in extreme environments and during food scarcity, offering an adaptive response. We identified genes linked to sheep fat deposition: MAT2B promotes fat synthesis by regulating cellular levels of S-adenosylmethionine (SAMe) [84]. TECRL participates in lipid production reactions [85]. HADH participates in β-oxidation and correlates with lipid content and droplet numbers in cells [86]; its knockout inhibits lipid storage.
This study utilized diverse selection signal scanning methods to reveal the distinctive evolutionary trajectories of indigenous sheep from the southern edge of the Taklamakan Desert. It identified candidate genes in the genome associated with wool follicle development, wool traits, environmental adaptability, disease resistance, reproduction, and high-altitude adaptation. These findings provide new germplasm resources and breeding strategies for sheep breeding and improvement, offering important references for other mammals in devising climate change coping strategies. In the future, we plan to conduct functional validation of these adaptive genes and further elucidate the molecular mechanisms underlying the adaptive evolution of sheep by integrating multiple omics data. Although this study detected genetic variations in four indigenous sheep breeds using high-throughput sequencing technology, limitations in genetic marker coverage may have hindered the comprehensive identification of all key genetic variations. However, with the advancement of high-throughput sequencing technology, our aim is to enhance sequencing depth, broaden the range of breeds, and augment population sizes comprehensively reveal the genetic diversity and evolutionary history of sheep populations.
Conclusions
The study revealed the genetic variability of native sheep breeds from the southern edge of the Taklimakan Desert, which provides new insights for the conservation of population in the future. Additionally, based on geographical and genetic relationships, the four native sheep breeds from the southern edge of the Taklimakan Desert and three sheep breeds from other countries were clustered into two groups, and HET based on geographical location were made further subdivision into three clusters using. Analysis of selection characteristics made a certain for various candidate genomic regions, which were associated with wool follicle development and wool traits, desert environmental adaptability, disease resistance, reproduction, and high-altitude adaptation. At the molecular level, we elucidated the genetic mechanisms underlying the adaptability of native sheep breeds from the southern edge of the Taklimakan Desert in the extreme environments. This provides new insights into the conservation and sustainable utilization of genetic resources in native sheep and offering important references for sheep and other mammals in devising climate change coping strategies.
Data availability
The datasets analysed during the current study are available in the OMIX, China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences repository, (https://ngdc.cncb.ac.cn/omix; GSA: OMIX006742).
Notes
References
Chessa B, Pereira F, Arnaud F, Amorim A, Goyache F, Mainland I, Kao RR, Pemberton JM, Beraldi D, Stear MJ, Alberti A, Pittau M, Iannuzzi L, Banabazi MH, Kazwala RR, Zhang YP, Arranz JJ, Ali BA, Wang Z, Uzun M, Palmarini M. Revealing the history of sheep domestication using retrovirus integrations. Volume 324. Science; 2009. pp. 532–6. (New York, N.Y.). 5926https://doi.org/10.1126/science.1170587.
Zeder MA. Domestication and early agriculture in the Mediterranean Basin: origins, diffusion, and impact. Proc Natl Acad Sci USA. 2008;105(33):11597–604. https://doi.org/10.1073/pnas.0801317105.
Yang F, He Q, Huang J, et al. Desert Environment and Climate Observation Network over the Taklimakan Desert. Bull Am Meteorol Soc. 2020;102(6):E1172–91. https://doi.org/10.1175/BAMS-D-20-0236.1.
Xu X, Wang Y, Wei W, Zhao T, Xu X. Summertime precipitation process and atmospheric water cycle over tarim basin under the specific large terrain background. Desert Oasis Meteorol. 2014;8(2):1–11. https://doi.org/10.3969/j.issn.1002-0799.2014.02.001.
Gui Dongwei Z, Fanjiang L, Zhen ZB. Characteristics of the clonal propagation of Alhagi Sparsifolia Shap. (Fabaceae) under different groundwater depths in Xinjiang, China. Rangel J. 2013;35:355–62. https://doi.org/10.1175/RJ13004.
Ma M, Yang X, He Q, Zhou C, Mamtimin A, Huo W, Yang F. Characteristics of dust devil and its dust emission in northern margin of the Taklimakan Desert. Aeolian Res. 2020;44:100579. https://doi.org/10.1016/j.aeolia.2020.100579.
Howden SM, Soussana JF, Tubiello FN, Chhetri N, Dunlop M, Meinke H. Adapting agriculture to climate change. Proc Natl Acad Sci USA. 2007;104(50):19691–6. https://doi.org/10.1073/pnas.0701890104.
Brito LF, Kijas JW, Ventura RV, Sargolzaei M, Porto-Neto LR, Cánovas A, Feng Z, Jafarikia M, Schenkel FS. Genetic diversity and signatures of selection in various goat breeds revealed by genome-wide SNP markers. BMC Genomics. 2017;18(1):229. https://doi.org/10.1186/s12864-017-3610-0.
Crossa J, Pérez-Rodríguez P, Cuevas J, Montesinos-López O, Jarquín D, de Los Campos G, Burgueño J, González-Camacho JM, Pérez-Elizalde S, Beyene Y, Dreisigacker S, Singh R, Zhang X, Gowda M, Roorkiwal M, Rutkoski J, Varshney RK. Genomic selection in plant breeding: methods, models, and perspectives. Trends Plant Sci. 2017;22(11):961–75. https://doi.org/10.1016/j.tplants.2017.08.011.
Abied A, Bagadi A, Bordbar F, Pu Y, Augustino SMA, Xue X, Xing F, Gebreselassie G, Mwacharo JHJM, Ma Y, Zhao Q. Genomic diversity, Population structure, and signature of selection in five Chinese native Sheep breeds adapted to Extreme environments. Genes. 2020;11(5):494. https://doi.org/10.3390/genes11050494.
Magee DA, Park SD, Scraggs E, Murphy AM, Doherty ML, Kijas JW, International Sheep Genomics Consortium, MacHugh DE. (2010). Technical note: High fidelity of whole-genome amplified sheep (Ovis aries) deoxyribonucleic acid using a high-density single nucleotide polymorphism array-based genotyping platform. Journal of animal science, 88(10), 3183–3186. https://doi.org/10.2527/jas.2009-2723.
Lei Z, Sun W, Guo T, Li J, Zhu S, Lu Z, Qiao G, Han M, Zhao H, Yang B, Zhang L, Liu J, Yuan C, Yue Y. Genome-wide selective signatures reveal candidate genes Associated with hair follicle development and wool shedding in Sheep. Genes. 2021;12(12):1924. https://doi.org/10.3390/genes12121924.
Cheng H, Zhang Z, Wen J, Lenstra JA, Heller R, Cai Y, Guo Y, Li M, Li R, Li W, He S, Wang J, Shao J, Song Y, Zhang L, Billah M, Wang X, Liu M, Jiang Y. Long divergent haplotypes introgressed from wild sheep are associated with distinct morphological and adaptive characteristics in domestic sheep. PLoS Genet. 2023;19(2):e1010615. https://doi.org/10.1371/journal.pgen.1010615.
Cao YH, Xu SS, Shen M, Chen ZH, Gao L, Lv FH, Xie XL, Wang XH, Yang H, Liu CB, Zhou P, Wan PC, Zhang YS, Yang JQ, Pi WH, Hehua E, Berry DP, Barbato M, Esmailizadeh A, Nosrati M, Li MH. Historical introgression from wild relatives enhanced climatic adaptation and resistance to Pneumonia in Sheep. Mol Biol Evol. 2021;38(3):838–55. https://doi.org/10.1093/molbev/msaa236.
Zhang CL, Liu C, Zhang J, Zheng L, Chang Q, Cui Z, Liu S. Analysis on the desert adaptability of indigenous sheep in the southern edge of Taklimakan Desert. Sci Rep. 2022;12(1):12264. https://doi.org/10.1038/s41598-022-15986-x.
Kim, J., Hanotte, O., Mwai, O. A., Dessie, T., Bashir, S., Diallo, B., Agaba, M.,Kim, K., Kwak, W., Sung, S., Seo, M., Jeong, H., Kwon, T., Taye, M., Song, K. D.,Lim, D., Cho, S., Lee, H. J., Yoon, D., Oh, S. J., … Kim, H. (2017). The genome landscape of indigenous African cattle. Genome biology, 18(1), 34. https://doi.org/10.1186/s13059-017-1153-y.
Zhang J, Zhang C-l, Tuersuntuohe M, Liu S. Population structure and selective signature of sheep around Tarim Basin. Front Ecol Evol. 2023;11:1146561. https://doi.org/10.3389/fevo.2023.1146561.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. https://doi.org/10.1086/519795.
Letunic I, Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49:W293–6. https://doi.org/10.1093/nar/gkab301.
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655–64. https://doi.org/10.1101/gr.094052.109.
Hohenlohe PA, Phillips PC, Cresko WA. Detect selection in natural populations: key concepts and methodological considerations. Int J Plant Sci. 2010;171(9):1059–71. https://doi.org/10.1086/656306. USING POPULATION GENOMICS TO.
Oleksyk TK, Smith MW, O’Brien SJ. Genome-wide scans for footprints of natural selection. Philos Trans R Soc Lond B Biol Sci. 2010;365(1537):185–205. https://doi.org/10.1098/rstb.2009.0219.
Sabeti, P. C., Varilly, P., Fry, B., Lohmueller, J., Hostetter, E., Cotsapas, C.,Xie, X., Byrne, E. H., McCarroll, S. A., Gaudet, R., Schaffner, S. F., Lander, E.S., International HapMap Consortium, Frazer, K. A., Ballinger, D. G., Cox, D. R.,Hinds, D. A., Stuve, L. L., Gibbs, R. A., Belmont, J. W., … Stewart, J. (2007). Genome-wide detection and characterization of positive selection in human populations. Nature,449(7164), 913–918. https://doi.org/10.1038/nature06250.
Szpiech ZA, Hernandez RD. Selscan: an efficient multithreaded program to perform EHH-based scans for positive selection. Mol Biol Evol. 2014;31(10):2824–7. https://doi.org/10.1093/molbev/msu211.
Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4(3):e72. https://doi.org/10.1371/journal.pbio.0040072.
Fischer MC, Rellstab C, Leuzinger M, Roumet M, Gugerli F, Shimizu KK, Holderegger R, Widmer A. Estimating genomic diversity and population differentiation - an empirical comparison of microsatellite and SNP variation in Arabidopsis halleri. BMC Genomics. 2017;18(1):69. https://doi.org/10.1186/s12864-016-3459-7.
Wei C, Wang H, Liu G, Wu M, Cao J, Liu Z, Liu R, Zhao F, Zhang L, Lu J, Liu C, Du L. Genome-wide analysis reveals population structure and selection in Chinese indigenous sheep breeds. BMC Genomics. 2015;16(1):194. https://doi.org/10.1186/s12864-015-1384-9.
BECKWITH CI. Empires of the Silk Road: a history of Central Eurasia from the bronze age to the Present. Princeton University Press; 2009. http://www.jstor.org/stable/j.ctt7sq0.
Lv, F. H., Cao, Y. H., Liu, G. J., Luo, L. Y., Lu, R., Liu, M. J., Li, W. R., Zhou,P., Wang, X. H., Shen, M., Gao, L., Yang, J. Q., Yang, H., Yang, Y. L., Liu, C. B.,Wan, P. C., Zhang, Y. S., Pi, W. H., Ren, Y. L., Shen, Z. Q., … Li, M. H. Whole-Genome Resequencing of Worldwide Wild and Domestic Sheep Elucidates Genetic Diversity, Introgression, and Agronomically Important Loci. Molecular biology and evolution. 2022;39(2):msab353. https://doi.org/10.1093/molbev/msab353.
Chen ZH, Xu YX, Xie XL, Wang DF, Aguilar-Gómez D, Liu GJ, Li X, Esmailizadeh A, Rezaei V, Kantanen J, Ammosov I, Nosrati M, Periasamy K, Coltman DW, Lenstra JA, Nielsen R, Li MH. Whole-genome sequence analysis unveils different origins of European and Asiatic Mouflon and domestication-related genes in sheep. Commun Biology. 2021;4(1):1307. https://doi.org/10.1038/s42003-021-02817-4.
Kominakis A, Tarsani E, Hager-Theodorides AL, Mastranestasis I, Hadjigeorgiou I. Clustering patterns mirror the geographical distribution and genetic history of Lemnos and Lesvos sheep populations. PLoS ONE. 2021;16(3):e0247787. https://doi.org/10.1371/journal.pone.0247787.
Ligda C, Altarayrah J, Georgoudis A. Genetic analysis of Greek sheep breeds using microsatellite markers for setting conservation priorities. Small Rumin Res Elsevier. 2009;83:42–8. https://doi.org/10.1016/j.smallrumres.2009.04.002.
Galbraith H. Fundamental hair follicle biology and fine fibre production in animals. Animal: Int J Anim Bioscience. 2010;4(9):1490–509. https://doi.org/10.1017/S175173111000025X.
Lv X, Gao W, Jin C, Wang L, Wang Y, Chen W, Zou S, Huang S, Li Z, Wang J, Sun W. Preliminary study on microR-148a and microR-10a in dermal papilla cells of Hu Sheep. BMC Genet. 2019;20(1):70. https://doi.org/10.1186/s12863-019-0770-8.
Li Y, Lv X, Wang S, Cao X, Yuan Z, Getachew T, Mwacharo JM, Haile A, Sun W. BMP7 functions to regulate proliferation of dermal papilla cells in Hu Sheep. Genes. 2022;13(2):201. https://doi.org/10.3390/genes13020201.
He J, Zhao B, Huang X, Fu X, Liu G, Tian Y, Wu C, Mao J, Liu J, Gun S, Tian K. Gene network analysis reveals candidate genes related with the hair follicle development in sheep. BMC Genomics. 2022;23(1):428. https://doi.org/10.1186/s12864-022-08552-2.
Arzik Y, Kizilaslan M, Behrem S, White SN, Piel LMW, Cinar MU. Genome-wide scan of wool production traits in Akkaraman Sheep. Genes. 2023;14(3):713. https://doi.org/10.3390/genes14030713.
Jin M, Lu J, Fei X, Lu Z, Quan K, Liu Y, Chu M, Di R, Wang H, Wei C. (2020). Genetic Signatures of Selection for Cashmere Traits in Chinese Goats. Animals: an open access journal from MDPI, 10(10), 1905. https://doi.org/10.3390/ani10101905.
Sun H, He Z, Xi Q, Zhao F, Hu J, Wang J, Liu X, Zhao Z, Li M, Luo Y, Li S. Lef1 and Dlx3 may facilitate the maturation of secondary hair follicles in the skin of Gansu Alpine Merino. Genes. 2022;13(8):1326. https://doi.org/10.3390/genes13081326.
Clavel C, Grisanti L, Zemla R, Rezza A, Barros R, Sennett R, Mazloom AR, Chung CY, Cai X, Cai CL, Pevny L, Nicolis S, Ma’ayan A, Rendl M. Sox2 in the dermal papilla niche controls hair growth by fine-tuning BMP signaling in differentiating hair shaft progenitors. Dev Cell. 2012;23(5):981–94. https://doi.org/10.1016/j.devcel.2012.10.013.
Langbein L, Rogers MA, Praetzel-Wunder S, Helmke B, Schirmacher P, Schweizer J. K25 (K25irs1), K26 (K25irs2), K27 (K25irs3), and K28 (K25irs4) represent the type I inner root sheath keratins of the human hair follicle. J Invest Dermatol. 2006;126(11):2377–86. https://doi.org/10.1038/sj.jid.5700494.
Bawden CS, McLaughlan C, Nesci A, Rogers G. A unique type I keratin intermediate filament gene family is abundantly expressed in the inner root sheaths of sheep and human hair follicles. J Invest Dermatol. 2001;116(1):157–66. https://doi.org/10.1046/j.1523-1747.2001.00215.x.
Liu Y, Zhang J, Xu Q, Kang X, Wang K, Wu K, Fang M. Integrated miRNA-mRNA analysis reveals regulatory pathways underlying the curly fleece trait in Chinese tan sheep. BMC Genomics. 2018;19(1):360. https://doi.org/10.1186/s12864-018-4736-4.
Cadieu E, Neff MW, Quignon P, Walsh K, Chase K, Parker HG, Vonholdt BM, Rhue A, Boyko A, Byers A, Wong A, Mosher DS, Elkahloun AG, Spady TC, André C, Lark KG, Cargill M, Bustamante CD, Wayne RK, Ostrander EA. Coat variation in the domestic dog is governed by variants in three genes. Volume 326. New York, N.Y.): Science; 2009. pp. 150–3. 5949https://doi.org/10.1126/science.1177808.
Bauer A, Hadji Rasouliha S, Brunner MT, Jagannathan V, Bucher I, Bannoehr J, Varjonen K, Bond R, Bergvall K, Welle MM, Roosje P, Leeb T. A second KRT71 allele in curly coated dogs. Anim Genet. 2019;50(1):97–100. https://doi.org/10.1111/age.12743.
Gandolfi B, Alhaddad H, Joslin SE, Khan R, Filler S, Brem G, Lyons LA. (2013). A splice variant in KRT71 is associated with curly coat phenotype of Selkirk Rex cats. Scientific reports, 3, 2000. https://doi.org/10.1038/srep02000.
Liu Y, Ding Y, Liu Z, Chen Q, Li X, Xue X, Pu Y, Ma Y, Zhao Q. Integration Analysis of Transcriptome and Proteome reveal the mechanisms of Goat wool bending. Front cell Dev Biology. 2022;10:836913. https://doi.org/10.3389/fcell.2022.836913.
Gong G, Fan Y, Li W, Yan X, Yan X, Zhang L, Wang N, Chen O, Zhang Y, Wang R, Liu Z, Jiang W, Li J, Wang Z, Lv Q, Su R. Identification of the key genes Associated with different hair types in the Inner Mongolia Cashmere Goat. Animals: Open Access J MDPI. 2022;12(11):1456. https://doi.org/10.3390/ani12111456.
Wang Z, Zhang H, Yang H, Wang S, Rong E, Pei W, Li H, Wang N. Genome-wide association study for wool production traits in a Chinese Merino sheep population. PLoS ONE. 2014;9(9):e107101. https://doi.org/10.1371/journal.pone.0107101.
Manunza A, Diaz JR, Sayre BL, Cozzi P, Bobbo T, Deniskova T, Dotsev A, Zinovieva N, Stella A. Discovering novel clues of natural selection on four worldwide goat breeds. Sci Rep. 2023;13(1):2110. https://doi.org/10.1038/s41598-023-27490-x.
Mwacharo JM, Kim ES, Elbeltagy AR, Aboul-Naga AM, Rischkowsky BA, Rothschild MF. Genomic footprints of dryland stress adaptation in Egyptian fat-tail sheep and their divergence from East African and western Asia cohorts. Sci Rep. 2017;7(1):17647. https://doi.org/10.1038/s41598-017-17775-3.
Zhao B, Luo H, Huang X, Wei C, Di J, Tian Y, Fu X, Li B, Liu GE, Fang L, Zhang S, Tian K. Integration of a single-step genome-wide association study with a multi-tissue transcriptome analysis provides novel insights into the genetic basis of wool and weight traits in sheep. Genet Selection Evolution: GSE. 2021;53(1):56. https://doi.org/10.1186/s12711-021-00649-8.
Zoccolillo M, Moia C, Comincini S, Cittaro D, Lazarevic D, Pisani KA, Wit JM, Bozzola M. Identification of novel genetic variants associated with short stature in a Baka pygmies population. Hum Genet. 2020;139(11):1471–83. https://doi.org/10.1007/s00439-020-02191-x.
Palygin O, Levchenko V, Ilatovskaya DV, Pavlov TS, Pochynyuk OM, Jacob HJ, Geurts AM, Hodges MR, Staruschenko A. Essential role of Kir5.1 channels in renal salt handling and blood pressure control. JCI Insight. 2017;2(18):e92331. https://doi.org/10.1172/jci.insight.92331.
Shelton EL, Ector G, Galindo CL, Hooper CW, Brown N, Wilkerson I, Pfaltzgraff ER, Paria BC, Cotton RB, Stoller JZ, Reese J. Transcriptional profiling reveals ductus arteriosus-specific genes that regulate vascular tone. Physiol Genom. 2014;46(13):457–66. https://doi.org/10.1152/physiolgenomics.00171.2013.
Lawson ND, Mugford JW, Diamond BA, Weinstein BM. Phospholipase C gamma-1 is required downstream of vascular endothelial growth factor during arterial development. Genes Dev. 2003;17(11):1346–51. https://doi.org/10.1101/gad.1072203.
Siddam AD, Gautier-Courteille C, Perez-Campos L, Anand D, Kakrana A, Dang CA, Legagneux V, Méreau A, Viet J, Gross JM, Paillard L, Lachke SA. The RNA-binding protein Celf1 post-transcriptionally regulates p27Kip1 and Dnase2b to control fiber cell nuclear degradation in lens development. PLoS Genet. 2018;14(3):e1007278. https://doi.org/10.1371/journal.pgen.1007278.
Wu J, Lin Z, Chen G, Luo Q, Nie Q, Zhang X, Luo W. Characterization of Chicken skin yellowness and exploration of genes involved in skin yellowness deposition in Chicken. Front Physiol. 2021;12:585089. https://doi.org/10.3389/fphys.2021.585089.
Lu N, Liu J, Xu M, Liang J, Wang Y, Wu Z, Xing Y, Diao F. CSMD3 is Associated with Tumor Mutation Burden and Immune Infiltration in Ovarian Cancer patients. Int J Gen Med. 2021;14:7647–57. https://doi.org/10.2147/IJGM.S335592.
Estrada-Reyes ZM, Rae O, Postley C, Jiménez Medrano MB, Gutiérrez L, J. D., Mateescu RG. Association study reveals Th17, Treg, and Th2 loci related to resistance to Haemonchus Contortus in Florida native sheep1. J Anim Sci. 2019;97(11):4428–44. https://doi.org/10.1093/jas/skz299.
Caldwell JM, Collins MH, Kemme KA, Sherrill JD, Wen T, Rochman M, Stucke EM, Amin L, Tai H, Putnam PE, Jiménez-Dalmaroni MJ, Wormald MR, Porollo A, Abonia JP, Rothenberg ME. Cadherin 26 is an alpha integrin-binding epithelial receptor regulated during allergic inflammation. Mucosal Immunol. 2017;10(5):1190–201. https://doi.org/10.1038/mi.2016.120.
Li J, Liu J, Campanile G, Plastow G, Zhang C, Wang Z, Cassandro M, Gasparrini B, Salzano A, Hua G, Liang A, Yang L. Novel insights into the genetic basis of buffalo reproductive performance. BMC Genomics. 2018;19(1):814. https://doi.org/10.1186/s12864-018-5208-6.
Du X, Yin H, Pan Z, Wu W, Shang P, Chamba Y, Li Q. BMP7 is a candidate gene for reproductive traits in Yorkshire sows. Anim Reprod Sci. 2020;221:106598. https://doi.org/10.1016/j.anireprosci.2020.106598.
Li X, Ye J, Han X, Qiao R, Li X, Lv G, Wang K. Whole-genome sequencing identifies potential candidate genes for reproductive traits in pigs. Genomics. 2020;112(1):199–206. https://doi.org/10.1016/j.ygeno.2019.01.014.
Herpin A, Englberger E, Zehner M, Wacker R, Gessler M, Schartl M. Defective autophagy through epg5 mutation results in failure to reduce germ plasm and mitochondria. FASEB Journal: Official Publication Federation Am Soc Experimental Biology. 2015;29(10):4145–61. https://doi.org/10.1096/fj.14-265462.
Esmaeili-Fard SM, Gholizadeh M, Hafezian SH, Abdollahi-Arpanahi R. Genes and pathways affecting Sheep Productivity traits: genetic parameters, Genome-Wide Association Mapping, and Pathway Enrichment Analysis. Front Genet. 2021;12:710613. https://doi.org/10.3389/fgene.2021.710613.
Boulanger G, Cibois M, Viet J, Fostier A, Deschamps S, Pastezeur S, Massart C, Gschloessl B, Gautier-Courteille C, Paillard L. Hypogonadism Associated with Cyp19a1 (Aromatase) posttranscriptional upregulation in Celf1 knockout mice. Mol Cell Biol. 2015;35(18):3244–53. https://doi.org/10.1128/MCB.00074-15.
E GX, Duan XH, Zhang JH, Huang YF, Zhao YJ, Na RS, Zhao ZQ, Ma YH, Chu MX, Basang WD, Zhu YB, An TW, Luo XL, Han YG, Zeng Y. Genome-wide selection signatures analysis of litter size in Dazu black goats using single-nucleotide polymorphism. 3 Biotech. 2019;9(9):336. https://doi.org/10.1007/s13205-019-1869-3.
Zhang J, Xu E, Ren C, Yan W, Zhang M, Chen M, Cardiff RD, Imai DM, Wisner E, Chen X. Mice deficient in Rbm38, a target of the p53 family, are susceptible to accelerated aging and spontaneous tumors. Proc Natl Acad Sci USA. 2014;111(52):18637–42. https://doi.org/10.1073/pnas.1415607112.
Hu DB, Li ZS, Ali I, Xu LJ, Fang NZ. Effect of potential role of p53 on embryo development arrest induced by H2O2 in mouse. In vitro cellular & developmental biology. Animal. 2017;53(4):344–53. https://doi.org/10.1007/s11626-016-0122-1.
Liang K, Yao L, Wang S, Zheng L, Qian Z, Ge Y, Chen L, Cheng X, Ma R, Li C, Jing J, Yang Y, Yu W, Xue T, Chen Q, Cao S, Ma J, Yao B. miR-125a-5p increases cellular DNA damage of aging males and perturbs stage-specific embryo development via Rbm38-p53 signaling. Aging Cell. 2021;20(12):e13508. https://doi.org/10.1111/acel.13508.
Wang H, Parra M, Conboy JG, Hillyer CD, Mohandas N, An X. Selective effects of protein 4.1 N deficiency on neuroendocrine and reproductive systems. Sci Rep. 2020;10(1):16947. https://doi.org/10.1038/s41598-020-73795-6.
Yang, J., Li, W. R., Lv, F. H., He, S. G., Tian, S. L., Peng, W. F., Sun, Y. W., Zhao,Y. X., Tu, X. L., Zhang, M., Xie, X. L., Wang, Y. T., Li, J. Q., Liu, Y. G., Shen,Z. Q., Wang, F., Liu, G. J., Lu, H. F., Kantanen, J., Han, J. L., … Liu, M. J. (2016).Whole-Genome Sequencing of Native Sheep Provides Insights into Rapid Adaptations to Extreme Environments. Molecular biology and evolution, 33(10), 2576–2592. https://doi.org/10.1093/molbev/msw129.
Bai, L., Liu, B., Ji, C., Zhao, S., Liu, S., Wang, R., Wang, W., Yao, P., Li, X.,Fu, X., Yu, H., Liu, M., Han, F., Guan, N., Liu, H., Liu, D., Tao, Y., Wang, Z., Yan,S., Florant, G., … Enqi Liu (2019). Hypoxic and Cold Adaptation Insights from the Himalayan Marmot Genome. iScience, 11, 519–530. https://doi.org/10.1016/j.isci.2018.11.034.
Mastrototaro G, Zaghi M, Massimino L, Moneta M, Mohammadi N, Banfi F, Bellini E, Indrigo M, Fagnocchi G, Bagliani A, Taverna S, Rohm M, Herzig S, Sessa A. TBL1XR1 ensures balanced neural development through NCOR complex-mediated regulation of the MAPK pathway. Front cell Dev Biology. 2021;9:641410. https://doi.org/10.3389/fcell.2021.641410.
Reyes-Nava NG, Yu HC, Coughlin CR 2nd, Shaikh TH, Quintana AM. Abnormal expression of GABAA receptor subunits and hypomotility upon loss of gabra1 in zebrafish. Biology open. 2020;9(4):bio051367. https://doi.org/10.1242/bio.051367.
Zhang T, Yang Y, Sima X. No association of GABRA1 rs2279020 and GABRA6 rs3219151 polymorphisms with risk of epilepsy and antiepileptic drug responsiveness in Asian and arabic populations: evidence from a meta-analysis with trial sequential analysis. Front Neurol. 2022;13:996631. https://doi.org/10.3389/fneur.2022.996631.
Grigorenko AP, Protasova MS, Lisenkova AA, Reshetov DA, Andreeva TV, Garcias GL, Roth M, Papassotiropoulos MDG, A., Rogaev EI. Neurodevelopmental syndrome with intellectual disability, Speech Impairment, and Quadrupedia Is Associated with glutamate receptor Delta 2 gene defect. Cells. 2022;11(3):400. https://doi.org/10.3390/cells11030400.
Keele, G. R., Prokop, J. W., He, H., Holl, K., Littrell, J., Deal, A., Francic, S.,Cui, L., Gatti, D. M., Broman, K. W., Tschannen, M., Tsaih, S. W., Zagloul, M., Kim,Y., Baur, B., Fox, J., Robinson, M., Levy, S., Flister, M. J., Mott, R., … Solberg Woods, L. C. (2018). Genetic Fine-Mapping and Identification of Candidate Genes and Variants for Adiposity Traits in Outbred Rats. Obesity (Silver Spring, Md.), 26(1),213–222. https://doi.org/10.1002/oby.22075.
Giudice J, Xia Z, Li W, Cooper TA. Neonatal cardiac dysfunction and transcriptome changes caused by the absence of Celf1. Sci Rep. 2016;6:35550. https://doi.org/10.1038/srep35550.
Blech-Hermoni Y, Sullivan CB, Jenkins MW, Wessely O, Ladd AN. CUG-BP, elav-like family member 1 (CELF1) is required for normal myofibrillogenesis, morphogenesis, and contractile function in the embryonic heart. Dev Dynamics: Official Publication Am Association Anatomists. 2016;245(8):854–73. https://doi.org/10.1002/dvdy.24413.
Davey KM, Parboosingh JS, McLeod DR, Chan A, Casey R, Ferreira P, Snyder FF, Bridge PJ, Bernier FP. Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition. J Med Genet. 2006;43(5):385–93. https://doi.org/10.1136/jmg.2005.036657.
Wachoski-Dark E, Zhao T, Khan A, Shutt TE, Greenway SC. Mitochondrial protein homeostasis and Cardiomyopathy. Int J Mol Sci. 2022;23(6):3353. https://doi.org/10.3390/ijms23063353.
Zhao C, Chen X, Wu W, Wang W, Pang W, Yang G. MAT2B promotes adipogenesis by modulating SAMe levels and activating AKT/ERK pathway during porcine intramuscular preadipocyte differentiation. Exp Cell Res. 2016;344(1):11–21. https://doi.org/10.1016/j.yexcr.2016.02.019.
Maglott D, Ostell J, Pruitt KD, Tatusova T. (2011). Entrez Gene: gene-centered information at NCBI. Nucleic acids research, 39(Database issue), D52–D57. https://doi.org/10.1093/nar/gkq1237.
Kerr AG, Sinha I, Dadvar S, Arner P, Dahlman I. Epigenetic regulation of diabetogenic adipose morphology. Mol Metabolism. 2019;25:159–67. https://doi.org/10.1016/j.molmet.2019.04.009.
Acknowledgements
We would like to thank everyone who contributed towards the article.
Funding
This study was funded by grants from the Natural Science Foundation of China (NO:32060743), Bintuan Science and Technology Program (NO: 2022CB001-09), Autonomous region rural revitalization industry development science and technology action(2022NC110), and Autonomous Region Agricultural Area High-efficiency Mutton Sheep Breeding and Promotion Technology System Project (NO: xjnqry-g-2310).
Author information
Authors and Affiliations
Contributions
Z.-P.H. and S.-D.L. conceived and directed this study. Z.-P.H. analyzed the data, with contributions from R.-Z.R. and W.Z. L.-L.Z. and J.-R.W. provided the samples or assisted during sample collection. C.-J.L. provided useful feedback and advice. Z.-P.H. and S.-D.L. wrote the paper. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This work was conducted in accordance with the standards set by the Ethics Committee of the College of Animal Science and Technology of Tarim University (SYXK2020-009) and approved by the Ethics Committee of the College of Animal Science and Technology of Tarim University. Written informed consent was obtained from the owners for the participation of their animals in this study.
Consent for publication
Not applicable.
Benefit sharing
The authors declare no benefit sharing.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Han, Zp., Yang, Rz., Zhou, W. et al. Population structure and selection signal analysis of indigenous sheep from the southern edge of the Taklamakan Desert. BMC Genomics 25, 681 (2024). https://doi.org/10.1186/s12864-024-10581-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10581-y