
Abstract
Background
Mycobacterium avium complex (MAC), including Mycobacterium intracellulare is a member of slow-growing mycobacteria and contributes to a substantial proportion of nontuberculous mycobacterial lung disease in humans affecting immunocompromised and elderly populations. Adaptation of pathogens in hostile environments is crucial in establishing infection and persistence within the host. However, the sophisticated cellular and molecular mechanisms of stress response in M. intracellulare still need to be fully explored. We aimed to elucidate the transcriptional response of M. intracellulare under acidic and oxidative stress conditions.
Results
At the transcriptome level, 80 genes were shown [FC] ≥ 2.0 and p < 0.05 under oxidative stress with 10 mM hydrogen peroxide. Specifically, 77 genes were upregulated, while 3 genes were downregulated. In functional analysis, oxidative stress conditions activate DNA replication, nucleotide excision repair, mismatch repair, homologous recombination, and tuberculosis pathways. Additionally, our results demonstrate that DNA replication and repair system genes, such as dnaB, dinG, urvB, uvrD2, and recA, are indispensable for resistance to oxidative stress. On the contrary, 878 genes were shown [FC] ≥ 2.0 and p < 0.05 under acidic stress with pH 4.5. Among these genes, 339 were upregulated, while 539 were downregulated. Functional analysis highlighted nitrogen and sulfur metabolism pathways as the primary responses to acidic stress. Our findings provide evidence of the critical role played by nitrogen and sulfur metabolism genes in the response to acidic stress, including narGHIJ, nirBD, narU, narK3, cysND, cysC, cysH, ferredoxin 1 and 2, and formate dehydrogenase.
Conclusion
Our results suggest the activation of several pathways potentially critical for the survival of M. intracellulare under a hostile microenvironment within the host. This study indicates the importance of stress responses in M. intracellulare infection and identifies promising therapeutic targets.
Similar content being viewed by others
Background
Non-tuberculous mycobacteria (NTM) are environmental mycobacteria other than M. tuberculosis complex and M. leprae, comprising more than 180 species [1]. Most NTM are non-pathogenic to humans, but some species cause infection in patients with immunocompromised conditions and structural lung disease [2,3,4]. The incidence and prevalence of NTM infections have steadily increased over the last several decades and emerged as a significant global public health concern [5,6,7,8]. Mycobacterium avium complex (MAC), including Mycobacterium avium and Mycobacterium intracellulare, is the most common causative agent of NTM-lung disease in the world and affects susceptible populations with certain risk factors such as bronchiectasis, chronic obstructive pulmonary disease, autoimmune disease, and aging [9].
Mycobacteria can adapt to stress conditions, including oxidative stress, nutrient starvation, pH change, and temperature, which elucidate its evolutional adaptation by a refined network of molecular mechanisms [10,11,12,13]. The production of reactive oxygen species (ROS) and the acidification of phagosomes are the critical mechanisms that macrophages employ to kill internalized pathogens during infections [14,15,16,17]. Consequently, mycobacteria have evolved defense mechanisms to protect themselves against host-induced stress [18,19,20]. Under oxidative stress conditions, two global transcription factors, OxyR and SoxRS, regulate the stress response in many bacterial species [21]. OxyR responds to peroxide stress, while SoxRS responds to superoxide stress [14]. However, pathogenic mycobacteria such as M. tuberculosis and M. leprae lack the general antioxidant mechanisms employed by most other intracellular bacterial pathogens [22]. Previous studies have reported that pathogenic mycobacteria exhibit multiple mutations within the oxyR gene, resulting in a dysfunctional protein [22]. Although M. tuberculosis lacks the functional oxyR gene, several other genes associated with the oxidative stress response, such as ahpC, katG, and furA, remain functional [23]. The presence of alternative detoxification pathways has the potential to act as a compensatory mechanism.
For example, Lu et al. investigated the proteomic profiling of Mycobacterium tuberculosis (Mtb) under oxidative stress, focusing on total cysteine thiols modification and S-sulfenylation modification [10]. They identified the differential expression of numerous proteins at total cysteine modification and S-sulfenylation modification levels under hydrogen peroxide exposure [10]. These cysteine-modified proteins were associated with oxidation–reduction, fatty acid synthesis, cell wall remodeling, and protein repair [10]. Recently, Yimcharoen et al. investigated the transcriptional response of drug susceptible and resistant Mtb strains after isoniazid exposure under stress conditions that mimic the host environment [13]. The expression of stress-response genes such as hspX, tgs1, icl1, and sigE and lipoarabinomannan (LAM) synthesis associated genes including pimB, mptA, mptC, dprE1, dprE2, and embC were highly differentiated between the drug-susceptible and resistant strains [13]. These findings suggest the role of stress response and LAM synthesis-associated genes may be pivotal for the adaptation and persistence of Mtb within the host. Similarly, Martini et al. showed that small non-coding RNA MTS1338 promoted distinct expression profiles for stress response in Mtb under macrophage-like stress conditions, suggesting stress-triggered small non-coding RNA enhances bacterial survival within the host by inducing global transcriptional changes [11].
Despite contradictory results, exposure to NTM from the environmental niche, such as soil and water, has been proposed as a source of infection. Tzou et al. demonstrated that the isolation rate of NTM from shower aerosols is higher in the NTM patient's homes than in control homes based on Washington and Oregon residents compared with age, sex, and geography-matched controls [24]. Furthermore, Reed et al. revealed that prolonged soil exposure is associated with M. avium complex exposure in a cross-sectional study in Florida by population-based random household survey [25]. Similarly, clinically dominant NTM species are present in patient’s potting soil [26]. Interestingly, NTM isolates from potting soil and patients showed similar restriction enzyme digestion patterns by PFGE [26]. On the contrary, Choi et al. reported that NTM species recovered from patients did not match in the showerheads [27]. NTM isolates from the environment and patients share similar characteristics, and elucidating the stress response of NTM isolates recovered from environmental sources may reveal the survival strategy within the host for successful infection. Therefore, we performed comprehensive transcriptional profiling of Mycobacterium intracellulare isolate recovered from soil exposed to oxidative and acidic stress conditions. We also identified differential gene groups and pathways in response to stress conditions. Our findings provide a better understanding of the molecular mechanisms for adaptation to the host-induced stress conditions in Mycobacterium intracellulare and possibly other MAC species.
Material and methods
Bacterial strains and growth conditions
Mycobacteria intracellulare S1-36 strain, that isolated from soil samples of South Korea in 2019 was used in this study. M. intracellulare S1-36 strain harbors 5.4 Mbp genome and showed similar genetic feature with M. intracellulare ATCC13950 strain through the whole genome sequencing analysis [28]. M. intracellulare S1-36 strain were cultured in Middlebrook 7H9 broth (MB7H9; Becton, Dickinson and Company) supplemented with 0.5% glycerol and 10% ODAC at 37 ℃ and 250 rpm.
In vitro stress conditions
The M. intracellulare S1-36 strain was inoculated onto 7H10 agar (Becton, Dickinson and Company), which was supplemented with 10% OADC, and then incubated at 37 °C for 3 weeks. A single colony of M. intracellulare was subsequently transferred to 10 mL of MB7H9 medium and incubated at 37 °C with shaking at 250 rpm until it reached an OD600 of 0.5. The mid-log phase M. intracellulare cultures were washed with 1 × PBS for 3 times and then transferred into 10 mL of Sauton’s media with either a final concentration of 10 mM hydrogen peroxide for oxidative stress or acidic Sauton’s media (pH 4.5) for acidic stress. Samples were incubated at 37 °C with shaking at 250 rpm for 16 h. Untreated M. intracellulare cultures were used as a control. All samples were prepared in biologically independent three biological replicates.
RNA extraction
We extracted total RNA from a culture of M. intracellulare using the RNeasy Plus Kits (Qiagen), according to the manufacturer's instructions with slight adjustments. In summary, following 16 h of exposure to stress conditions, the samples were centrifuged at 4000 rpm at 20 °C for 20 min. Subsequently, we subjected them to three washes with 1 × PBS and then resuspended them in 2 mL of RNAprotect Bacteria Reagent from Qiagen. To stabilize the transcriptional profile, we incubated the samples at room temperature for 5 min. Afterward, we centrifuged the samples at 4000 rpm at 20 °C for 20 min and resuspended them in 1 mL of RLT buffer containing 300 μL of 0.1 mm zirconia beads (BioSpec). The samples, inclusive of mycobacterial cells, underwent lysis at 4500 rpm for 45 s, repeated three times using the Precellys 24 homogenizer (Bertin Technologies). Following lysis, we centrifuged the samples at 13,000 rpm for 5 min, and subsequently transferred 700 μL of the supernatant to a 2 mL tube. We proceeded with the remaining RNA extraction steps as per the established protocol.
Library preparation and sequencing
The purified RNA samples were dispatched to Macrogen (Seoul, South Korea). We assessed the integrity of all RNA samples using an Agilent 2100 Bioanalyzer (Agilent Technologies, Waldbronn, Germany). Notably, all RNA samples displayed RNA integrity numbers greater than or equal to 8.6. We performed rRNA removal using the NEBNext rRNA Depletion kit and constructed sequencing libraries using the TruSeq Stranded Total RNA Library Prep Gold Kit by Illumina. The generated libraries were subjected to gel purification and were validated by evaluating size, purity, and concentration using the Agilent Bioanalyzer. For library quantification, we followed the qPCR Quantification Protocol Guide utilizing KAPA Library Quantification kits for Illumina Sequencing platforms. Subsequently, we conducted sequencing on an Illumina NextSeq, generating paired-end reads (2 × 101 bp). Image decomposition and quality value calculations were carried out using the modules within the Illumina pipeline. Macrogen conducted all procedures related to next-generation sequencing analysis.
Sequencing data analysis
FastQC was employed to assess the quality of sequences, and only sequences with a Phred quality score ≥ 30 were retained for further analysis. The sequencing reads were then aligned to the reference genome M. intracellulare ATCC13950 [29] using the Bowtie aligner. Following read mapping, read counts for each gene in each sample were extracted based on the gene annotations specific to the corresponding species using the HTSeq program. Differential expression analysis was carried out using DESeq, applying the criteria of |fold change|≥ 2 and a raw p-value from the nbinomWaldTest < 0.05.
Pathway enrichment analysis
We conducted pathway enrichment analysis, which identifies biologically relevant pathways by assessing the overlap between genes of interest and specific gene sets or pathways from the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database [30]. We employed Fisher’s exact test to identify statistically significant KEGG pathways, and corrected p-values were calculated using the Benjamini and Hochberg false discovery rate algorithm.
RNA-seq data validation
To validate the RNA-seq data, we selected nine differentially expressed genes (DEGs) for quantitative real-time PCR (qRT-PCR). The synthesis of cDNA was performed following the manufacturer’s instructions. In brief, 16 µl of samples containing 500 ng of RNA were incubated at 70 ℃ for 5 min to dissociate secondary structure within the RNA sample and chill the tube immediately on ice. Subsequently, we added 4 µl of reverse transcription master mix (ELPIS biotech, Korea) and incubate for 60 min at 37 ℃. Finally, the samples were incubated for 5 min at 94 ℃ to stop reaction. The cDNA samples were diluted for 1:10 with nuclease free water and used for further analysis. We performed qRT-PCR using a SsoFast Eva Green Supermix (Bio-Rad) and Rotor-Gene Q real-time PCR cycler (Qiagen). Amplification was performed for 40 cycles at 95 °C for 10 s, followed by 30 s at 62 °C with fluorescence detected during the extension step. The relative gene expression level was calculated by the 2−ΔΔCt method using beta subunit of RNA polymerase (rpoB) as a reference gene. The sequences of primers for qPCR experiment used in present study are listed in Table 1.
Results and discussion
In the present study, we investigated the transcriptomic changes of environmental M. intracellulare isolate under acidic and oxidative stress conditions that mimic the stress induced by the host environment. Previous studies have investigated pathogenic mycobacteria's transcriptional and proteomic profiles under various stressors, such as hypoxia, nutrient deprivation, changes in pH, and oxidative stress [31,32,33,34,35,36,37,38,39]. However, these prior investigations predominantly centered on M. tuberculosis rather than the MAC. Consequently, our understanding of the transcriptome of MAC, particularly in the context of M. intracellulare under host-induced stress conditions, still needs to be improved. This study represents the first effort to elucidate the specific transcripts expressed by M. intracellulare when exposed to acidic and oxidative stress conditions.
Macrophages are the first-line defense in the host immune response against intracellular bacteria, including M. tuberculosis and MAC [40, 41]. Once encountered, macrophages recognize and engulf bacteria via interaction between pathogen-associated molecular patterns and pattern recognition receptors [42]. Upon phagocytosis, various antimicrobial mechanisms are activated within the macrophages. The maturation of phagosomes into phagolysosomes induces its acidification modulated by the Abl tyrosine kinase and vacuolar-type ATPase [43]. This process is required to activate degradative enzymes, including hydrolase and cathepsins, showing optimal activity at low pH. [43]. Also, activated macrophages produce various antimicrobial effector molecules, such as antimicrobial peptides, lipid mediators, and oxygen and nitrogen radicals, to sterilize engulfed pathogens [44]. However, pathogenic mycobacteria can endure host-induced stress condition and survive within the macrophages by arresting the maturation of phagosome and lysosomal fusion [44].
Summary of RNA-sequencing data
The RNA samples' concentration ranged from 43.3 to 102.3 ng/µl. Also, all RNA samples showed an RNA integrity number score of ≥ 8.6 and rRNA ratio of > 1.0. RNA-seq library preparation was conducted with extracted total RNA from M. intracellulare under oxidative and acidic stress conditions. RNA-seq produced 32.3 to 36.8 million total reads per library. FastQC analyzed the quality of sequences, and sequences with a phred quality score ≥ 30 were used for further analysis. After eliminating the reads with low-quality bases and adapter sequences by the Trimmomatic program, the remaining sequences were 31.6 to 36.1 million reads per library (Table 2). Bowtie aligner mapped trimmed readds with reference genome M. intracellulare ATCC13950 (ASM27712v1). The processed reads ranged from 31.5 to 36 million reads per library and the rate of mapped reads with its reference genome ranged from 86.1 to 88.1% (Table 2). Collectively, a high-quality cDNA library was constructed and used for further RNA-seq data analysis.
Multidimensional scaling
Multidimensional scaling analysis plot showed the variability of the samples between control, acidic stress, and oxidative stress groups (Fig. 1A). The results revealed a distinct clustering of the three biological replicates of RNA samples belonging to each group. These data suggest that all the variations caused by biological replicates have been normalized, and each group was separately clustered.

Transcriptional profiling of M. intracellulare under oxidative and acidic stress conditions. The transcript data samples were analyzed from M. intracellulare cultures consisting of three groups as follows: Con (control): M. intracellulare cultures in pH 7.0 Sauton’s media, O (oxidative stress): M. intracellulare cultures in pH 7.0 Sauton’s media treated with 10 mM hydrogen oxide, A (acidic stress): M. intracellulare cultures in pH 4.5 Sauton’s media. Three biological replicates were used for analysis. A Multidimensional scaling plots of samples from transcript data samples. Three biological replicates have similar expression patterns in all experimental groups. B Cluster heatmap of differentially expressed genes (|fold change|≥ 2 and raw p < 0.05) in nine transcript data samples. C Numbers of differentially expressed genes in RNA-seq. The differentially expressed genes were defined as |fold change|≥ 2 and raw p < 0.05. D Venn diagram showing the shared genes that were significantly up- or down-regulated in M. intracellulare culture under oxidative and acidic stress conditions. Filter was set at |fold change|≥ 2 and raw p < 0.05
Differentially expressed genes (DEGs) in M. intracellulare under acidic and oxidative stress
We conducted transcriptomic analysis under oxidative and acidic stress conditions. The DESeq2 analysis showed that 998 differentially expressed genes (DEGs) were significantly expressed in M. intracellulare exposed to acidic and oxidative stress conditions compared to the control group, filtered by |fold change|≥ 2.0 and raw p-value < 0.05 (Fig. 1B). 878 DEGs were observed in the acidic stress group. Among them, 339 DEGs were significantly upregulated, while 539 DEGs were significantly downregulated (Fig. 1C). On the contrary, relatively small numbers of DEGs were observed in the oxidative stress group. In total, 80 DEGs were expressed in the oxidative stress group compared to the control group (Fig. 1C). Among them, 77 DEGs were significantly upregulated, while three DEGs were significantly downregulated in the oxidative stress group compared to the control group (Fig. 1C).
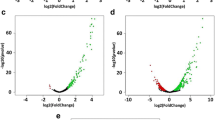
A Venn diagram showed 48 DEGs were overlapped between two groups (Fig. 1D). Among them, 46 DEGs were upregulated, while two DEGs were downregulated (Fig. 1D). Also, 293 and 537 DEGs were uniquely upregulated and downregulated in acidic stress group, respectively (Fig. 1D). Furthermore, 31 and one DEGs were uniquely upregulated and downregulated in oxidative stress group, respectively (Fig. 1D). Subsequently, we produced volcano plots analyzing the fold changes in expression with the corresponding raw p-values (Fig. 2). Volcano plot showed differentially expressed transcripts in acidic and oxidative stress conditions compared to the untreated control (Fig. 2A and B). Classification of these 48 DEGs by their functional categories based on Mycobrowser showed that the most relevant functional categories were “information pathways” (12.5%), “insertion seqs and phages” (10.4%), “intermediary metabolism and respiration” (6.3%), and “virulence, detoxification, adaptation” (6.2%). Description of the overlapped transcripts between acidic and oxidative stress conditions were listed in Table 3.

Volcano plots of M. intracellulare transcriptome under oxidative and acidic stress conditions. A Volcano plots comparing transcriptional levels between acidic stress group compared to control group. Yellow dots indicate an expression level change of fold change ≥ 2 and raw p < 0.05. Blue dots indicate an expression level change of fold change ≤ -2 and raw p < 0.05. Gray dots indicate no significant expression level change. B Volcano plots comparing transcriptional levels between oxidative stress group compared to control group. Yellow dots indicate an expression level change of fold change ≥ 2 and raw p < 0.05. Blue dots indicate an expression level change of fold change ≤ -2 and raw p < 0.05. Gray dots indicate no significant expression level change
We observed transcriptional changes in general stress in the present study, indicating genes differentially expressed under both stress conditions. A total of 48 genes were differentially expressed under both stress conditions. Among 48 DEGs, 46 genes were upregulated, whereas two were downregulated. The upregulated genes are classified into various categories, such as [intermediary metabolism and respiration], [information pathways], [virulence, detoxification, adaptation], [insertion seqs and phages], [cell wall and cell processes], and [regulatory proteins]. Also, several genes were classified into [unknown] and [conserved hypotheticals]. The genes encoding molecular chaperones such as groEL1, groEL2, and groES were upregulated under both stress conditions. Mycobacteria have two types of molecular chaperones, such as GroEL1 and GroEL2, and a single groES gene, which is combined with groEL1 at the transcriptional level [45]. Previous studies demonstrated that GroEL1 and GroEL2 proteins are upregulated under heat shock, thiol-specific oxidative stress, and macrophage infection [46,47,48]. Furthermore, GroEL1 and GroEL2 induce cytokine production in human PBMC, suggesting that these proteins are essential virulence factors in mycobacterial infection [49]. Also, the mycobacterial GroEL1 protein is involved in biofilm formation. Ojha et al. showed that GroEL1 promotes the formation of mature biofilms by modulating mycolic acid biosynthesis [50]. M. smegmatis groEL1 deletion mutant showed normal planktonic growth but could not produce biofilm [50]. Besides GroEL proteins, GroES proteins provoke immune responses, such as T cell proliferation and immunoglobulin response in tuberculosis and leprosy [51,52,53]. In M. avium complex, the role of molecular chaperones, such as GroEL1, GroEL2, and GroES in pathogenesis remains undiscovered. The construction of genetic mutants to elucidate the role of general stress response genes is needed in further investigation.
RNA-seq data validation
We tested nine DEGs' expression levels by RT-qPCR to validate the RNA-seq data. The mRNA levels of nine DEGs such as narH, narI, narJ, narU, groEL1, groEL2, dinB, mmpS, and groES, were upregulated in the acidic stress condition, consistent with the RNA-seq data. However, their expression levels were varied among the different DEGs. For instance, expression level of respiratory nitrate reductase complex including narH, narI, narJ, and narU was increased in acidic stress condition, we only observed 2.32-to-2.78-fold upregulation in the RT-qPCR result (Fig. 3). Similarly, the mRNA levels of molecular chaperones such as groEL1, groEL2, and groES were upregulated for 3.47-to-fourfold in acidic stress condition, we only detected 2.42-to-2.47-fold upregulation in the RT-qPCR analysis (Fig. 3). The difference in sensitivity and dynamic range between RNA-seq and qRT-PCR could explain the difference of fold change between two methods.

Validation of RNA-seq data by quantitative RT-PCR. The relative gene expression level of nine DEGs in acidic stress condition was normalized to the rpoB expression level relative to control group by the 2−ΔΔCT method
KEGG enrichment analysis
In this work, we performed KEGG enrichment analysis to provide a deeper insight into the biological mechanisms of up and downregulated DEGs. The result of KEGG enrichment analysis showed that target genes were significantly enriched in pathways belonging to metabolism (36 pathways, 78.2%), genetic information processing (seven pathways, 15.2%), environmental information processing (two pathways, 4.3%), and organismal systems (one pathway, 2.2%) (Fig. 3A). In detail, acidic stress condition activated several pathways such as two-component system, ABC transporters, valine, leucine, and isoleucine degradation, nitrogen metabolism, sulfur metabolism, fatty acid degradation, tryptophan metabolism, butanoate metabolism, lysine degradation, glycerolipid metabolism, pantothenate and CoA biosynthesis, oxidative phosphorylation, arginine and proline metabolism, terpenoid backbone biosynthesis, tuberculosis, starch, and sucrose metabolism, C5-branched dibasic acid metabolism, selenocompound metabolism, biosynthesis of unsaturated fatty acids, and valine, leucine, and isoleucine biosynthesis (Fig. 4B). Furthermore, seven pathways including mismatch repair, homologous recombination, DNA replication, nucleotide excision repair, aminoacyl-tRNA biosynthesis, RNA degradation, and tuberculosis were activated under oxidative stress condition (Fig. 4C). The enriched pathways contain different number of DEGs ranging from 2 to 20. Heatmaps of DEGs belong to enriched pathways were presented in supplementary Fig. 1 and 2.

KEGG enrichment analysis of M. intracellulare transcriptome under oxidative and acidic stress conditions. A KEGG enrichment pathways that are differentially expressed in the entire experimental groups filtered by p < 0.05. B Top 20 KEGG enrichment pathways from acidic stress group compared to control group. C Top 7 KEGG enrichment pathways from oxidative stress group compared to control group. Size of circle indicates the number of significant genes and color of circle indicates p value as presented in figure
Next, we selected the key genes potentially involved in the adaptation under acidic and oxidative stress conditions from the top 20 terms of KEGG pathways. A total of 28 and 13 genes were selected from acidic and oxidative stress conditions, respectively (Table 4). Under acidic stress conditions, most of selected DEGs were associated with nitrogen and sulfur metabolism. Also, several genes that involved in potassium transport, membrane stress, and heat shock stress (Table 4). On the contrary, oxidative stress conditions induces the gene expression profiles associated with DNA replication, nucleotide excision repair processes, and liporarabinomannan biosynthesis (Table 4). Overall, these findings indicate that the stress-responsive differentially expressed genes (DEGs) in M. intracellulare play a key role in diverse metabolic processes, including nitrogen and sulfur metabolism during acidic stress, as well as involvement in DNA repair, cell wall maintenance, and remodeling when facing oxidative stress.
We observed a clear distinct activated pathways between the acidic and oxidative stress conditions. Specifically, under acidic stress conditions, nitrogen and sulfur metabolisms emerged as major pathways in the M. intracellulare transcriptome. Nitrogen metabolism is a crucial biological pathway that holds particular significance in the pathogenesis of mycobacteria, notably M. tuberculosis [54]. The adaptation of mycobacteria to host-induced stresses, such as acidic pH and nutrient deprivation within macrophages, is closely associated with nitrogen metabolism [55]. Ammonium is an important molecule in the core nitrogen metabolism of most bacteria, facilitating the biosynthesis of glutamate and glutamine, both of which are primary nitrogen donors [54]. Bacterial ammonium assimilation typically involves two pathways: a low-affinity pathway regulated by glutamate dehydrogenase (GDH) and a high-affinity pathway regulated by glutamine synthetase and glutamine oxoglutarate aminotransferase (GOGAT) [54]. In mycobacteria, the glutamine synthetase and GOGAT pathways play a significant role in ammonium assimilation, whereas the GDH pathway is primarily involved in glutamate catabolism [56, 57]. Nitrate is transported into the mycobacterial cell from the extracellular environment via the NarK2 transporter and subsequently reduced to nitrite by the nitrate reductase operon (NarGHJI) [54]. Nitrites are further converted into ammonium by the nitrite reductase enzymes NirB and NirD [54]. In our study, we observed a significant upregulation of narGHJI in the M. intracellulare transcriptome, along with increased expression of the nitrite reductase enzymes (NirB and NirD) under acidic stress conditions (depicted in Fig. 5).

Transcriptomic pathways in M. intracellulare that exhibit differential expression in response to acidic stress. A The nitrogen metabolism pathways in M. intracellulare are presented here and show differential expression under acidic stress. We have depicted gene expression associated with nitrogen metabolism, with the colors of gene IDs indicating their expression levels as described in the figure. The gene expression levels of differentially expressed genes identified under acidic stress were compared with the corresponding transcripts detected under untreated control growth conditions. B The sulfur metabolism pathways in M. intracellulare are presented here and show differential expression under acidic stress. We have depicted gene expression associated with nitrogen metabolism, with the colors of gene IDs indicating their expression levels as described in the figure. The gene expression levels of differentially expressed genes identified under acidic stress were compared with the corresponding transcripts detected under untreated control growth conditions
Previous research has highlighted the close relationship between the nitrate reductase system and the evolutionary success of "modern" M. tuberculosis lineages, which exhibit enhanced virulence and infectivity compared to other M. tuberculosis complex species [58]. Furthermore, the multimeric nitrate reductase complex NarGHIJ expression was significantly upregulated in human lung granulomas derived from tuberculosis patients [59]. Importantly, nitrate respiration facilitated by NarGHJI results in the accumulation of nitrite, which can exert a toxic effect on bacterial cells under acidic pH conditions due to its conversion into nitric oxide, thereby exhibiting antimicrobial activity [60].
Notably, advantage of nitrate and nitrite reductase systems is their capacity to produce ammonium, serving as a buffer in acidic environments. We showed that M. intracellulare upregulates NirBD nitrite reductase and nitrite export proteins, such as NarK3 and NarU, under acidic stress conditions (See Fig. 4). Similar findings were observed in the hypoxic culture of M. tuberculosis. Akhtar et al. showed that the expression of NirBD was significantly upregulated at both the transcriptional and protein levels in a THP-1 cell-based in vitro dormancy model [61]. Furthermore, Malm et al. provided evidence that NarGHJI and NirBD in M. tuberculosis facilitate the assimilatory reduction of nitrate and nitrite, respectively, with GlnR as a transcriptional modulator for NirBD [62]. The upregulation of NirBD confers several advantages to mycobacteria, including the provision of ammonium for pH regulation in acidic environments and the reduction of nitric oxide toxicity. Further investigations are warranted to elucidate the role of nitrate and nitrite reductase systems in MAC, with a focus on intracellular survival mechanisms.
Previous studies suggest that sulfur metabolism and sulfur-containing metabolites play a pivotal role in the pathogenesis of mycobacteria [63, 64]. Sulfur-containing metabolites derived from mycobacteria influence bacterial infectivity and pathogenicity [65,66,67]. One unique major component of the cell wall, glycolipid Sulfolipid-1, induces the expression of cytokines in human tuberculosis patients [66]. Additionally, reduced sulfur-containing metabolites, such as cysteine, methionine, and coenzyme A, participate in the synthesis of essential biomolecules like proteins, lipids, and mycothiol [63]. Among these molecules, coenzyme A is a critical element in lipid metabolism, responsible for maintaining and modifying mycobacterial cell walls [68]. Also, mycothiol serves as an intracellular reducing agent that regulates cellular redox status, providing protection to bacterial cells by detoxifying electrophilic compounds, reactive oxygen and nitrogen species, as well as antibiotics [69]. In contrast, the sulfate molecule of M. tuberculosis, menaquinone S881, exerts a negative regulatory effect on bacterial virulence in mouse models [67]. The sulfate assimilation pathways are responsible for biosynthesis of these sulfur-containing metabolites in M. tuberculosis.
The bacterial sulfate assimilation pathway involves a series of enzymatic reactions responsible for the uptake and processing of inorganic sulfate from the host (see Fig. 5). In Mycobacterium intracellulare, this pathway initiates with the active transport of extracellular sulfate, followed by its conversion into adenosine 5′-phosphosulfate (APS) through the catalytic activity of ATP sulfurylase (as shown in Fig. 5). APS can be further phosphorylated by APS kinase, resulting in the production of 3′-phosphoadenosine 5′-phosphosulfate (PAPS), which serves as the universal sulfate donor within the bacterial cell. PAPS can be subsequently converted into sulfite by PAPS reductase and then serves as a substrate for various enzymes, including formate dehydrogenase and sulfite reductases (depicted in Fig. 5). These collective reactions constitute the sulfation branch of the sulfate assimilation pathway in M. intracellulare.
In the present study, we observed a significant upregulation of genes associated with sulfur metabolism. The expression of sulfate adenylyltransferase subunit 1 and 2 (OCU_15790 and 15,800) was significantly upregulated in acidic stress condition (see Fig. 4). Also, significant upregulation of PAPS reductase (OCU_18650) was observed. Furthermore, several DEGs involved in assimilatory sulfate reduction pathway including formate dyhydrogenase (OCU_20490), ferredoxin 1 (OCU_21500), and ferredoxin 2 (OCU_18660) were upregulated under acidic stress. The orthologue of sulfate adenylyltransferase subunit 1 and 2 from M. tuberculosis was upregulated within the macrophages and in vitro stationary phase growth [70,71,72]. Upregulation of the assimilatory sulfate reduction pathway indicates the accumulation of sulfide under acidic stress conditions.
Sulfide serves as a substrate for biosynthesis of cysteine and then cysteine can be converted into hydrogen sulfide (H2S) via various enzymes [73]. In mammalian cells, three type of H2S producing enzymes have been identified as follows: cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfurtransferase [73]. The presence of homologues for CBS and CSE in M. intracellulare genome suggest that M. intracellulare has the capacity to generate H2S. Although H2S, initially thought to be an intermediate metabolite of sulfur metabolism produced by bacteria, previous studies have reported that H2S plays important physiological roles, such as modulating the host immune response and maintaining redox homeostasis in many bacterial species [64, 74, 75]. Therefore, H2S can influence the intra- and extracellular environments of bacterial pathogens during infection, potentially favoring persistence of pathogen. The transcriptional response to acid stress may not be directly linked to oxidative stress. Nevertheless, when exposed to low pH, genes responsible for oxidative stress have been observed to undergo significant upregulation [76]. It is plausible that a low pH disrupts the electron transfer chain, leading to the generation of superoxide [77]. Superoxide, in turn, can initiate the formation of other reactive oxygen species, potentially inducing oxidative stress [77]. Consequently, the upregulation of assimilatory sulfate reduction pathways under acidic stress suggests the promotion of resistance to secondary oxidative stress through H2S production. Further studies are required to clarify the role of H2S in MAC pathogenesis, with a focus on modulating host immune responses and metabolism.
Under oxidative stress conditions, seven KEGG pathways were significantly activated. Among the activated pathways, all of them belonged to the genetic information processing category, except for the "tuberculosis" pathway (Fig. 4). Specifically, DNA replication associated genes such as OCU_00740 (replicative DNA helicase, DnaB), OCU_03510 (DNA polymerase III subunit epsilon, dinG), OCU_22700 (hypothetical protein), and OCU_04360 (DNA-directed DNA polymerase III subunit delta) were upregulated. Also, DNA repair associated genes, including OCU_41260 (endonuclease), OCU_12600 (DNA-3-methyladenine glycosylase I), OCU_29740 (excinuclease ABC subunit B, uvrB), OCU_39940 (ATP-dependent DNA helicase, uvrD2), OCU_39980 (Probable ATP-dependent DNA helicase), OCU_34590 (recombinase A, recA), OCU_04800 (DNA repair protein RadA), and OCU_33030 (holliday junction resolvase) were upregulated (Fig. 6).

Transcriptomic pathways in M. intracellulare that exhibit differential expression in response to oxidative stress. Several pathways such as DNA replication A, Base excision repair B, Nucleotide excision repair C, and Mismatch repair D in M. intracellulare are presented here and show differential expression under acidic stress. We have depicted gene expression associated with nitrogen metabolism, with the colors of gene IDs indicating their expression levels as described in the figure. The gene expression levels of differentially expressed genes identified under acidic stress were compared with the corresponding transcripts detected under untreated control growth conditions
Several DNA repair systems that counteract oxidative-induced mutagenesis have been described in various bacterial species, including M. tuberculosis. Kelley et al. revealed that DnaB helicase plays a crucial role in the transition to a dormant state in M. smegmatis under oxidative stress by modulating intein splicing [78]. Furthermore, DinG unwinds G4 DNA structures, which are frequently present in the mycobacterial genome and play a critical role in the regulation of mycobacterial gene expression [79]. In that regard, targeting G4 DNA structures with G4-DNA specific ligands can be a therapeutic strategy for MAC infection. UvrB and UvrC are key components of the nucleotide excision repair system in bacteria and are associated with bacterial virulence. Previous studies have shown that nucleotide excision repair genes modulate mycobacterial survival within the host. Oxidative stress results in significant upregulation of numerous DNA repair system genes such as recA, dinB, uvrB, lexA, radA, and helicase in M. smegmatis, suggesting their universal role for stress response in mycobacteria [80]. The expression of several uvr genes, including uvrB, was upregulated in M. tuberculosis within human macrophages [81]. Moreover, Darwin and Nathan provided evidence that the M. tuberculosis uvrB mutant exhibited a significant reduction in bacterial load within bone marrow macrophages and mouse models [82]. The recombinase A, encoded by recA gene contributes to M. tuberculosis survival by suppressing of the mitogen-activated protein kinase activity in THP-1-derived macrophages in vitro [83]. In addition, the recA deletion mutant of M. bovis BCG was more susceptible to DNA damage but showed a similar bacterial load in the BALB/c mouse model [84].
Conclusion
In conclusion, we provide detailed insights into the transcriptomic stress response of M. intracellulare strain S1-36 under oxidative and acid stress conditions. Notably, exposure to acidic stress resulted in prominent changes in the transcriptome. Our results provide evidence for the importance of nitrogen and sulfur metabolism genes in the acidic stress response, including narGHIJ, nirBD, narU, narK3, cysND, cysC, cysH, ferredoxin 1 and 2, and formate dehydrogenase. Additionally, our results demonstrate that DNA replication and repair system genes, such as dnaB, dinG, urvB, uvrD2, radA, and recA, are indispensable for resistance to oxidative stress. Further reverse-genetics approaches, including gene silencing, targeted gene disruption, and transposon-mediated mutagenesis, are required to validate our predictions based on RNA-seq.
Availability of data and materials
All raw RNA-seq reads and processed files of transcriptome sequencing analyzed in this study are available in NCBI with the GEO accession number GSE244264 under the project number PRJNA1021764.
References
Koh WJ. Nontuberculous Mycobacteria-Overview. Microbiol Spectr. 2017;5(1). https://doi.org/10.1128/microbiolspec.TNMI7-0024-2016.
Dhillon SS, Watanakunakorn C. Lady Windermere syndrome: middle lobe bronchiectasis and Mycobacterium avium complex infection due to voluntary cough suppression. Clin Infect Dis. 2000;30(3):572–5. https://doi.org/10.1086/313726.
Wallace JM, Hannah JB. Mycobacterium avium complex infection in patients with the acquired immunodeficiency syndrome. A clinicopathologic study. Chest. 1988;93(5):926–32. https://doi.org/10.1378/chest.93.5.926.
Zoumot Z, Boutou AK, Gill SS, van Zeller M, Hansell DM, Wells AU, et al. Mycobacterium avium complex infection in non-cystic fibrosis bronchiectasis. Respirology. 2014;19(5):714–22. https://doi.org/10.1111/resp.12287.
Blakney RA, Ricotta EE, Frankland TB, Honda S, Zelazny A, Mayer-Barber KD, et al. Incidence of Nontuberculous Mycobacterial Pulmonary Infection, by Ethnic Group, Hawaii, USA, 2005–2019. Emerg Infect Dis. 2022;28(8):1543–50. https://doi.org/10.3201/eid2808.212375.
Dahl VN, Molhave M, Floe A, van Ingen J, Schon T, Lillebaek T, et al. Global trends of pulmonary infections with nontuberculous mycobacteria: a systematic review. Int J Infect Dis. 2022;125:120–31. https://doi.org/10.1016/j.ijid.2022.10.013.
Lee H, Myung W, Koh WJ, Moon SM, Jhun BW. Epidemiology of Nontuberculous Mycobacterial Infection, South Korea, 2007–2016. Emerg Infect Dis. 2019;25(3):569–72. https://doi.org/10.3201/eid2503.181597.
Park SC, Kang MJ, Han CH, Lee SM, Kim CJ, Lee JM, et al. Prevalence, incidence, and mortality of nontuberculous mycobacterial infection in Korea: a nationwide population-based study. BMC Pulm Med. 2019;19(1):140. https://doi.org/10.1186/s12890-019-0901-z.
Daley CL. Mycobacterium avium Complex Disease. Microbiol Spectr. 2017;5(2). https://doi.org/10.1128/microbiolspec.TNMI7-0045-2017.
Lu Y, Chen H, Wang P, Pang J, Lu X, Li G, et al. Identification and Quantification of S-Sulfenylation Proteome of Mycobacterium tuberculosis under Oxidative Stress. Microbiol Spectr. 2023;11(2):e0338622. https://doi.org/10.1128/spectrum.03386-22.
Martini BA, Grigorov AS, Skvortsova YV, Bychenko OS, Salina EG, Azhikina TL. Small RNA MTS1338 Configures a Stress Resistance Signature in Mycobacterium tuberculosis. Int J Mol Sci. 2023;24(9):7928. https://doi.org/10.3390/ijms24097928.
Schwarz MGA, Correa PR, Almeida PSL, Mendonca-Lima L. Mycobacterium bovis BCG dodecin gene codes a functional protein despite of a start codon mutation. Tuberculosis (Edinb). 2023;143:102400. https://doi.org/10.1016/j.tube.2023.102400.
Yimcharoen M, Saikaew S, Wattananandkul U, Phunpae P, Intorasoot S, Tayapiwatana C, et al. Mycobacterium tuberculosis Adaptation in Response to Isoniazid Treatment in a Multi-Stress System That Mimics the Host Environment. Antibiotics (Basel). 2023;12(5):852. https://doi.org/10.3390/antibiotics12050852.
Ezraty B, Gennaris A, Barras F, Collet JF. Oxidative stress, protein damage and repair in bacteria. Nat Rev Microbiol. 2017;15(7):385–96. https://doi.org/10.1038/nrmicro.2017.26.
Huang L, Nazarova EV, Russell DG. Mycobacterium tuberculosis: Bacterial Fitness within the Host Macrophage. Microbiol Spectr. 2019;7(2). https://doi.org/10.1128/microbiolspec.BAI-0001-2019.
Lopez-Perez W, Sai K, Sakamachi Y, Parsons C, Kathariou S, Ninomiya-Tsuji J. TAK1 inhibition elicits mitochondrial ROS to block intracellular bacterial colonization. Proc Natl Acad Sci U S A. 2021;118(25):e2023647118. https://doi.org/10.1073/pnas.2023647118.
Tur J, Pereira-Lopes S, Vico T, Marin EA, Munoz JP, Hernandez-Alvarez M, et al. Mitofusin 2 in Macrophages Links Mitochondrial ROS Production, Cytokine Release, Phagocytosis, Autophagy, and Bactericidal Activity. Cell Rep. 2020;32(8):108079. https://doi.org/10.1016/j.celrep.2020.108079.
Arumugam P, Shankaran D, Bothra A, Gandotra S, Rao V. The MmpS6-MmpL6 Operon Is an Oxidative Stress Response System Providing Selective Advantage to Mycobacterium tuberculosis in Stress. J Infect Dis. 2019;219(3):459–69. https://doi.org/10.1093/infdis/jiy526.
Rai R, Singh V, Mathew BJ, Singh AK, Chaurasiya SK. Mycobacterial response to an acidic environment: protective mechanisms. Pathog Dis. 2022;80(1):ftac032. https://doi.org/10.1093/femspd/ftac032.
Zulauf KE, Sullivan JT, Braunstein M. The SecA2 pathway of Mycobacterium tuberculosis exports effectors that work in concert to arrest phagosome and autophagosome maturation. PLoS Pathog. 2018;14(4):e1007011. https://doi.org/10.1371/journal.ppat.1007011.
Sherman DR, Sabo PJ, Hickey MJ, Arain TM, Mahairas GG, Yuan Y, et al. Disparate responses to oxidative stress in saprophytic and pathogenic mycobacteria. Proc Natl Acad Sci U S A. 1995;92(14):6625–9. https://doi.org/10.1073/pnas.92.14.6625.
Deretic V, Song J, Pagan-Ramos E. Loss of oxyR in Mycobacterium tuberculosis. Trends Microbiol. 1997;5(9):367–72. https://doi.org/10.1016/S0966-842X(97)01112-8.
Nambi S, Long JE, Mishra BB, Baker R, Murphy KC, Olive AJ, et al. The Oxidative Stress Network of Mycobacterium tuberculosis Reveals Coordination between Radical Detoxification Systems. Cell Host Microbe. 2015;17(6):829–37. https://doi.org/10.1016/j.chom.2015.05.008.
Tzou CL, Dirac MA, Becker AL, Beck NK, Weigel KM, Meschke JS, et al. Association between Mycobacterium avium Complex Pulmonary Disease and Mycobacteria in Home Water and Soil. Ann Am Thorac Soc. 2020;17(1):57–62. https://doi.org/10.1513/AnnalsATS.201812-915OC.
Reed C, von Reyn CF, Chamblee S, Ellerbrock TV, Johnson JW, Marsh BJ, et al. Environmental risk factors for infection with Mycobacterium avium complex. Am J Epidemiol. 2006;164(1):32–40. https://doi.org/10.1093/aje/kwj159.
De Groote MA, Pace NR, Fulton K, Falkinham JO 3rd. Relationships between Mycobacterium isolates from patients with pulmonary mycobacterial infection and potting soils. Appl Environ Microbiol. 2006;72(12):7602–6. https://doi.org/10.1128/AEM.00930-06.
Choi JY, Sim BR, Park Y, Yong SH, Shin SJ, Kang YA. Identification of nontuberculous mycobacteria isolated from household showerheads of patients with nontuberculous mycobacteria. Sci Rep. 2022;12(1):8648. https://doi.org/10.1038/s41598-022-12703-6.
SJ Kim and HS Yoo. Whole genome sequencing of Mycobacterium intracellulare isolated from South Korea. https://www.ncbi.nlm.nih.gov/nuccore/CP076381 . 2022. Accessed.
SJ Kim and HS Yoo. Whole genome sequencing of Mycobacterium intracellulare ATCC 13950. https://www.ncbi.nlm.nih.gov/nuccore/CP076382 . 2022. Accessed.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. https://doi.org/10.1073/pnas.0506580102.
Alebouyeh S, Cardenas-Pestana JA, Vazquez L, Prados-Rosales R, Del Portillo P, Sanz J, et al. Iron deprivation enhances transcriptional responses to in vitro growth arrest of Mycobacterium tuberculosis. Front Microbiol. 2022;13:956602. https://doi.org/10.3389/fmicb.2022.956602.
Barrientos OM, Langley E, Gonzalez Y, Cabello C, Torres M, Guzman-Beltran S. Mycobacterium tuberculosis whiB3 and Lipid Metabolism Genes Are Regulated by Host Induced Oxidative Stress. Microorganisms. 2022;10(9):1821. https://doi.org/10.3390/microorganisms10091821.
Birhanu AG, Gomez-Munoz M, Kalayou S, Riaz T, Lutter T, Yimer SA, et al. Proteome Profiling of Mycobacterium tuberculosis Cells Exposed to Nitrosative Stress. ACS Omega. 2022;7(4):3470–82. https://doi.org/10.1021/acsomega.1c05923.
Del Portillo P, Garcia-Morales L, Menendez MC, Anzola JM, Rodriguez JG, Helguera-Repetto AC, et al. Hypoxia Is Not a Main Stress When Mycobacterium tuberculosis Is in a Dormancy-Like Long-Chain Fatty Acid Environment. Front Cell Infect Microbiol. 2018;8:449. https://doi.org/10.3389/fcimb.2018.00449.
Dow A, Sule P, O’Donnell TJ, Burger A, Mattila JT, Antonio B, et al. Zinc limitation triggers anticipatory adaptations in Mycobacterium tuberculosis. PLoS Pathog. 2021;17(5):e1009570. https://doi.org/10.1371/journal.ppat.1009570.
Martini MC, Zhou Y, Sun H, Shell SS. Defining the Transcriptional and Post-transcriptional Landscapes of Mycobacterium smegmatis in Aerobic Growth and Hypoxia. Front Microbiol. 2019;10:591. https://doi.org/10.3389/fmicb.2019.00591.
Singh S, Goswami N, Tyagi AK, Khare G. Unraveling the role of the transcriptional regulator VirS in low pH-induced responses of Mycobacterium tuberculosis and identification of VirS inhibitors. J Biol Chem. 2019;294(26):10055–75. https://doi.org/10.1074/jbc.RA118.005312.
Tiwari S, van Tonder AJ, Vilcheze C, Mendes V, Thomas SE, Malek A, et al. Arginine-deprivation-induced oxidative damage sterilizes Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2018;115(39):9779–84. https://doi.org/10.1073/pnas.1808874115.
Vilcheze C, Yan B, Casey R, Hingley-Wilson S, Ettwiller L, Jacobs WR Jr. Commonalities of Mycobacterium tuberculosis Transcriptomes in Response to Defined Persisting Macrophage Stresses. Front Immunol. 2022;13:909904. https://doi.org/10.3389/fimmu.2022.909904.
Shamaei M, Mirsaeidi M. Nontuberculous Mycobacteria, Macrophages, and Host Innate Immune Response. Infect Immun. 2021;89(8):e0081220. https://doi.org/10.1128/IAI.00812-20.
Upadhyay S, Mittal E, Philips JA. Tuberculosis and the art of macrophage manipulation. Pathog Dis. 2018;76(4):fty037. https://doi.org/10.1093/femspd/fty037.
Chai Q, Wang L, Liu CH, Ge B. New insights into the evasion of host innate immunity by Mycobacterium tuberculosis. Cell Mol Immunol. 2020;17(9):901–13. https://doi.org/10.1038/s41423-020-0502-z.
Weiss G, Schaible UE. Macrophage defense mechanisms against intracellular bacteria. Immunol Rev. 2015;264(1):182–203. https://doi.org/10.1111/imr.12266.
Prasla Z, Sutliff RL, Sadikot RT. Macrophage Signaling Pathways in Pulmonary Nontuberculous Mycobacteria Infections. Am J Respir Cell Mol Biol. 2020;63(2):144–51. https://doi.org/10.1165/rcmb.2019-0241TR.
Kong TH, Coates AR, Butcher PD, Hickman CJ, Shinnick TM. Mycobacterium tuberculosis expresses two chaperonin-60 homologs. Proc Natl Acad Sci U S A. 1993;90(7):2608–12. https://doi.org/10.1073/pnas.90.7.2608.
Dosanjh NS, Rawat M, Chung JH, Av-Gay Y. Thiol specific oxidative stress response in Mycobacteria. FEMS Microbiol Lett. 2005;249(1):87–94. https://doi.org/10.1016/j.femsle.2005.06.004.
Monahan IM, Betts J, Banerjee DK, Butcher PD. Differential expression of mycobacterial proteins following phagocytosis by macrophages. Microbiology (Reading). 2001;147(Pt 2):459–71. https://doi.org/10.1099/00221287-147-2-459.
Stewart GR, Wernisch L, Stabler R, Mangan JA, Hinds J, Laing KG, et al. Dissection of the heat-shock response in Mycobacterium tuberculosis using mutants and microarrays. Microbiology (Reading). 2002;148(Pt 10):3129–38. https://doi.org/10.1099/00221287-148-10-3129.
Lewthwaite JC, Coates AR, Tormay P, Singh M, Mascagni P, Poole S, et al. Mycobacterium tuberculosis chaperonin 60.1 is a more potent cytokine stimulator than chaperonin 60.2 (Hsp 65) and contains a CD14-binding domain. Infect Immun. 2001;69(12):7349–55. https://doi.org/10.1128/IAI.69.12.7349-7355.2001.
Ojha A, Anand M, Bhatt A, Kremer L, Jacobs WR Jr, Hatfull GF. GroEL1: a dedicated chaperone involved in mycolic acid biosynthesis during biofilm formation in mycobacteria. Cell. 2005;123(5):861–73. https://doi.org/10.1016/j.cell.2005.09.012.
Barnes PF, Mehra V, Rivoire B, Fong SJ, Brennan PJ, Voegtline MS, et al. Immunoreactivity of a 10-kDa antigen of Mycobacterium tuberculosis. J Immunol. 1992;148(6):1835–40.
Mehra V, Bloom BR, Bajardi AC, Grisso CL, Sieling PA, Alland D, et al. A major T cell antigen of Mycobacterium leprae is a 10-kD heat-shock cognate protein. J Exp Med. 1992;175(1):275–84. https://doi.org/10.1084/jem.175.1.275.
Rojas RE, Segal-Eiras A. Immunoglobulin G response against 10-kDa and 65-kDa heat-shock proteins in leprosy patients and their household contacts. FEMS Immunol Med Microbiol. 1996;15(4):189–98. https://doi.org/10.1111/j.1574-695X.1996.tb00085.x.
Gouzy A, Poquet Y, Neyrolles O. Nitrogen metabolism in Mycobacterium tuberculosis physiology and virulence. Nat Rev Microbiol. 2014;12(11):729–37. https://doi.org/10.1038/nrmicro3349.
Borah K, Beyss M, Theorell A, Wu H, Basu P, Mendum TA, et al. Intracellular Mycobacterium tuberculosis Exploits Multiple Host Nitrogen Sources during Growth in Human Macrophages. Cell Rep. 2019;29(11):3580–91 e4. https://doi.org/10.1016/j.celrep.2019.11.037.
Amon J, Titgemeyer F, Burkovski A. A genomic view on nitrogen metabolism and nitrogen control in mycobacteria. J Mol Microbiol Biotechnol. 2009;17(1):20–9. https://doi.org/10.1159/000159195.
Viljoen AJ, Kirsten CJ, Baker B, van Helden PD, Wiid IJ. The role of glutamine oxoglutarate aminotransferase and glutamate dehydrogenase in nitrogen metabolism in Mycobacterium bovis BCG. PLoS One. 2013;8(12):e84452. https://doi.org/10.1371/journal.pone.0084452.
Goh KS, Rastogi N, Berchel M, Huard RC, Sola C. Molecular evolutionary history of tubercle bacilli assessed by study of the polymorphic nucleotide within the nitrate reductase (narGHJI) operon promoter. J Clin Microbiol. 2005;43(8):4010–4. https://doi.org/10.1128/JCM.43.8.4010-4014.2005.
Fenhalls G, Stevens L, Moses L, Bezuidenhout J, Betts JC, Helden Pv P, et al. In situ detection of Mycobacterium tuberculosis transcripts in human lung granulomas reveals differential gene expression in necrotic lesions. Infect Immun. 2002;70(11):6330–8. https://doi.org/10.1128/IAI.70.11.6330-6338.2002.
Yoon SS, Coakley R, Lau GW, Lymar SV, Gaston B, Karabulut AC, et al. Anaerobic killing of mucoid Pseudomonas aeruginosa by acidified nitrite derivatives under cystic fibrosis airway conditions. J Clin Invest. 2006;116(2):436–46. https://doi.org/10.1172/JCI24684.
Akhtar S, Khan A, Sohaskey CD, Jagannath C, Sarkar D. Nitrite reductase NirBD is induced and plays an important role during in vitro dormancy of Mycobacterium tuberculosis. J Bacteriol. 2013;195(20):4592–9. https://doi.org/10.1128/JB.00698-13.
Malm S, Tiffert Y, Micklinghoff J, Schultze S, Joost I, Weber I, et al. The roles of the nitrate reductase NarGHJI, the nitrite reductase NirBD and the response regulator GlnR in nitrate assimilation of Mycobacterium tuberculosis. Microbiology (Reading). 2009;155(Pt 4):1332–9. https://doi.org/10.1099/mic.0.023275-0.
Bhave DP, Muse WB 3rd, Carroll KS. Drug targets in mycobacterial sulfur metabolism. Infect Disord Drug Targets. 2007;7(2):140–58. https://doi.org/10.2174/187152607781001772.
Hatzios SK, Bertozzi CR. The regulation of sulfur metabolism in Mycobacterium tuberculosis. PLoS Pathog. 2011;7(7):e1002036. https://doi.org/10.1371/journal.ppat.1002036.
Gangadharam PR, Cohn ML, Middlebrook G. Infectivity, Pathogenicity and sulpholipid fraction of some Indian and British strains of tubercle bacilli. Tubercle. 1963;44:452–5. https://doi.org/10.1016/s0041-3879(63)80087-2.
Gilleron M, Stenger S, Mazorra Z, Wittke F, Mariotti S, Bohmer G, et al. Diacylated sulfoglycolipids are novel mycobacterial antigens stimulating CD1-restricted T cells during infection with Mycobacterium tuberculosis. J Exp Med. 2004;199(5):649–59. https://doi.org/10.1084/jem.20031097.
Mougous JD, Senaratne RH, Petzold CJ, Jain M, Lee DH, Schelle MW, et al. A sulfated metabolite produced by stf3 negatively regulates the virulence of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2006;103(11):4258–63. https://doi.org/10.1073/pnas.0510861103.
Takayama K, Wang C, Besra GS. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin Microbiol Rev. 2005;18(1):81–101. https://doi.org/10.1128/CMR.18.1.81-101.2005.
Newton GL, Buchmeier N, Fahey RC. Biosynthesis and functions of mycothiol, the unique protective thiol of Actinobacteria. Microbiol Mol Biol Rev. 2008;72(3):471–94. https://doi.org/10.1128/MMBR.00008-08.
Hampshire T, Soneji S, Bacon J, James BW, Hinds J, Laing K, et al. Stationary phase gene expression of Mycobacterium tuberculosis following a progressive nutrient depletion: a model for persistent organisms? Tuberculosis (Edinb). 2004;84(3–4):228–38. https://doi.org/10.1016/j.tube.2003.12.010.
Pinto R, Tang QX, Britton WJ, Leyh TS, Triccas JA. The Mycobacterium tuberculosis cysD and cysNC genes form a stress-induced operon that encodes a tri-functional sulfate-activating complex. Microbiology (Reading). 2004;150(Pt 6):1681–6. https://doi.org/10.1099/mic.0.26894-0.
Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, et al. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J Exp Med. 2003;198(5):693–704. https://doi.org/10.1084/jem.20030846.
Kies PJ, Hammer ND. A Resourceful Race: Bacterial Scavenging of Host Sulfur Metabolism during Colonization. Infect Immun. 2022;90(5):e0057921. https://doi.org/10.1128/iai.00579-21.
Shatalin K, Nuthanakanti A, Kaushik A, Shishov D, Peselis A, Shamovsky I, et al. Inhibitors of bacterial H(2)S biogenesis targeting antibiotic resistance and tolerance. Science. 2021;372(6547):11369–1175. https://doi.org/10.1126/science.abd8377.
Shatalin K, Shatalina E, Mironov A, Nudler E. H2S: a universal defense against antibiotics in bacteria. Science. 2011;334(6058):986–90. https://doi.org/10.1126/science.1209855.
Mols M, Abee T. Primary and secondary oxidative stress in Bacillus. Environ Microbiol. 2011;13(6):1387–94. https://doi.org/10.1111/j.1462-2920.2011.02433.x.
Mols M, Abee T. Bacillus cereus responses to acid stress. Environ Microbiol. 2011;13(11):2835–43. https://doi.org/10.1111/j.1462-2920.2011.02490.x.
Kelley DS, Lennon CW, Li Z, Miller MR, Banavali NK, Li H, et al. Mycobacterial DnaB helicase intein as oxidative stress sensor. Nat Commun. 2018;9(1):4363. https://doi.org/10.1038/s41467-018-06554-x.
Thakur RS, Desingu A, Basavaraju S, Subramanya S, Rao DN, Nagaraju G. Mycobacterium tuberculosis DinG is a structure-specific helicase that unwinds G4 DNA: implications for targeting G4 DNA as a novel therapeutic approach. J Biol Chem. 2014;289(36):25112–36. https://doi.org/10.1074/jbc.M114.563569.
Li X, Wu J, Han J, Hu Y, Mi K. Distinct responses of Mycobacterium smegmatis to exposure to low and high levels of hydrogen peroxide. PLoS One. 2015;10(7):e0134595. https://doi.org/10.1371/journal.pone.0134595.
Graham JE, Clark-Curtiss JE. Identification of Mycobacterium tuberculosis RNAs synthesized in response to phagocytosis by human macrophages by selective capture of transcribed sequences (SCOTS). Proc Natl Acad Sci U S A. 1999;96(20):11554–9. https://doi.org/10.1073/pnas.96.20.11554.
Darwin KH, Nathan CF. Role for nucleotide excision repair in virulence of Mycobacterium tuberculosis. Infect Immun. 2005;73(8):4581–7. https://doi.org/10.1128/IAI.73.8.4581-4587.2005.
Szulc-Kielbik I, Brzezinska M, Kielbik M, Brzostek A, Dziadek J, Kania K, et al. Mycobacterium tuberculosis RecA is indispensable for inhibition of the mitogen-activated protein kinase-dependent bactericidal activity of THP-1-derived macrophages in vitro. FEBS J. 2015;282(7):1289–306. https://doi.org/10.1111/febs.13219.
Sander P, Papavinasasundaram KG, Dick T, Stavropoulos E, Ellrott K, Springer B, et al. Mycobacterium bovis BCG recA deletion mutant shows increased susceptibility to DNA-damaging agents but wild-type survival in a mouse infection model. Infect Immun. 2001;69(6):3562–8. https://doi.org/10.1128/IAI.69.6.3562-3568.2001.
Acknowledgements
We are grateful to the Department of Infectious Diseases, College of Veterinary Science, Seoul National University for providing valuable bacterial sample (Mycobacteria intracellulare S1-36 strain).
Funding
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2021R1I1A2045131) and the Ministry of Science and ICT (NRF- 2021R1C1C2012177).
Author information
Authors and Affiliations
Contributions
Hyun-Eui Park: Writing – original draft, review & editing, Visualization, Investigation, Formal analysis. Jeong-Ih Shin, Kyu-Min Kim, Jeong-Gyu Choi, Won-Jun An, Minh Phuong Trinh, and Kyeong-Min Kang: Investigation, Formal analysis. Jung-Wan Yoo, Jung-Hyun Byun, Kon-Ho Lee, Myung Hwan Jung, Hyung-Lyun Kang, Seung Cheol Baik, and Woo-Kon Lee: Writing – review & editing, Resources. Min-Kyoung Shin: Writing – original draft, Supervision, Resources, Project administration, Funding acquisition, Conceptualization.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary Figure 1 and 2. Heatmaps of differentially expressed genes belong to the top 20 and 7 enriched KEGG pathways under acidic and oxidative stress conditions revealed by transcriptome profiling.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Park, HE., Kim, KM., Shin, JI. et al. Prominent transcriptomic changes in Mycobacterium intracellulare under acidic and oxidative stress. BMC Genomics 25, 376 (2024). https://doi.org/10.1186/s12864-024-10292-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10292-4